Larissa Tetsch, Maisach
Indikationsgebiet
Die spinale Muskelatrophie (SMA) ist eine vererbbare, fortschreitende Muskelschwäche, die zu einem allmählichen Verlust der Bewegungsfähigkeit führt und mit einer meist deutlich reduzierten Lebenserwartung einhergeht. Ursächlich ist ein Verlust beider Kopien des Gens „Survival of motor neuron 1“ (SMN1), der in 95 % der Fälle durch eine homozygote Deletion des langen Arms von Chromosom 5 (5q) zustande kommt. Der Mangel an SMN1-Protein führt zu einer Degeneration der Motoneurone, die die Muskelfasern ansteuern.
Die häufigsten Ausprägungsformen der SMA sind eine früh einsetzende, infantile Form, bei der die ersten Symptome innerhalb der ersten Lebensmonate auftreten (Typ I), und eine spät einsetzende Form, bei der die Kinder zunächst zumindest frei sitzen können (Typ II) oder sogar laufen lernen (Typ III). Die motorischen Fähigkeiten gehen bei den spät einsetzenden Formen jedoch mit fortschreitender Krankheit wieder verloren. Die Lebenserwartung bei der vorherrschenden infantilen Form beträgt meist nur wenige Jahre.
Pharmakologie
Pharmakodynamik (Wirkungsmechanismus)
Risdiplam ist ein niedermolekularer Arzneistoff („small molecule“), der als Spleißmodifikator der Prä-mRNA des SMN2-Gens wirkt. Neben den beiden Genkopien von SMN1 besitzen die meisten Menschen mehrere Genkopien von SMN2, das sich von SMN1 nur in einem einzigen Nukleotid unterscheidet. Dieser einzelne Basenaustausch verändert jedoch das Spleißmuster der Prä-mRNA: In der reifen mRNA fehlt Exon 7, sodass kein funktionelles SMN2-Protein mehr entstehen kann. Risdiplam korrigiert das Spleißmuster derart, dass SMN2-Proteine gebildet werden, die die Funktion des fehlenden SMN1-Proteins zumindest teilweise kompensieren können. Damit ähnelt der Wirkungsmechanismus dem bereits seit 2017 zur Behandlung der SMA zugelassenen Wirkstoffs Nusinersen. Risdiplam hat jedoch den Vorteil, dass es nicht wie Nusinersen intrathekal appliziert werden muss, sondern oral verabreicht werden kann. Vor allem bei Patienten mit häufigen Begleiterscheinungen der SMA wie Skoliose oder Atembeschwerden ist die intrathekale Applikation oft schwierig. In klinischen Studien führte Risdiplam zu einer Verdopplung der Menge an SMN-Protein im Blut nach vier Wochen Behandlungszeit. Der Effekt blieb über die gesamte Behandlungszeit von mindestens zwölf Monaten bestehen.
Pharmakokinetik
Die Pharmakokinetik von Risdiplam war zwischen 0,6 und 18 mg annähernd dosislinear, wobei Körpergewicht und Alter der Probanden einen signifikanten Einfluss ausübten. Die geschätzte Exposition (mittlere AUC0–24h) bei Patienten mit infantiler SMA und einem Alter von zwei bis sieben Monaten betrug bei einer therapeutischen Dosis von 0,2 mg/kg 1930 ng × h/ml, bei Patienten mit spät einsetzender SMA (Alter 2 bis 25 Jahre) bei einer therapeutischen Dosis von 0,25 oder 0,5 mg/kg 2070 ng × h/ml (Tab. 1). Im nüchternen Zustand wurde Risdiplam schnell resorbiert; die maximale Plasmakonzentration wurde nach 1 bis 4 Stunden erreicht. Eine gleichzeitige Nahrungsaufnahme hatte keinen nachteiligen Einfluss.
Tab 1. Pharmakokinetische Parameter von Risdiplam [4]
|
Parameter |
|
|
Mittlere absolute orale Bioverfügbarkeit |
81 % |
|
Mittlere maximale Gesamt-Plasmakonzentration (Cmax) |
|
|
tmax nach oraler Mehrfachgabe |
1–4 h |
|
Mittlere AUC |
|
|
Plasmaeiweißbindung |
89 % |
|
Mittleres Verteilungsvolumen |
98 l im zentralen Kompartiment; 93 l für das periphere Volumen |
|
Ausscheidung über Fäzes |
53 % der Dosis |
|
Ausscheidung über Urin |
28 % der Dosis |
|
Scheinbare systemische Clearance |
2,6 l/h |
|
Scheinbare Eliminationshalbwertszeit |
Ca. 50 h |
|
Steady State |
7–14 Tage nach 1x täglicher Gabe |
AUC: Area under the curve; SMA: spinale Muskelatrophie (SMA); tmax: Zeit bis zur maximalen Plasmakonzentration
Die Verstoffwechselung erfolgt hauptsächlich über die Flavin-Monooxygenasen 1 und 3 (FMO1 und 3) sowie über Cytochrom P450 (CYP) 1A1, 2J2, 3A4 und 3A7. Die gleichzeitige zweimal tägliche Anwendung des starken CYP3A-Inhibitors Itraconazol hatte dennoch keinen signifikanten Einfluss auf die Pharmakokinetik von Risdiplam. Der Wirkstoff ist kein Substrat des humanen Multidrug Resistance Proteins 1 (MDR1).
Klinische Ergebnisse
Daten zur Wirksamkeit
Die Zulassung von Risdiplam basiert auf zwei klinischen Studien, die jeweils Wirksamkeit, Sicherheit, Pharmakokinetik und Pharmakodynamik bei Patienten mit infantiler SMA (FIREFISH) oder spät einsetzender SMA (SUNFISH) untersucht haben.
FIREFISH-Studie
FIREFISH ist eine zweiteilige, offene klinische Studie der Phase II/III mit Patienten, die an infantiler SMA (Typ I) mit nachgewiesenem Defekt beider SMN1-Genkopien leiden [2]. Teil 1 der Studie war als Dosisfindungsphase konzipiert, in Teil 2 untersuchte man die Wirksamkeit bei der in Teil 1 festgelegten Dosierung. Patienten konnten nur an einem der Studienteile teilnehmen. Der primäre Wirksamkeitsendpunkt war die Fähigkeit, nach zwölf Behandlungsmonaten mindestens fünf Sekunden lang ohne Unterstützung zu sitzen, gemessen anhand von Testelement 22 der Grobmotorik-Skala der Bayley Scales of Infant and Toddler Development (BSID-III; Kasten).
Infokasten: Scores zur Einschätzung der Schwere neuromuskulärer Erkrankungen (Auswahl)
- BSID-III (Bayley Scales of Infant and Toddler Development): beurteilt die frühkindliche Entwicklung (1–42 Monate) in Kognition, Sprache und Motorik; der Untertest zur Grobmotorik bei SMA umfasst 72 Items; max. erreichbarer Gesamt-Score: 72
- CHOP-INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders): beurteilt bei Kleinkindern (1–38 Monate) die motorischen Fähigkeiten auf Basis aktiver und passiver Reflexe anhand von 16 Items, max. erreichbarer Gesamt-Score: 64
- HINE-2 (Hammersmith Infant Neurological Examination Module 2): Sequenzieller Test, mit dem Veränderungen neurologischer, entwicklungs- und verhaltensbiologischer Parameter bei Kleinkindern (2–24 Monate) beurteilt werden können; prüft acht Meilensteine der motorischen Entwicklung; max. erreichbarer Gesamt-Score: 26
- MFM-32 (Motor Function Measurement): beurteilt die Motorik speziell bei Patienten mit neuromuskulären Erkrankungen inkl. SMA und kann auch kleine Änderungen der motorischen Fähigkeiten erfassen; altersabhängig kommen zwei verschiedene Varianten zum Einsatz (MFM-20 bei 2- bis 7-Jährigen; MFM-32 bei 6- bis 62-Jährigen; umfasst 32 Items; max. erreichbarer Gesamt-Score: 96
- RULM (Revised Upper Limb Modul): beurteilt die Funktion der oberen Extremitäten bei Patienten mit SMA Typ II oder Typ III (≥30 Monate); umfasst 19 Items; max. erreichbarer Gesamt-Score: 37
An Teil 2 der Studie nahmen 41 Patienten teil. Das mediane Alter beim Einsetzen der Symptome betrug 1,5 Monate und beim Eintritt in die Studie 5,3 Monate. Zum Ausgangszeitpunkt lag die durchschnittliche Punktzahl des CHOP-INTEND (CHildren’s Hospital of Philadelphia Infant Test for Neuromuscular Disease) bei 22, des HINE-2 (Hammersmith Infant Neurological Examination Module 2) bei 1,0.
Von den behandelten Kindern erreichten 12/41 (29 %) den primären Endpunkt (90%-Konfidenzintervall [KI] 18 %–43 %), und rund 90 % konnten den CHOP-INTEND um mindestens vier Punkte erhöhen (Abb. 1). Auf der HINE-2-Skala zeigten sich bei 32/41 (78 %) der Patienten Verbesserungen. Nach den zwölf Behandlungsmonaten lebten 38 der 41 Studienteilnehmer (93 %) noch, 35 (85 %) ohne dauerhafte Beatmung und 34 (83 %) waren nicht auf eine künstliche Ernährung angewiesen. Unbehandelte Patienten mit SMA Typ I wären dagegen nie in der Lage alleine zu sitzen, und nur ein Viertel von ihnen würde ohne dauerhafte Beatmung ein Alter von mehr als 14 Monaten erreichen.
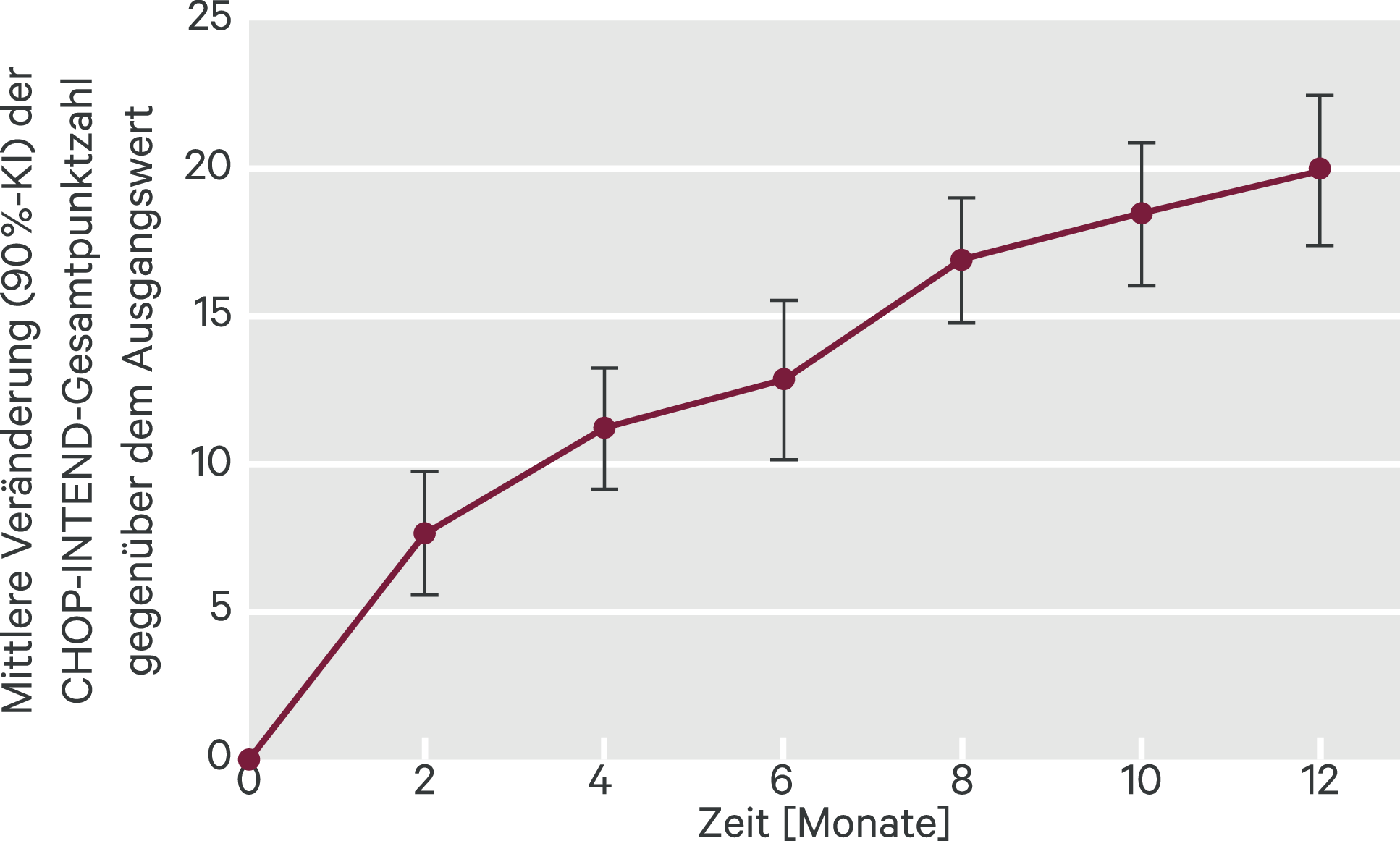
Abb. 1. FIREFISH Teil 2: Mittlere Veränderung der CHOP-INTEND-Gesamtpunktzahl gegenüber dem Ausgangswert (mod. nach [4]) KI: Konfidenzintervall
In Teil 1 der Studie wurden 21 Patienten mit einem medianen Alter von 6,7 Monaten eingeschlossen [1]. Von diesen Patienten erhielten 17 die therapeutische Dosis, die für Teil 2 ausgewählt wurde. Nach einer Behandlungszeit von zwölf Monaten konnten 41 % der Probanden fünf Sekunden selbstständig sitzen. Nach einem weiteren Jahr erreichten weitere 3 Patienten den primären Endpunkt. Nach dem ersten Jahr waren 90 % der Studienteilnehmer ohne dauerhafte Beatmung am Leben, nach zwei Jahren immerhin noch 17/21 (81 %). Drei Patienten starben während der Behandlung, ein weiterer 3,5 Monate nach Absetzen der Behandlung.
SUNFISH
SUNFISH ist eine multizentrische Studie mit Patienten im Alter von 2 bis 25 Jahren, die an SMA Typ II oder Typ III leiden [3]. Wie bei FIREFISH war der erste Teil als Dosisfindungsphase konzipiert, der zweite als randomisierte, doppelblinde, Placebo-kontrollierte Bestätigungsphase. Der primäre Endpunkt war die Veränderung der MFM-32-Gesamtpunktzahl (Motor Function Measure) nach zwölf Monaten (Kasten).
In Teil 2 der Studie wurden 180 nicht gehfähige Patienten eingeschlossen, von denen mehr als 75 % an SMA Typ II litten [5]. Die Probanden wurden nach Alter stratifiziert und 2 : 1 in eine Wirkstoff- und eine Placebo-Gruppe randomisiert. Das mediane Alter zu Studienbeginn betrug neun Jahre. Zum Ausgangszeitpunkt hatten die Patienten eine MFM-32-Punktzahl von 46,1 sowie eine Punktzahl von 20,1 im Revised Upper Limb Module (RULM). Nach zwölf Monaten hatte sich die MFM-32-Gesamtpunktzahl in der Risdiplam-Gruppe im Unterschied zur Placebo-Gruppe verbessert (Abb. 2). Der Unterschied betrug 1,55 Punkte auf der MFM-32-Skala (95%-KI 0,30–2,81) und 1,59 Punkte auf der RULM-Skala (95%-KI 0,55–2,62). Die Verbesserung wurde als klinisch bedeutsam und statistisch signifikant eingestuft. Nach Ende der 12-monatigen Behandlungsphase erhielten 117 Patienten weiterhin Risdiplam und zeigten eine anhaltende Verbesserung der motorischen Funktion [6]. Die mittlere Veränderung gegenüber dem Ausgangspunkt betrug für MFM-32 1,83 (95%-KI 0,74–2,92) und für RULM 2,79 (95%-KI 1,94–3,64).
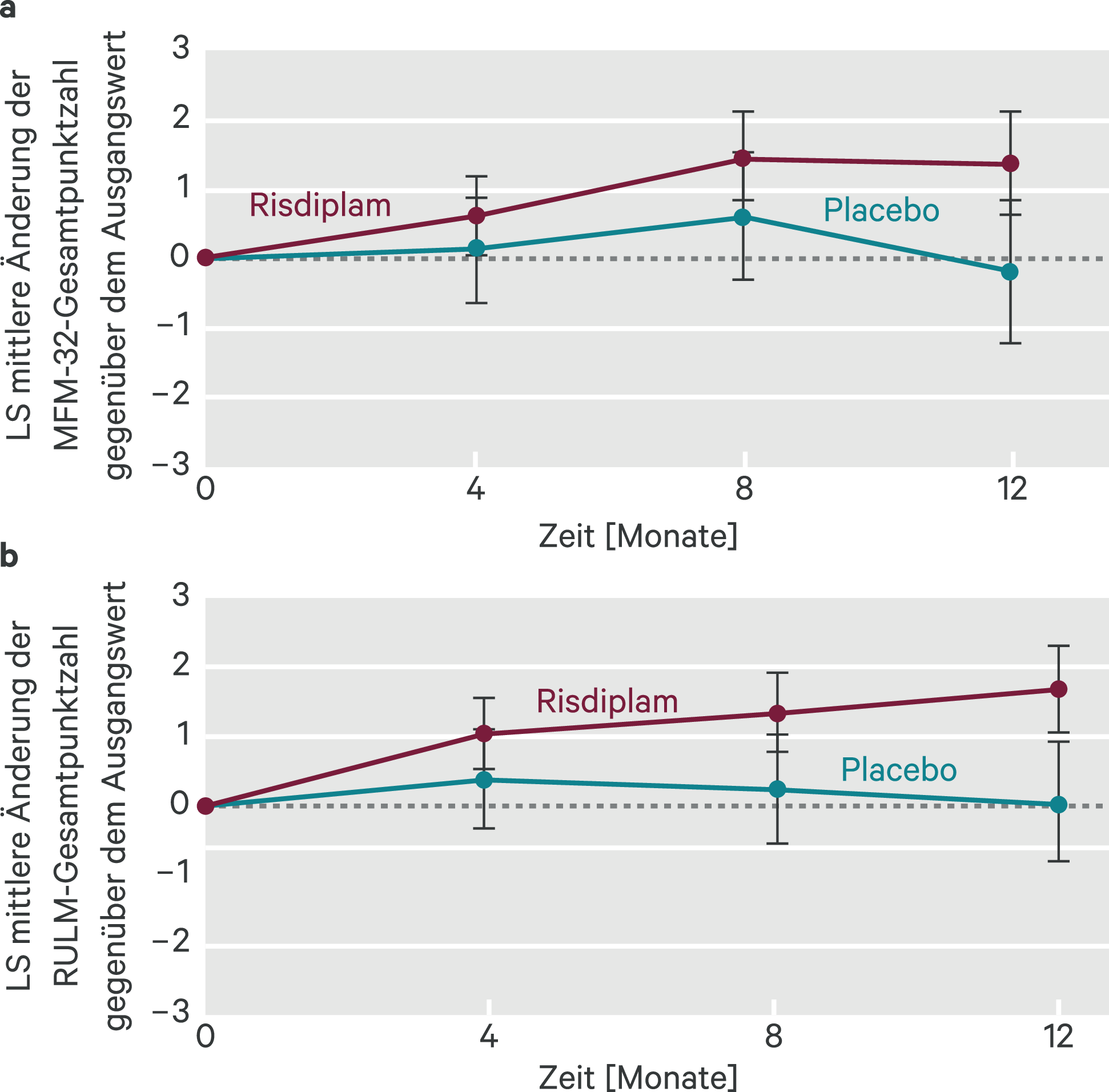
Abb. 2. SUNFISH Teil 2: mittlere Änderung der MFM-32-Gesamtpunktzahl (a) und der RULM-Gesamtpunktzahl (b) gegenüber dem Ausgangswert im Verlauf von 12 Monaten (a); Differenz der Mittelwerte der kleinsten Fehlerquadrate (LS) für die Änderung der RULM-Punktzahl gegenüber dem Ausgangswert (95-%-Konfidenzintervall; mod. nach [4])
In Teil 1 wurden 51 Patienten eingeschlossen, von denen sieben gehfähig waren. Die anhand des MFM-32 gemessenen motorischen Fähigkeiten verbesserten sich unter Risdiplam nach einem Jahr mit einer mittleren Zunahme von 2,7 Punkten (95%-KI 1,5–3,8). Diese als klinisch bedeutsam bewertete Verbesserung blieb unter Behandlung bis zu zwei Jahre lang erhalten.
Daten zur Verträglichkeit
Ein Überblick über die in den klinischen Studien unter Risdiplam am häufigsten aufgetretenen, unerwünschten Wirkungen findet sich in Tabelle 2. Sowohl bei Patienten mit infantiler (48,4 %) als auch mit spät einsetzender SMA (21,7 %) war Fieber die häufigste unerwünschte Wirkung. Bei der infantilen Form folgten Ausschlag (27,4 %) und Durchfall (16,1 %), bei der spät einsetzenden Form Kopfschmerzen (20 %), Durchfall und Ausschlag (jeweils 16,7 %). Die Nebenwirkungen klangen bei fortgesetzter Behandlung im Allgemeinen wieder ab.
Tab 2. Die häufigsten Nebenwirkungen unter Risdiplam [4]
|
Häufigkeit |
Nebenwirkung |
|
|
Infantile SMA |
Spät einsetzende SMA |
|
|
Sehr häufig (≥ 1/10) |
|
|
|
Häufig (≥ 1/100 bis < 1/10) |
|
|
SMA: spinale Muskelatrophie
Weitere Vorsichtsmaßnahmen
Tierexperimentelle Studien haben eine Reproduktionstoxizität gezeigt. Daher sollte vor Beginn einer Therapie mit Risdiplam bei gebärfähigen Patientinnen eine Schwangerschaft ausgeschlossen werden. Weibliche Patienten müssen während der Therapie und bis einen Monat nach Therapieende eine sichere Verhütungsmethode anwenden. Bei männlichen Patienten sollte bis mindestens vier Monate nach Therapieende zuverlässig verhütet werden. Die Behandlung kann die männliche Fertilität beeinträchtigen. Bei männlichen Patienten mit Kinderwunsch sollte vor Beginn der Therapie eine Spermienkonservierung in Betracht gezogen werden. Für eine Beeinträchtigung der weiblichen Fertilität gibt es keine Hinweise. Da über eine potenzielle gesundheitsschädigende Wirkung von Risdiplam für den gestillten Säugling nichts bekannt ist und bei Ratten der Wirkstoff in die Muttermilch ausgeschieden wird, wird empfohlen, während der Behandlung mit Risdiplam nicht zu stillen.
Daten zu Wechselwirkungen
Risdiplam und sein Metabolit M1 hemmen die Multidrug and Toxin Extrusion Transporter 1 und 2-K (MATE1 und MATE2-K). Somit kann Risdiplam die Plasmakonzentration von Arzneimitteln wie Metformin, die über den Transporter eliminiert werden, erhöhen. Bei gleichzeitiger Anwendung sollten deshalb die arzneimittelbezogenen Toxizitäten überwacht und gegebenenfalls eine Dosisreduzierung des gleichzeitig angewendeten Arzneimittels in Betracht gezogen werden.
Das Potenzial für synergistische Effekte bei gleichzeitiger Anwendung von Risdiplam und retinotoxischen Arzneimitteln wurde nicht untersucht. Bei gleichzeitiger Anwendung von Wirkstoffen mit erwiesener oder vermuteter Retinotoxizität ist deshalb Vorsicht geboten.
Dosierung, Einsatz, und Handhabung
Dosierung
Risdiplam wird einmal täglich zur gleichen Tageszeit nach einer Mahlzeit eingenommen. Die empfohlene tägliche Dosis berechnet sich nach Alter und Körpergewicht. Bis zu einem Alter von 2 Jahren liegt die Dosis bei 0,2 mg/kg, ab einem Alter von 2 Jahren bei 0,25 mg/kg – solange, bis ein Körpergewicht von 20 kg erreicht ist. Patienten mit mehr als 20 kg Körpergewicht erhalten die tägliche Höchstdosis von 5 mg. Eine versäumte Dosis ist schnellstmöglich nachzuholen, sofern nicht mehr als sechs Stunden seit dem geplanten Einnahmezeitpunkt vergangen sind. Ist dies nicht gegeben, wird die Dosis übersprungen und die Einnahme mit der regulär geplanten nächsten Dosis fortgesetzt.
Besondere Patientengruppen
Auf Basis der vorhandenen Daten ist bei älteren Patienten über 65 Jahre, bei Patienten mit Nierenfunktionsstörung sowie bei Patienten mit leichten und mittleren Leberfunktionsstörungen keine Dosisanpassung erforderlich. Patienten mit schweren Leberfunktionsstörungen und Kinder unter zwei Monaten wurden nicht untersucht.
Interessenkonflikterklärung
Es bestehen keine Interessenkonflikte.
Literatur
1. Baranello G, Darras BT, Day JW, Deconinck N, et al. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med 2021;384 : 915–23.
2. ClinicalTrials.gov. NCT02908685. https://clinicaltrials.gov/ct2/show/NCT02908685 (Zugriff am 03. Mai 2021).
3. ClinicalTrials.gov. NCT02913482. www.clinicaltrials.gov/ct2/show/NCT02913482 (Zugriff am 03. Mai 2021).
4. European Medicine Agency (EMA). EPAR (Zusammenfassung der Merkmale des Arzneimittels). www.ema.europa.eu/en/documents/product-information/evrysdi-epar-product-information_de.pdf (Zugriff am 03. Mai 2021).
5. Mercuri E, Barisic N, Boespflug-Tanguy O, Deconinck N, et al. SUNFISH Part 2: Efficacy and safety of risdiplam (RG7916) in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA) (1260). Neurology 2020;94(15 Supplement).
6. Oskoui M, Day JW, Deconinck N, Mazzone E, et al. SUNFISH Part 2: 24-month efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA) (2240). Neurology 2021;96(15 Supplement).
Modifizierter Nachdruck aus Arzneimitteltherapie 2021;39:289–93.
Dr. Larissa Tetsch, Steinröselweg 9, 82 216 Maisach, E-Mail: larissa.tetsch@gmail.com
Risdiplam
Spinal muscular atrophy (SMA) is a heritable, progressive disease. Most often, it shows first symptoms within the first months of life. SMA is one of the main causes of infant death. Causative for the muscular atrophy is a degeneration of motor neurons due to a lack of functional SMN protein. Since May 2017, the antisense oligonucleotide Nusinersen is approved in the European Union as the first treatment for SMA. Nusinersen is injected into the liquor, acts by changing splicing of the SMN2 pre-messenger RNA and thereby increases the amount of functional SMN protein. Since 25th of February 2021 Risdiplam is approved in the European Union. It acts in the same way as Nusinersen but is applied orally. In the clinical studies which led to approval for the therapy of patients with infantile and later onset SMA, FIREFISH and SUNFISH, risdiplam had a positive influence on motorical functions and time of event-free survival of the participants. The most frequent adverse reactions were diarrhoea, rash, fever and headache, which generally resolved despite ongoing treatment.
Key words: Risdiplam; spinal muscular atrophy; SMA
Psychopharmakotherapie 2021; 28(05):201-204