Holger Petri, Bad Wildungen*
Sulfonylharnstoffe und Glinide
Sulfonylharnstoffe wie Glibenclamid und Glimepirid werden hauptsächlich über das Cytochrom-P450(CYP)-Isoenzym 2C9 verstoffwechselt [5, 6]. In einer Studie wurde der Einfluss auf die Plasmaspiegel von Glimepirid durch den starken CYP2C9-Hemmer Fluconazol (Abb. 1) untersucht. Im Ergebnis stieg die AUC (Fläche unter der Konzentrations-Zeit-Kurve) des oralen Antidiabetikums um das 2,4-Fache [12]. Auch moderate CYP2C9-Inhibitoren wie Cotrimoxazol können die insulinotropen Wirkungen von Glibenclamid und Glimepirid mit dem Risiko von Hypoglykämien verstärken [23]. Das Antibiotikum erhöht bei Patienten unter Glibenclamid-Therapie die Wahrscheinlichkeit für starke Unterzuckerungen um das 6-Fache [9]. Bei einer Therapie mit den beiden Sulfonylharnstoffen Glibenclamid und Glipizid (in Deutschland nicht im Handel) scheint in Kombination mit bestimmten Antiinfektiva, unabhängig von deren pharmakologischen Eigenschaften, generell ein erhöhtes Hypoglykämie-Risiko zu bestehen [15]. Neben Cotrimoxazol sind Clarithromycin und Levofloxacin besonders betroffen [22].
CYP2C9 wird polymorph exprimiert. Träger des CYP2C9*3-Allels haben einen verlangsamten Stoffwechsel. Bei homozygoten Trägern (Poor Metabolizer) beträgt die Gesamtclearance für Glibenclamid und Glimepirid verglichen mit normalen Metabolisierern nur 20% [11]. Diese Patienten sollten wie heterozygote Träger (Intermediate Metabolizer) geringere Standarddosen erhalten und engmaschig auf dosisabhängige Nebenwirkungen überwacht werden [11]. Die Interaktionstabelle (Tab. 1) enthält Angaben zur Häufigkeit von CYP2C9-Polymorphismen.
Der Metabolismus des Sulfonylharnstoffs Gliquidon erfolgt CYP-unabhängig [7].
Nateglinid wird über CYP2C9 abgebaut. Fluconazol erhöht die AUC des Glinids um 48%, der moderate CYP2C9-Hemmer Sulfinpyrazon (in Deutschland nicht zugelassen) um 28% [14, 18]. Hingegen wird Repaglinid in erster Linie über CYP2C8 und nachgeordnet über CYP3A4 verstoffwechselt. Der starke CYP2C8-Hemmer Gemfibrozil steigert die AUC von Repaglinid um das 8,1-Fache, Itraconazol als starker CYP3A4-Hemmer die Exposition nur um das 1,4-Fache. In der Kombination Gemfibrozil und Itraconazol erhöht sich die AUC um das 19,4-Fache, wobei sich die Halbwertszeit des normalerweise kurzwirksamen Glinids von 1,3 Stunden auf 6,1 Stunden verlängert [13]. Somit wird der Abbau von Repaglinid über CYP3A4 dann von Bedeutung, wenn der Metabolismus über CYP2C8 vermindert wird. Dies kann durch CYP2C8-Inhibitoren erfolgen. Aber auch langsame Metabolisierer von CYP2C8-Substraten sollten erhöhte Repaglinid-Plasmakonzentrationen aufweisen, insbesondere bei Kombination mit CYP3A4-Hemmern. Untersuchungen zeigen, dass die unter der kaukasischen Bevölkerung häufig vorkommende Allelvariante CYP2C8*3 bei Substraten wie Amodiaquin und Paclitaxel zu einer Erhöhung der Plasmaspiegel führen kann. Jedoch bleiben bei Einnahme von Repaglinid in therapeutischen Dosen die AUC-Werte unverändert, in niedriger Dosis erhöht sich sogar die Clearance [3].
Thiazolidindione (Glitazone)
Pioglitazon ist das zurzeit einzige in Deutschland zugelassene Thiazolidindion. Es ist Substrat von CYP2C8. Gemfibrozil steigert die Exposition von Pioglitazon um mehr als das 3-Fache, während der CYP2C8-Induktor Rifampicin zu einer Senkung des AUC-Werts auf etwa die Hälfte führt. Es sollten die Blutzuckerspiegel bei An- und Absetzen der CYP2C8-Modulatoren überwacht werden [4, 21]. Bei Probanden mit dem CYP2C8*3-Allel zeigt sich eine erhöhte Clearance von Pioglitazon gegenüber Trägern des Wildtyp-Allels [3].
DPP-4-Inhibitoren (Gliptine) und SGLT-2-Inhibitoren (Gliflozine)
Saxagliptin ist Substrat von CYP3A4. Der moderate CYP3A4-Inhibitor Diltiazem erhöhte in einer Studie die AUC des Gliptins um 109%, der starke CYP3A4-Hemmer Ketoconazol um 145% [16]. Die Dosis des oralen Antidiabetikums sollte daher in Kombination mit starken CYP3A4-Hemmern auf 2,5 mg pro Tag begrenzt werden [16, 17]. Diese Empfehlung findet sich in der deutschen Fachinformation nicht wieder [8]. Der starke CYP3A4-Induktor Rifampicin reduzierte in einer weiteren Untersuchung die Exposition der Muttersubstanz um 76%. Der Anteil des aktiven Metaboliten und die Hemmung der Plasma-DPP-4-Aktivität blieben insgesamt aber unbeeinflusst. Die Autoren empfehlen daher keine Dosisanpassung in Kombination mit Rifampicin [24]. Gemäß Fachinformation wird eine sorgfältige Blutzuckerkontrolle empfohlen [8].
Sitagliptin wird überwiegend unverändert renal eliminiert [25]. Die beiden SGLT-2-Inhibitoren Dapagliflozin und Empagliflozin werden unabhängig von CYP-Enzymen metabolisiert [10, 20].
Metformin und Alpha-Glucosidasehemmer
Metformin aus der Gruppe der Biguanide und die beiden Alpha-Glucosidasehemmer Acarbose und Miglitol besitzen kein Potenzial für klinisch relevante pharmakokinetische Wechselwirkungen [1, 2, 19].
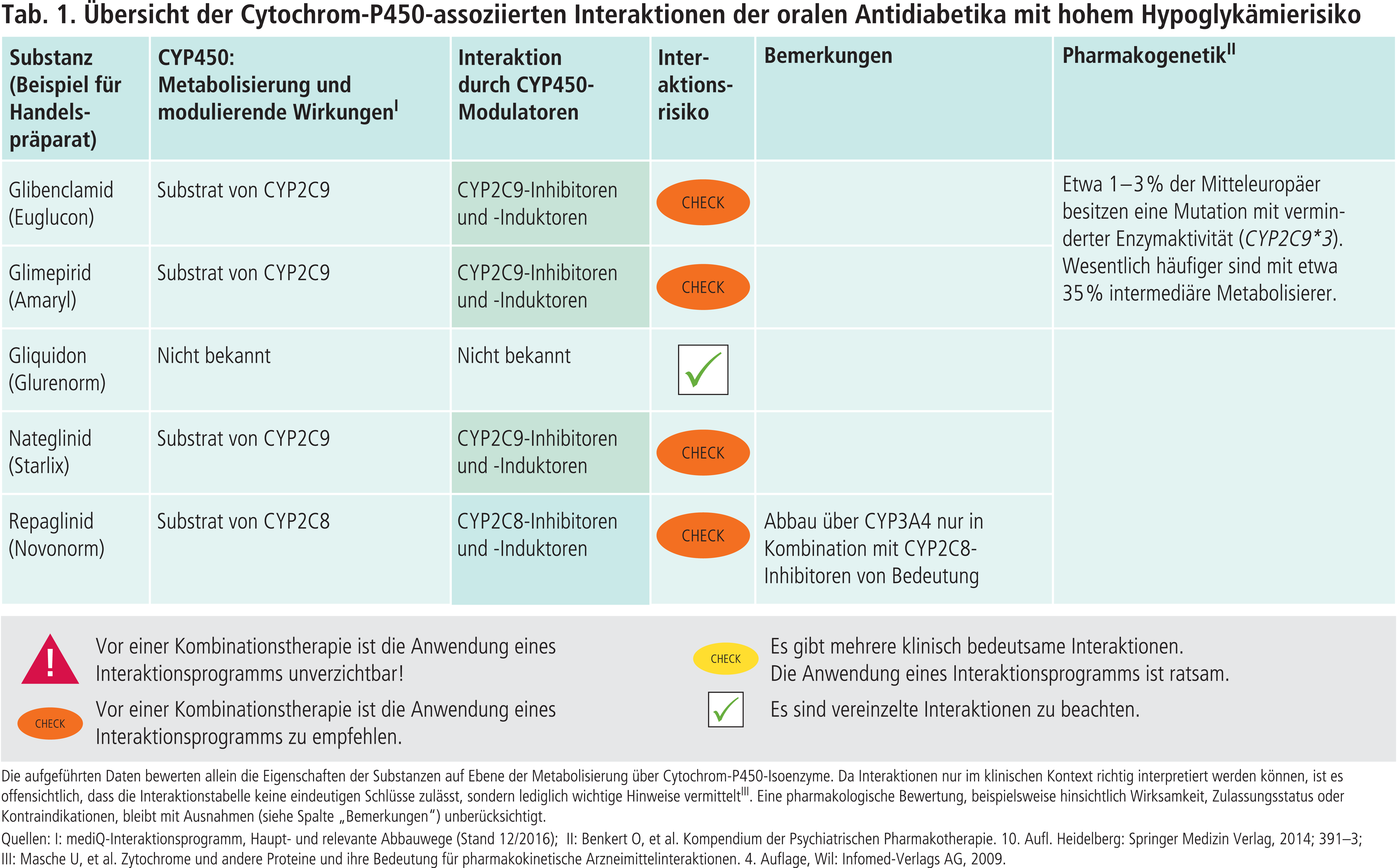

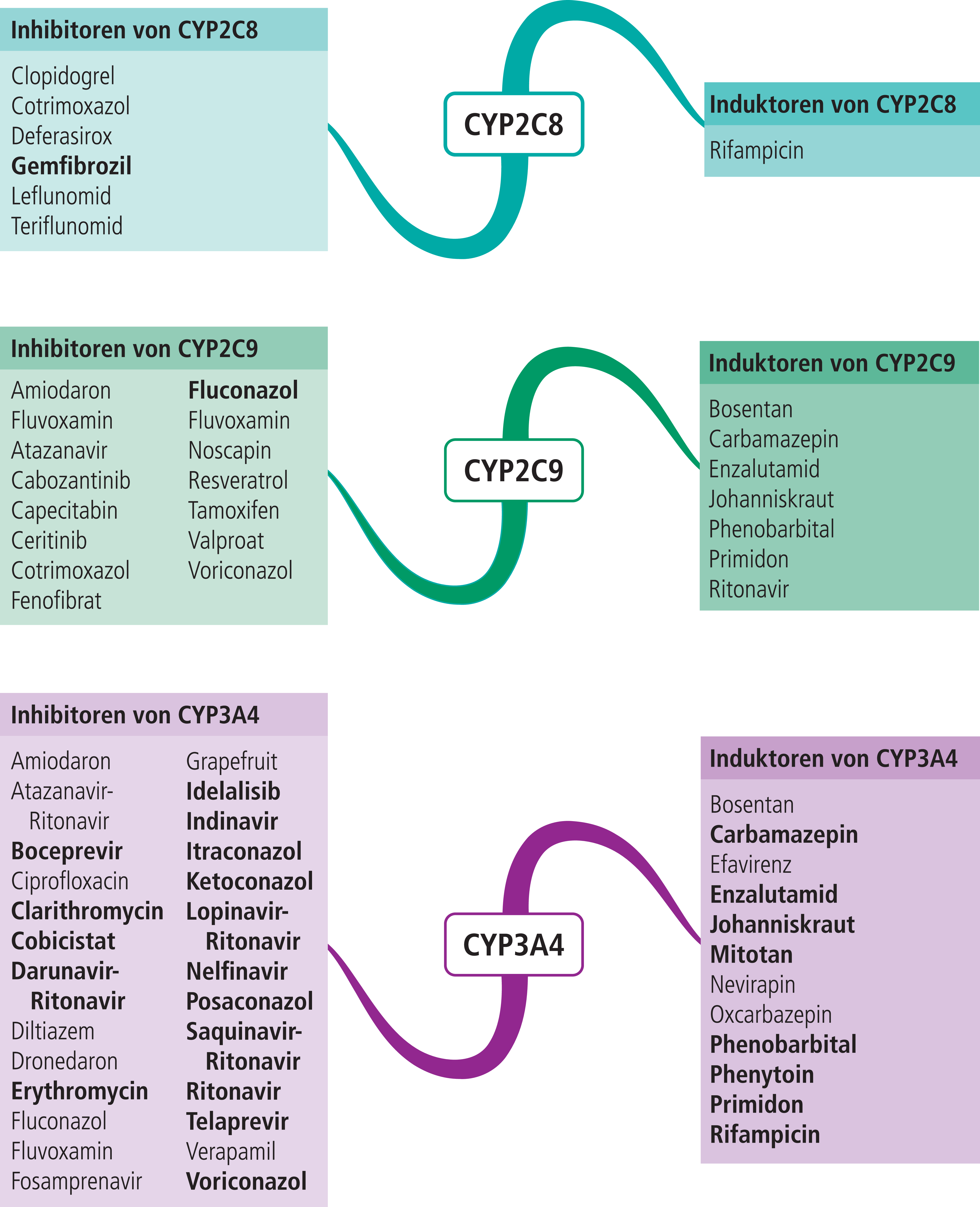
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C8, 2C9 und 3A4 (Stand: 12/2016) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Ahr HJ, Boberg M, Brendel E, et al. Pharmacokinetics of miglitol. Absorption, distribution, metabolism, and excretion following administration to rats, dogs, and man. Arzneimittelforschung 1997;47:734–45.
2. Ahr HJ, Boberg M, Krause HP, et al. Pharmacokinetics of acarbose. Part I: Absorption, concentration in plasma, metabolism and excretion after single administration of [14C]acarbose to rats, dogs and man. Arzneimittelforschung 1989;39:1254–60.
3. Aquilante CL, Niemi M, Gong L, et al. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 8. Pharmacogenet Genomics 2013;23:721–8.
4. Deng LJ, Wang F, Li HD. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol 2005;61:831–6.
5. Fachinformation Amaryl®. Stand: Dezember 2015.
6. Fachinformation Euglucon®. Stand: Juni 2016.
7. Fachinformation Glurenorm®. Stand: Dezember 2015.
8. Fachinformation Onglyza®. Stand: April 2016.
9. Juurlink DN, Mamdani M, Kopp A, et al. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA 2003;289:1652–8.
10. Kasichayanula S, Liu X, Lacreta F, et al. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin Pharmacokinet 2014;53:17–27.
11. Kirchheiner J, Roots I, Goldammer M, et al. Effect of genetic polymorphisms in cytochrome P450 (CYP) 2C9 and CYP2C8 on the pharmacokinetics of oral antidiabetic drugs. Clin Pharmacokinet 2005;44:1209–25.
12. Niemi M, Backman JT, Neuvonen M, et al. Effects of fluconazole and fluvoxamine on the pharmacokinetics and pharmacodynamics of glimepiride. Clin Pharmacol Ther 2001;69:194–200.
13. Niemi M, Backman JT, Neuvonen M, et al. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 2003;46:347–51.
14. Niemi M, Neuvonen M, Juntti-Patinen L, et al. Effect of fluconazole on the pharmacokinetics and pharmacodynamics of nateglinide. Clin Pharmacol Ther 2003;74:25–31.
15. Parekh TM, Raji M, Lin YL, et al. Hypoglycemia after antimicrobial drug prescription for older patients using sulfonylureas. JAMA Intern Med 2014;174:1605–12.
16. Patel CG, Li L, Girgis S. Two-way pharmacokinetic interaction studies between saxagliptin and cytochrome P450 substrates or inhibitors: simvastatin, diltiazem extended-release, and ketoconazole. Clin Pharmacol 2011;3:13–25.
17. Prescribing information Onglyza. Stand: April 2016.
18. Sabia H, Sunkara G, Liqueros-Saylan M, et al. Effect of a selective CYP2C9 inhibitor on the pharmacokinetics of nateglinide in healthy subjects. Eur J Clin Pharmacol 2004;60:407–12.
19. Scheen AJ. Drug interactions of clinical importance with antihyperglycaemic agents: an update. Drug Saf 2005;28:601–31.
20. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet 2014;53:213–25.
21. Scheen AJ. Pharmacokinetic interactions with thiazolidinediones. Clin Pharmacokinet 2007;46:1–12.
22. Schelleman H, Bilker WB, Brensinger CM, et al. Anti-infectives and risk of severe hypoglycemia in glipizide and glyburide users. Clin Pharmacol Ther 2010;88:214–22.
23. Tirkkonen T, Heikkilä P, Huupponen R, et al. Potential CYP2C9-mediated drug-drug interactions in hospitalized type 2 diabetes mellitus patients treated with the sulphonylureas glibenclamide, glimepiride or glipizide. J Intern Med 2010;268:359–66.
24. Upreti VV, Boulton DW, Li L, et al. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of saxagliptin, a dipeptidyl peptidase-4 inhibitor, in healthy subjects. Br J Clin Pharmacol 2011;72:92–102.
25. Vincent SH, Reed JR, Bergman AJ, et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos 2007;35:533–8.
*Nachdruck aus Krankenhauspharmazie 2017;38:42–6.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2017; 24(01)