Holger Petri, Bad Wildungen*
Abirateronacetat
Abirateronacetat ist ein CYP17-Inhibitor. In der Folge wird die Testosteronbiosynthese gehemmt und damit die Androgensynthese sowohl in den Nebennieren, den Hoden und den Tumorzellen blockiert [13]. Abirateronacetat ist ein Prodrug, das durch Esterasen rasch zu Abirateron hydrolysiert wird. Abirateron wird über CYP3A4 metabolisiert. Als Substrat der Sulfotransferase 2A1 (SULT2A1) erfolgt der primäre Abbau jedoch über eine Phase-II-Reaktion. Die Metaboliten sind pharmakologisch inaktiv [3]. Der starke CYP3A4-Induktor Rifampicin (Abb. 1) senkte in einer klinischen Studie mit gesunden Probanden die AUC (Fläche unter der Konzentrations-Zeit-Kurve) des Antiandrogens um 55 %. Der starke CYP3A4-Inhibitor Ketoconazol (Abb. 1) erhöhte den AUC-Wert dagegen nur um 15 %, was als nicht klinisch relevant betrachtet wird [3, 4]. Die Diskrepanz bei den AUC-Änderungen erklärt sich durch die zusätzlichen induktiven Effekte von Rifampicin auf die Expression der Sulfotransferase [3]. Die Kombination mit CYP3A4-Induktoren sollte dennoch vermieden werden, es sei denn, es gibt keine Alternativen [10]. Die US-amerikanische Fachinformation empfiehlt bei notwendiger Komedikation eine Verdopplung der Tagesdosis durch eine zweimal tägliche Einnahme anstelle der einmal täglichen [17].
Der starke CYP3A4-Induktor Apalutamid reduzierte bei Patienten mit metastasiertem kastrationsresistentem Prostatakarzinom (mCRPC) die Bioverfügbarkeit von Abirateron durchschnittlich um 25 % [16]. In einem Fallbericht wird über einen Patienten berichtet, der unter Abiraterontherapie wegen einer neu aufgetretenen Trigeminusneuralgie auf Anordnung eines Neurologen die Einnahme von Carbamazepin begonnen hatte. Das Koanalgetikum musste nach einem Monat wegen Nebenwirkungen abgesetzt werden. Der AUC-Wert und der Abirateron-Talspiegel waren bei Kombination um 67 % bzw. 61 % niedriger als einen Monat nach Beendigung der gemeinsamen Einnahme. Carbamazepin induziert, wie Rifampicin, sowohl CYP3A4 und SULT2A1 [2]. Vor diesem Hintergrund erscheinen Dosiserhöhungen von Abirateron nur bei gleichzeitiger Induktion von SULT2A1 und CYP3A4 gerechtfertigt. Risikobehaftete Kombinationen sollten vorzugsweise durch ein therapeutisches Drug-Monitoring (TDM) begleitet werden. Zumindest sollten die Talspiegel einen empfohlenen Schwellenwert nicht unterschreiten, weil die PSA(Prostata-spezifisches Antigen)-Antwort mit der Höhe der Abirateron-Talspiegel korreliert [2].
Abirateron hemmt CYP2D6 und CYP2C8. Patienten hatten unter Abirateron-Einnahme eine 2,9-fach höhere Exposition des CYP2D6-Testsubstrats Dextromethorphan. Der AUC-Wert von Pioglitazon, einem CYP2C8-Testsubstrat, stieg durch Abirateron bei gesunden Probanden um 46 % [3]. Vorsicht ist besonders geboten bei Arzneimitteln, deren Abbau primär über CYP2D6 erfolgt wie Atomoxetin, Metoprolol und Nortriptylin.
Apalutamid
Apalutamid ist ein selektiver Androgenrezeptor(AR)-Inhibitor, der direkt an die Liganden-bindende Domäne des AR bindet. Apalutamid verhindert die nukleare Translokation des AR, hemmt die DNA-Bindung und die AR-vermittelte Transkription und zeigt dabei keine agonistische Aktivität am Androgenrezeptor. Apalutamid wird im Steady State gleichermaßen über CYP2C8 oder CYP3A4 verstoffwechselt. Es entsteht mit N-Desmethylapalutamid ein pharmakologisch wirksamer Metabolit, der in vitro ein Drittel der Aktivität der Muttersubstanz hat. Der starke CYP2C8-Hemmer Gemfibrozil (Abb. 1) erhöhte die Exposition der aktiven Bestandteile (Summe aus Apalutamid und aktivem Metaboliten) um 43 %. Dies erfordert bei akzeptabler Verträglichkeit keine Dosisreduktion. Durch den starken CYP3A4-Hemmer Itraconazol (Abb. 1) wurde die AUC der aktiven Bestandteile kaum verändert. Es wird angenommen, dass moderate und schwache Inhibitoren von CYP2C8 und CYP3A4 sich nicht auf die Bioverfügbarkeit auswirken. Es wird auch nicht erwartet, dass starke CYP2C8- und CYP3A4-Induktoren (Abb. 1) die Exposition der aktiven Bestandteile klinisch relevant beeinflussen, sodass keine In-vivo-Studien durchgeführt worden sind. Apalutamid beschleunigt zudem seinen eigenen Metabolismus durch Auto-Induktion [8].
Apalutamid fördert den Metabolismus anderer Arzneimittel durch Induktion der Isoenzyme CYP2C9, CYP2C19 und CYP3A4. In einer Arzneimittelwechselwirkungsstudie sanken die AUC-Werte der Testsubstrate S-Warfarin (CYP2C9) um 46 %, von Omeprazol (CYP2C19) um 85 % und von Midazolam (CYP3A4) um 92 %. Auch kann gemäß In-vitro-Daten die Bioverfügbarkeit von CYP2B6-Substraten reduziert sein. Weitere Interaktionen sind durch die Induktion von Arzneistofftransportern möglich. So sanken die Exposition von Fexofenadin, einem P-Glykoprotein(P-gp)-Substrat, um 30 %, sowie von Rosuvastatin, einem Substrat des Breast Cancer Resistance-Proteins (BCRP) und des organischen Anion-Transporterpeptids 1B1 (OATP1B1) um 41 % [8].
Bicalutamid
Bicalutamid ist ein nichtsteroidales Antiandrogen der ersten Generation. Es hat im Vergleich zu Flutamid eine längere Halbwertszeit und liegt als Razemat vor [5]. Der Stoffwechsel führt über direkte Glucuronidierung und oxidative Reaktionen zu inaktiven Metaboliten. Daten aus klinischen Studien zu möglichen CYP-Interaktionen liegen nicht vor. Da gemäß In-vitro-Untersuchungen R-Bicalutamid die Aktivität von CYP3A4 hemmt, wurde in einer klinischen Studie der Einfluss auf die Exposition von Midazolam getestet. Obwohl sich die AUC im Durchschnitt nur um 27 % erhöhte, kann diese bis im Einzelfall zu 90 % steigen, was bei CYP3A4-Substraten mit enger therapeutischer Breite von Relevanz sein kann [5, 7].
Cyproteronacetat
Cyproteronacetat ist ein steroidales Antiandrogen und besitzt gestagene Eigenschaften. Wie andere Steroidhormonone ist es ein Substrat von CYP3A4. Somit sind Veränderungen der Exposition durch CYP3A4-Modulatoren möglich. Klinische Studien mit starken CYP3A4-Modulatoren sind jedoch nicht durchgeführt worden.
Die in der Antiandrogen-Behandlung des Prostatakarzinoms notwendig hohen Dosen Cyproteronacetat können gemäß In-vitro-Inhibitionsuntersuchungen eine Hemmung von CYP2C8, 2C9, 2C19, 3A4 und 2D6 zur Folge haben und sind bei Kombination mit Substraten mit enger therapeutischer Breite zu berücksichtigen [6]. In einem Fallbericht wird das Auftreten einer Rhabdomyolyse bei einem 66-jährigen Patienten unter Cyproteronacetat und Simvastatin beschrieben, die sich nach Absetzen beider Substanzen zurückbildete. Obwohl für Cyproteronacetat bislang kein klinisch relevanter hemmender Einfluss auf CYP3A gezeigt werden konnte, kann als Interaktionsmechanismus eine Hemmung des Simvastatin-Metabolismus nicht ausgeschlossen werden [1].
Darolutamid
Darolutamid ist ein nichtsteroidaler Androgenrezeptor-Antagonist mit spezifischen Hemmeigenschaften, wodurch es sich von anderen bekannten Androgenrezeptor-Antagonisten unterscheidet und möglicherweise bei Resistenz gegenüber Apalutamid und Enzalutamid wirksam ist [15]. Darolutamid ist in Deutschland noch nicht als Fertigarzneimittel zugelassen. Darolutamid und sein aktiver Metabolit Keto-Darolutamid sind Substrate von CYP3A4. Der starke CYP3A4-Inhibitor Itraconazol (Abb. 1) erhöhte in einer klinischen Studie mit gesunden Probanden die AUC von Darolutamid und Keto-Darolutamid um das 1,75- bzw. 1,8-Fache. Der starke CYP3A4-Induktor Rifampicin senkte die Bioverfügbarkeit von Muttersubstanz und Metabolit auf 28 % bzw. 25 % [Clinical Study Synopsis Bayer, Juni 2018]. Als Modulator weist Darolutamid jedoch kein Potenzial für pharmakokinetische Wechselwirkungen auf [21].
Enzalutamid
Enzalutamid ist ein Androgenrezeptorantagonist der zweiten Generation. Enzalutamid bindet effektiv an den Androgenrezeptor und bewirkt so eine verminderte Translokation des Rezeptors in den Zellkern. Zudem unterdrückt es die Aktivierung von Zielgenen der Tumorgenese [13]. Enzalutamid wird primär über CYP2C8 und nachgeordnet über CYP3A4 metabolisiert. Es werden zwei Hauptmetaboliten gebildet, von denen N-Demethyl-Enzalutamid pharmakologisch aktiv ist [3, 9]. Der moderate CYP2C8- und starke CYP3A4-Induktor Rifampicin reduzierte die AUC von Enzalutamid und seines aktiven Metaboliten um 37 %, was keine Dosisanpassung erforderlich macht. Unter Kombination mit dem starken CYP3A4-Hemmer Ketoconazol erhöhte sich die Exposition nur um das 1,3-Fache. Der starke CYP2C8-Inhibitor Gemfibrozil (Abb. 1) steigerte die Bioverfügbarkeit der therapeutischen Fraktion um das 2,2-Fache [3]. Dies erfordert eine Reduktion der Tagesdosis des Antiandrogens auf die Hälfte [9].
Enzalutamid beschleunigt durch Induktion von CYP2C9, CYP219 und CYP3A4 die Clearance anderer Arzneimittel. So sank in einer klinischen Studie die Exposition der Testsubstrate S-Warfarin (CYP2C9) um 56 %, von Omeprazol (CYP2C19) um 70 % und von Midazolam (CYP3A4) um 86 % [3]. Die Halbwertszeit von Enzalutamid beträgt knapp sechs Tage. Somit stellt sich das Steady State mit Verzögerung ein und das volle Induktionspotenzial zeigt sich nach Beginn der Einnahme erst nach einem Monat [9]. Deshalb sollten Patienten, die Arzneimittel einnehmen, die Substrate von CYP2C9, CYP2C19 und CYP3A4 sind, in den ersten Wochen der kombinierten Einnahme auf einen sukzessiven Wirkungsverlust oder eine Wirkungsverstärkung bei aktiven Metaboliten engmaschig beobachtet werden [9]. Nach Beendigung der Enzalutamid-Therapie ist von einer mehrwöchigen Deinduktionszeit auszugehen.
Flutamid
Flutamid ist wie Bicalutamid ein nichtsteroidales Antiandrogen der ersten Generation. Flutamid wird hauptsächlich über CYP1A2 zu dem bei Prostatakrebs wirksamen Hauptmetaboliten 2-Hydroxyflutamid verstoffwechselt. Eine verminderte CYP1A2-Enzymaktivität scheint die Bildung von hepatotoxischen Metaboliten nach Flutamid-Gabe zu beeinflussen [14, 18]. Das Risiko ist besonders erhöht, wenn bei gleichzeitigem Mangel an Glutathion die Glutathion-S-Transferase bei der Detoxifikation reaktiver Zwischenprodukte nicht ausreichend aktiv ist [12].
QT-Zeit-Verlängerung unter Androgen-Deprivationstherapie
Die Androgen-Deprivationstherapie erhöht das Risiko kardialer Nebenwirkungen, besonders bei Prostatakarzinom-Patienten mit Herzinsuffizienz [11, 20]. Zu den Risikofaktoren für eine Torsade-de-pointes(TdP)-Arrhythmie zählen die Anwendung mehrerer QT-Zeit-verlängernder Arzneimittel und erhöhte Plasmaspiegel [19].
Zu diesen metabolisch bedingten AUC-Erhöhungen kann es durch hemmende Effekte (Abirateronacetat) oder nach Absetzen von Induktoren und der daraus resultierenden Deinduktion kommen (Apalutamid und Enzalutamid). Dies kann eine bedarfsweise Dosisanpassung von Komedikamenten mit TdP-Potenzial bedingen.
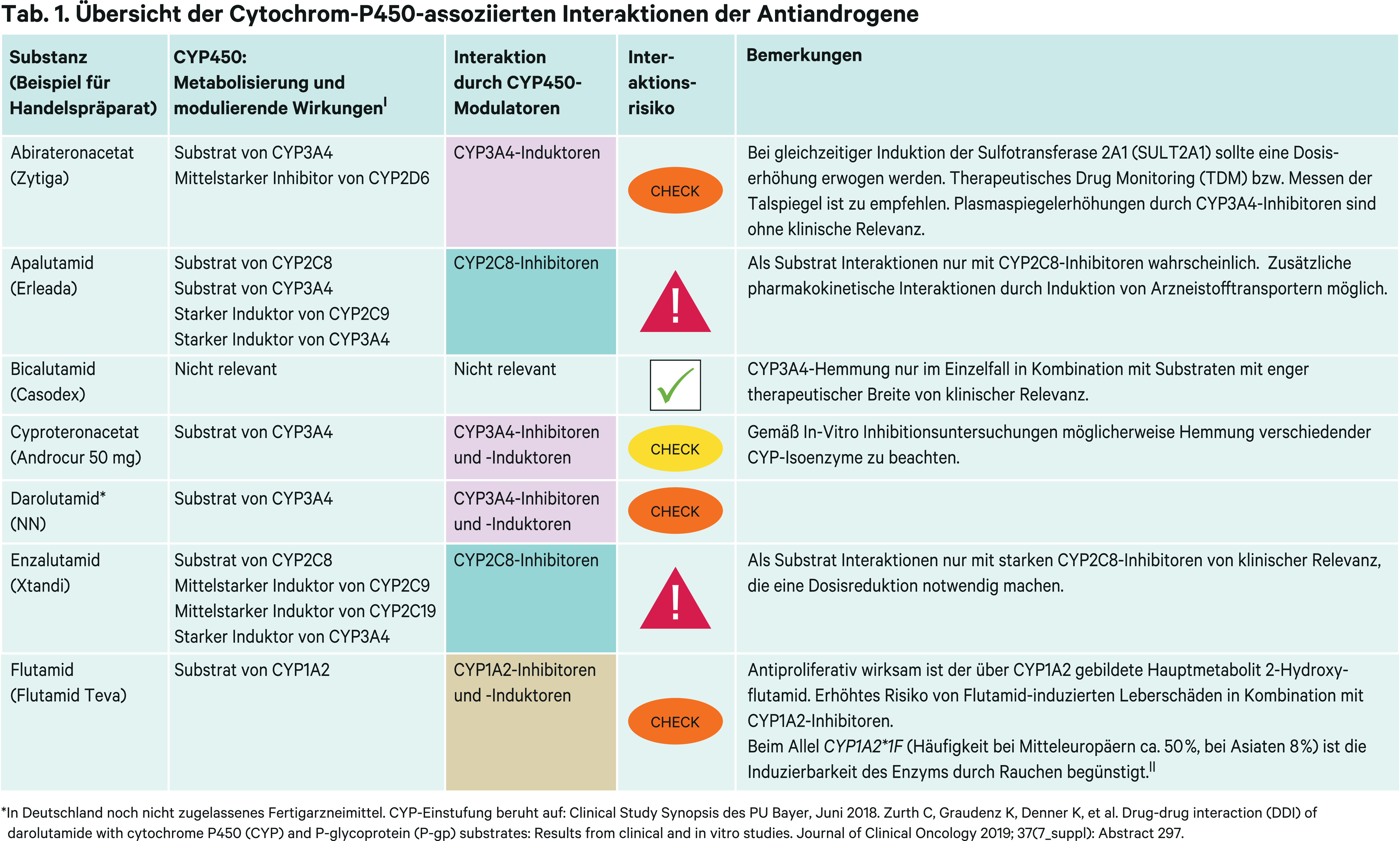

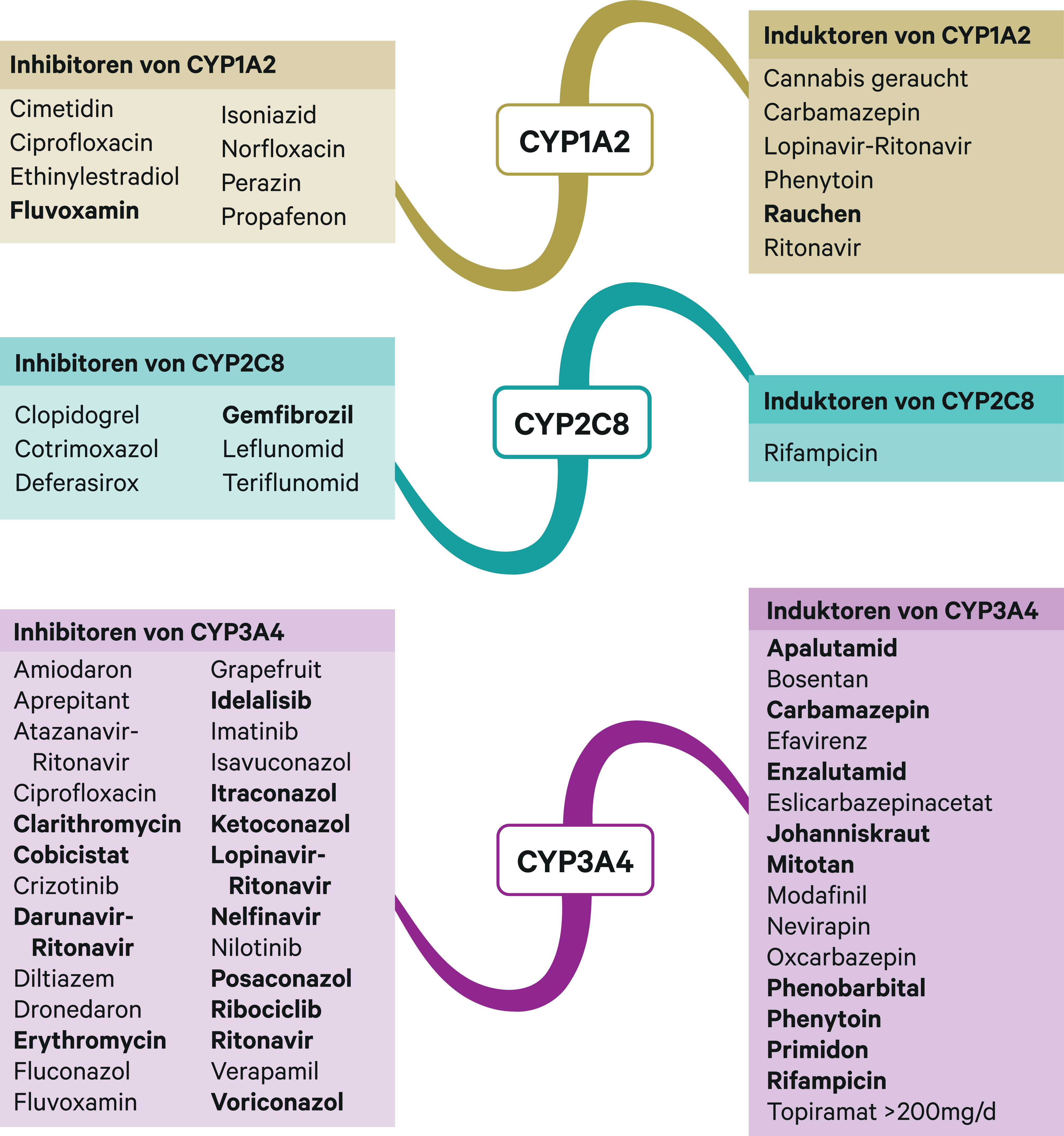
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2C8 und 3A4 (Stand 6/2019) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Ahamed E, Bissett D. Rhabdomyolysis in prostate cancer – caution in prescribing cyproterone acetate with statins. Clin Oncol (R Coll Radiol) 2004;8:528–9.
2. Benoist GE, van der Doelen MJ, Ter Heine R, et al. A clinically relevant decrease in abiraterone exposure associated with carbamazepine use in a patient with castration-resistant metastatic prostate cancer. Br J Clin Pharmacol 2018;84:1064–67.
3. Benoist GE, Hendriks RJ, Mulders PF, et al. Pharmacokinetic aspects of the two novel oral drugs used for metastatic castration-resistant prostate cancer: Abiraterone acetate and enzalutamide. Clin Pharmacokinet 2016;55:1369–80.
4. Bernard A, Vaccaro N , Acharya M, et al. Impact on abiraterone pharmacokinetics and safety: Openlabel drug-drug interaction studies with ketoconazole and rifampicin. Clin Pharmacol Drug Dev 2015:63–73.
5. Cockshott ID. Bicalutamide: clinical pharmacokinetics and metabolism. Clin Pharmacokinet 2004;43:855–78.
6. Fachinformation Androcur® 50 mg. Stand: Oktober 2018.
7. Fachinformation Casodex®. Stand: Januar 2018.
8. Fachinformation Erleada®. Stand: Januar 2019.
9. Fachinformation Xtandi®. Stand: Oktober 2018.
10. Fachinformation Zytiga®. Stand: November 2017.
11. Gagliano-Jucá T, Travison TG, Kantoff PW, et al. Androgen deprivation therapy is associated with prolongation of QTc interval in men with prostate cancer. J Endocr Soc 2018;2:485–96.
12. Giorgetti R, di Muzio M, Giorgetti A, et al. Flutamide-induced hepatotoxicity: ethical and scientific issues. Eur Rev Med Pharmacol Sci 2017;21(1 Suppl):69–77.
13. Knipper S, de Santis M, Grimm MO, et al. Aktuelle medikamentöse Therapiekonzepte des metastasierten Prostatakarzinoms. Onkologe 2019;25:343–51.
14. Kronabel D. Möglicher Einfluss von Lebensmitteln auf die Metabolisierung von Flutamid. Deutsche Zeitschrift für Onkologie 2013;45:163–6.
15. Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep 2015;5:12007.
16. Posades EM, Chi KN de Wit R, et al. Pharmacokinetics and safetey of ARN-509 with abiraterone acetate and prednisone in patients with metastatic castration-resistant prostata cancer. Poster presented at: The American Association for Cancer Research Annual Meeting; April 18–22; Philadelphia, PA.
17. Prescribing information Zytiga®. Stand April 2018.
18. Shet MS, McPhaul M, Fisher CW, et al. Metabolism of the antiandrogenic drug (flutamide) by human CYP1A2. Drug Metab Dispos 1997;25:1298–303.
19. Wenzel-Seifert K, Ostermeier CP, Omar NB, et al. Unerwünschte kardiovaskuläre Wirkungen von Psychopharmaka. Psychopharmkotherapie 2013;20:148–57.
20. Ziehr DR, Chen MH, Zhang D, et al. Association of androgen-deprivation therapy with excess cardiac-specific mortality in men with prostate cancer. BJU Int 2015;116:358–65.
21. Zurth C, Graudenz K, Denner K, et al. Drug-drug interaction (DDI) of darolutamide with cytochrome P450 (CYP) and P-glycoprotein (P-gp) substrates: Results from clinical and in vitro studies. Journal of Clinical Oncology 2019; 37(7 Suppl):297.
*korrigierter Nachdruck aus Krankenhauspharmazie 2019;40:367–72.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2019; 26(04)