Dieter Angersbach, Norbert Banik und Siegfried Schön, München
Obwohl heute zahlreiche Antidepressiva verfügbar sind, besteht ein großer medizinischer Bedarf nach wirksameren und verträglicheren Therapiealternativen. Eine Voraussetzung für die Zulassung solcher Medikamente ist der eindeutige Nachweis ihrer Wirksamkeit und Verträglichkeit. Sowohl vom wissenschaftlichen als auch gesellschaftlichen Standpunkt wäre es unvertretbar, Substanzen anzuwenden, die diesen Ansprüchen nicht genügen.
Zulassungsstudien, in denen die Prüfsubstanz nur mit einem bereits zugelassenen Verum verglichen wird (als so genannte Nicht-Unterlegenheits-Studien), liefern keinen ausreichenden Wirksamkeitsbeweis. In mehr als der Hälfte (52%) aller Vergleichsstudien waren später zugelassene Antidepressiva im Vergleich zu Plazebo unwirksam [7].
Studien, in denen die Prüfsubstanz nur mit einem zugelassenen Antidepressivum verglichen wird und die den Nachweis gestatten, ob die Prüfsubstanz mindestens so wirksam ist wie das zugelassene Antidepressivum, lassen daher darüber im Unklaren, ob eine über einen Plazebo-Effekt hinausreichende antidepressive Wirksamkeit erreicht wurde. Ein nur auf der Basis von Vergleichen mit zugelassenen Arzneimitteln entwickeltes Antidepressivum könnte wirksam oder unwirksam sein.
Aus diesem Grund fordern sowohl die amerikanische FDA [4] als auch die europäische EMEA [3] Daten aus Plazebo-kontrollierten Studien.
In den USA werden seit vielen Jahren Plazebo-kontrollierte Antidepressiva-Studien durchgeführt. So sind bis auf wenige Ausnahmen alle Plazebo-kontrollierten Paroxetin-Studien in den USA durchgeführt worden. In Europa können solche Studien ebenfalls durchgeführt werden. Die ethische Rechtfertigung dieser Studien ist jedoch weiterhin umstritten und die Diskussion über ihre Notwendigkeit wird kontrovers geführt. Als bedenklich eingestuft werden die vermeintlich unwirksame Behandlung und hohe Suizidgefährdung der Patienten im Plazebo-Arm [6]. Eine Auswertung von FDA-Daten zeigte jedoch [8], dass die Suizidalität in der Plazebo-Gruppe nicht höher ist als in der Verum-Gruppe. Die Daten zeigen ebenfalls, dass eine Plazebo-Behandlung in klinischen Studien gut wirksam ist [10] und der Wirkungsunterschied zum Antidepressivum geringer ist, als man aufgrund der klinischen Erfahrungen annehmen würde. Die hohe Response in der Plazebo-Gruppe wird auf die besondere pflegerische und ärztliche Zuwendung in solchen Studien zurückgeführt.
Die World Medical Association (WMA) stellt in der neuesten Überarbeitung der Helsinki-Deklaration [11] klar, dass sie Plazebo-kontrollierte Studien für ethisch vertretbar hält, wenn eine Plazebo-Kontrolle aus methodischen Gründen notwendig ist, um die Wirksamkeit und Verträglichkeit einer Therapie zu beurteilen.
Mithilfe einer Umfrage im Jahr 2002 in mehreren europäischen Ländern sollte nun beurteilt werden, ob und in wie weit es auch in Europa möglich ist, Plazebo-kontrollierte Antidepressiva-Studien durchzuführen und zur Entwicklung neuer Antidepressiva beizutragen.
Methodik
Es wurde ein Fragebogen an 973 Kliniken und Praxen in 21 europäischen Ländern verschickt, in dem die Teilnehmer (niedergelassene Psychiater, Psychiater in Klinikambulanzen) zur Teilnahmemöglichkeit an zwei Plazebo-kontrollierten Doppelblindstudien befragt wurden. Die Fragebogen wurden an Universitätskliniken (in Deutschland an alle) und an solche niedergelassenen Psychiater gesendet, die sich bereits zuvor an psychiatrischen Studien von der Firma GlaxoSmithKline (GSK) beteiligt hatten. Die regionalen Krankenhäuser (in Deutschland in der Regel psychiatrische Bezirkskrankenhäuser) wurden nach ihrer subjektiven Bekanntheit bei den GSK-Niederlassungen der jeweiligen Länder selektiert.
Bei einer Studie (Design 1) handelte es sich um eine vierarmige Studie mit drei festen Dosen der fiktiven Prüfsubstanz und der Plazebo-Gruppe (Phase-II-Dosisfindungsstudie). Die andere Studie (Design 2) war eine dreiarmige Studie mit jeweils flexiblen Dosen der fiktiven Prüfsubstanz, einer (nicht benannten) aktiven Vergleichssubstanz in fester Dosierung und Plazebo (Phase-III-Zulassungsstudie). Bei Design 1 bekommen alle Patienten eine Behandlung mit Plazebo oder der (eventuell unwirksamen) Testsubstanz, während bei Design 2 ein Drittel der Patienten ein zugelassenes (und damit als wirksam vorauszusetzendes) Antidepressivum erhält.
Ein- und Ausschlusskriterien waren für beide Studien identisch. Die wichtigsten Einschlusskriterien waren:
Ambulante Patienten
Alter wenigstens 18 Jahre
Schriftliches Einverständnis
Depressive Erkrankung nach DSM(Diagnostic and statistical manual of mental disorders)-IV
Depressive Episode nach DSM-IV
Bestehen der Episode seit wenigstens acht Wochen
MADRS(Montgomery-Asberg depression scale)-Score von 25 beim Screening
Die Teilnehmer wurden gebeten, folgende Fragen zu beantworten:
Interesse und Möglichkeit der Teilnahme?
Würde die Ethikkommission zustimmen?
Wie viele Patienten könnten in sechs Monaten gescreent werden?
Gründe für eine Teilnahme?
Gründe für eine Ablehnung?
Weitere Fragen betrafen Vorbereitungszeit, Studienerfahrung, insbesondere Erfahrung mit Plazebo-kontrollierten Studien, und persönliche Einschätzung der Notwendigkeit und Rechtfertigung von Plazebo-kontrollierten Studien.
Ergebnisse
Von 973 europaweit verschickten Fragebogen wurden 331 zurückgesandt (Gesamt-Antwortquote: 34%), die meisten davon aus:
Frankreich (n=128; landesspezifische Antwortquote: 24,4%),
Deutschland (n=78; 32,1%),
Belgien (n=19; 29,7%) und
Spanien (n=19; 73,1%).
Von den antwortenden Zentren oder Ärzten war über die Hälfte an einer Teilnahme interessiert, an Design 2 mit 60% etwas mehr als an Design 1 (55%). Der Anteil der Interessenten an Design 1 in den vier teilnehmerstärksten Ländern variierte von 47% (Belgien) bis 68% (Spanien). Die Unterschiede zwischen den Behandlungseinrichtungen waren gering (Abb. 1). Das Interesse variierte bei Design 1 zwischen 47% (regionale Krankenhäuser), 52% (Universitätskliniken) und 58% (niedergelassene Ärzte) und bei Design 2 zwischen 53% (regionale Krankenhäuser) und 62% (niedergelassene Ärzte).
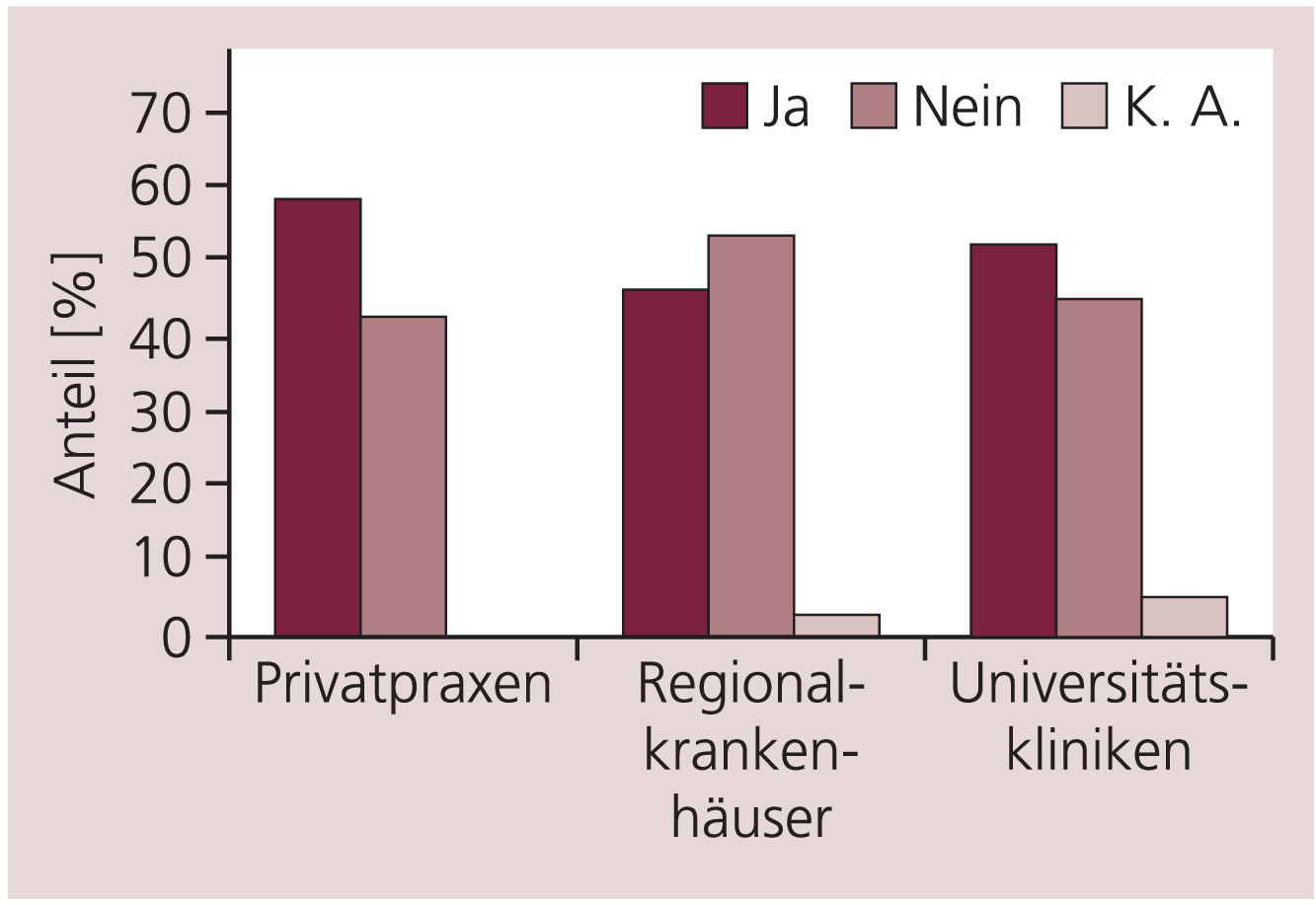
Abb. 1. Interesse an der Teilnahme an Plazebo-kontrollierten Antidepressiva-Studien in Abhängigkeit von den Behandlungseinrichtungen. Beurteilt wurde Design 1 (vierarmige Studie mit drei festen Dosen der Prüfsubstanz und der Plazebo-Gruppe).
Alle Interessierten hatten bereits an Antidepressiva-Studien (mit oder ohne Plazebo-Kontrolle) teilgenommen.
Die Erwartung eines positiven Votums durch die Ethikkommissionen war generell gering (Abb. 2). Nur 28% (Design 1) und 36% (Design 2) aller Teilnehmer erwarteten ein positives Votum. Jeweils mehr als die Hälfte, insbesondere die nicht interessierten Teilnehmer, machte keine Angaben oder konnte das Votum nicht einschätzen. Am zuversichtlichsten waren die Universitätskliniken: Ein positives Votum erwarten etwa 40% (Design 1) und 44% (Design 2). Am wenigsten zuversichtlich waren die niedergelassenen Teilnehmer: Nur 20 beziehungsweise 29% von ihnen erwartet ein positives Votum.
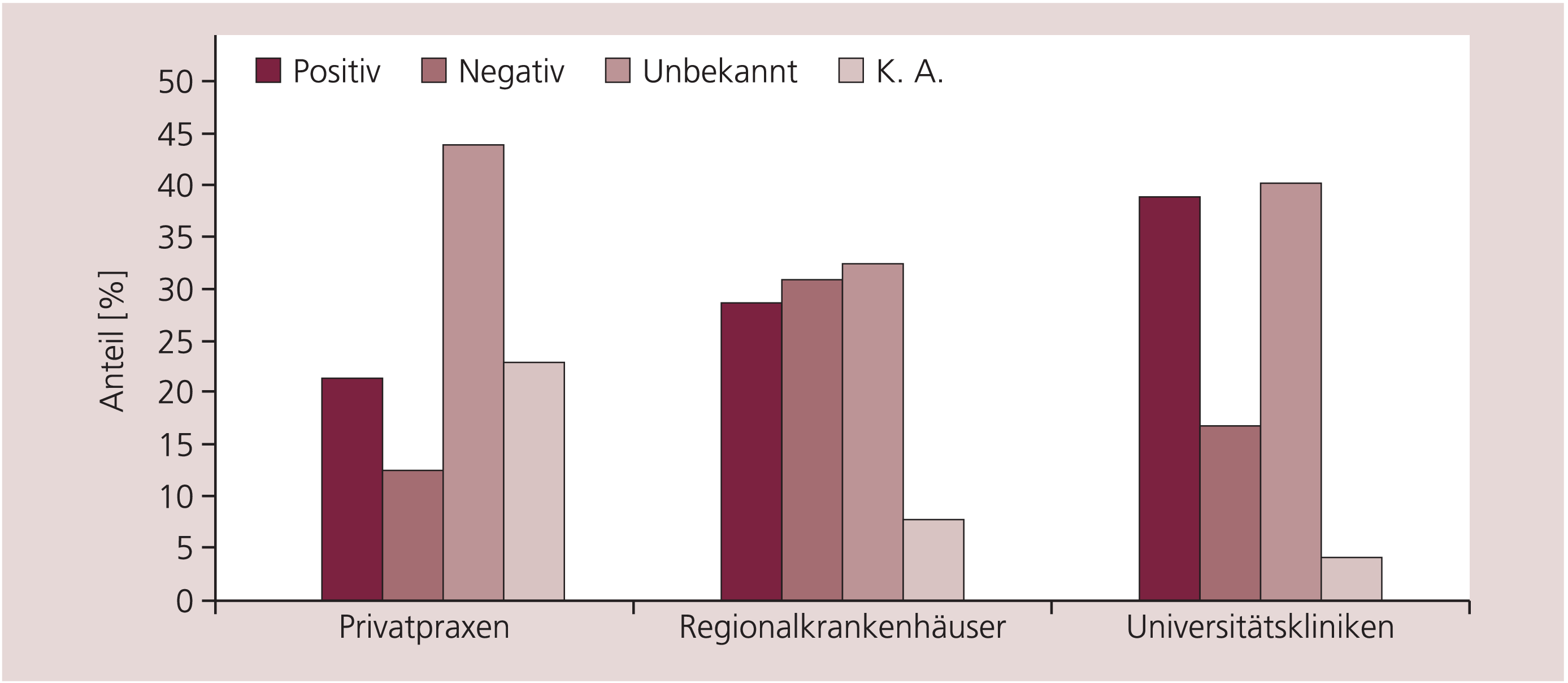
Abb. 2. Erwartetes Ethikvotum aller befragten Behandlungseinrichtungen, unabhängig von ihrem Interesse an der Teilnahme bei Plazebo-kontrollierten Antidepressiva-Studien. Beurteilt wurde Design 1 (vierarmige Studie mit drei festen Dosen der Prüfsubstanz und der Plazebo-Gruppe).
Zentren, die teilnehmen würden
Von den Teilnehmern mit Interesse an einer Studienteilnahme erwarteten nur 50% (Design 1) bis 60% (Design 2) ein positives Votum (Abb. 3). Von besonderem Interesse war nun die Einschätzung der Gruppe der Ärzte, die sowohl Interesse an der Teilnahme hatten als auch ein positives Votum erwarteten, wie viele Patienten sie innerhalb von sechs Monaten screenen könnten. Für eine Studie mit dem Design 1 würden europaweit 76 Zentren zwischen 7 und 11 Patienten überprüfen können. Danach könnten nach Einschätzung dieser Gruppe etwa 710 Patienten rekrutiert werden.
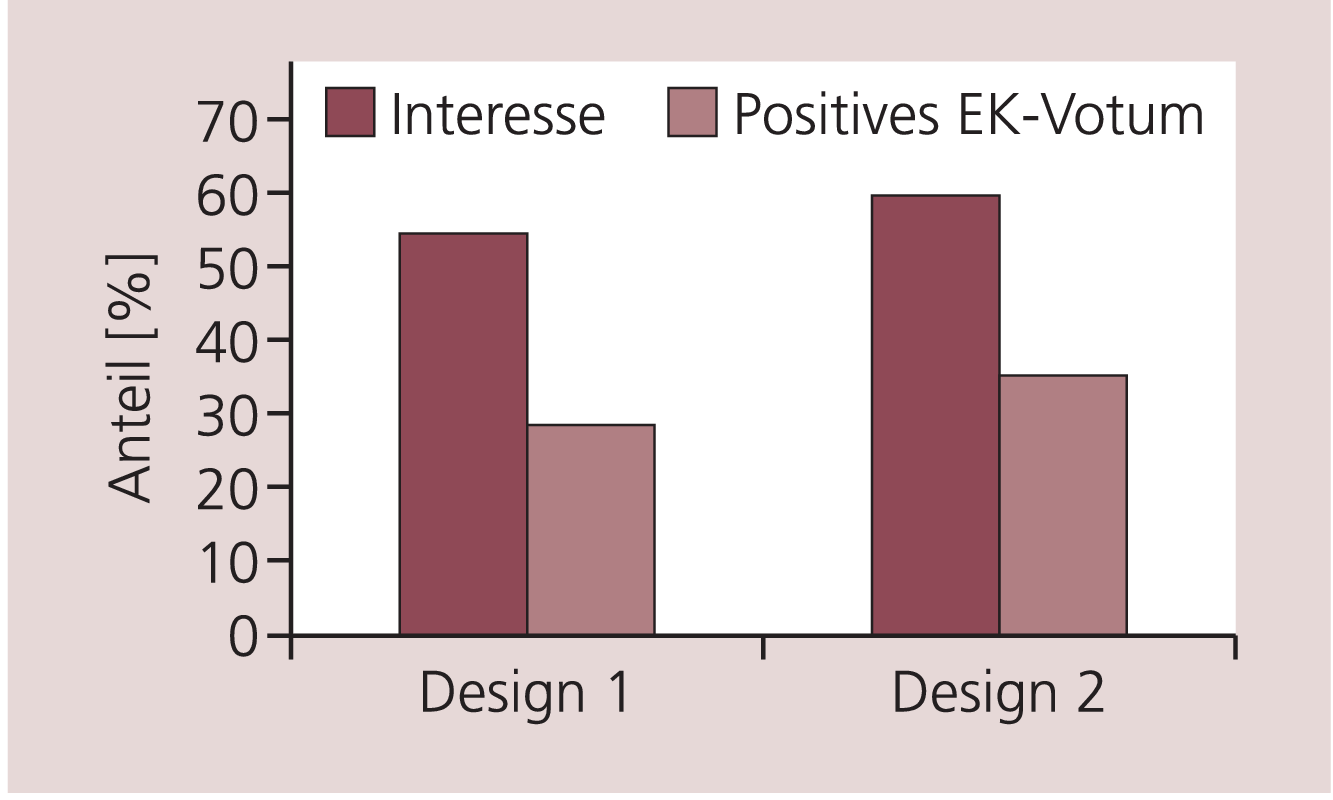
Abb. 3. Anteil der an der Teilnahme an Plazebo-kontrollierten Antidepressiva-Studien interessierten Prüfzentren und Anteil dieser Zentren, die ein positives Ethikvotum (EK-Votum) erwarten.
An einer Studie mit dem Design 2 (Prüfsubstanz gegen Antidepressivum und Plazebo) würden sich mehr Zentren beteiligen als an der Studie mit festen Dosen der Prüfsubstanz. 99 Zentren würden zwischen 7 und 13 Patienten einschließen und etwa 1200 Patienten screenen.
Als einer der Gründe für das Interesse an einer Teilnahme wurde am häufigsten „Interesse an der Forschung“ angekreuzt (93%). Für über 80% der Interessierten war auch die Notwendigkeit Plazebo-kontrollierter Studien und der Entwicklung neuer Medikamente ein wesentlicher Grund. Weiterhin wurde darauf hingewiesen, dass die Studien zur Verbesserung des Behandlungsstandards beitragen können. Häufig wurden auch ökonomische Gründe genannt.
Als Wünsche an den Sponsor wurde von den Universitätskliniken am häufigsten die Mitarbeit am Studiendesign genannt. Die niedergelassenen Ärzte wünschten sich naturgemäß, dass die Studien mit ambulanten Patienten durchgeführt werden. Zwischen 35 und 50% der drei Behandlungseinrichtungen würden lieber Patienten einschließen, die nicht zu schwer krank sind (HAMD[Hamilton depression scale]-17-Score < 24). Weitere Wünsche betrafen ein engmaschiges Monitoring der Patienten, die psychotrope Begleitmedikation (Anxiolytika und Sedativa sollten erlaubt sein) oder ein gutes Training für das Studienpersonal.
Zentren, die eine Teilnahme ablehnen
Häufige Gründe für die Ablehnung der Teilnahme waren die ablehnende Haltung der Ethikkommission und der Klinikverwaltung. Viele Ärzte (60% der Niedergelassenen der Gruppe der Nicht-Interessierten) glauben, dass sich keine Patienten bereitfinden werden, sich im Rahmen von Plazebo-kontrollierten Studien behandeln zu lassen. Bemerkenswert ist, dass relativ viele Universitätskliniken Plazebo-kontrollierte Studien für unethisch (30% dieser Gruppe) oder für unnötig halten (37%). Weitere Gründe wurden genannt, wie fehlende Kapazität, zu befürchtende Schwierigkeiten mit der Krankenkasse oder auch mangelnde Erfahrung mit Plazebo-kontrollierten Studien.
Diskussion
Diese Umfrage wurde nicht mit dem Ziel durchgeführt, ein nach Marktforschungskriterien valides Bild der Beurteilung Plazebo-kontrollierter Antidepressiva-Studien und der Beteiligung von Ärzten an solchen Studien zu erhalten. Ebenso wenig sollte damit Überzeugungsarbeit pro Plazebo geleistet werden. Die Umfrage war vielmehr als Entscheidungshilfe konzipiert, ob GSK solche Studien für Europa mit Aussicht auf Erfolg planen kann oder aber mit erheblichen Problemen rechnen muss und daher Zulassungsstudien für Antidepressiva besser in nicht-europäischen Ländern belassen sollte. Diese Frage könnte, wenn sie auch von anderen forschenden Firmen gestellt wird, den europäischen Forschungsstandort berühren.
Die Mitarbeit der angeschriebenen Ärzte war mit einer Rücklaufquote von 34% eher höher als erwartet und die Umfrage kann damit auch als ausreichende Basis für eine Planungsentscheidung angesehen werden. Bei unverbindlichen Umfragen, beispielsweise im Rahmen einer einfachen Marktforschung, liegt die Rücklaufquote bei etwa 10%. Die befriedigende Rücklaufquote bei dieser Umfrage wurde möglicherweise durch eine (kleine) Honorierung und durch Erinnerungstelefonate erreicht.
Es ist ermutigend für die Planung Plazebo-kontrollierter Studien, dass sich mehr als die Hälfte der Ärzte beteiligen würde. Ein Unsicherheitsfaktor in der Beurteilung der Machbarkeit dieser Studien ist die mögliche Haltung der Klinikverwaltung und insbesondere der Ethikkommissionen. Die Umfrage hat gezeigt, dass vielen Teilnehmern die Haltung ihrer Ethikkommission zu Plazebo-kontrollierten Studien unbekannt war und dass offensichtlich zu diesen Studien noch keine konkreten Erfahrungen in der Zusammenarbeit mit der Kommission vorlagen.
In dieser Situation wäre es sinnvoll, dass Sponsoren, Prüfärzte, wissenschaftlich arbeitende Psychiater, Mitglieder von Ethikkommissionen und Vertreter der Zulassungsbehörde gemeinsam über wissenschaftliche und ethische Fragen beraten, so dass die Prüfprotokolle Plazebo-kontrollierter Studien höchsten Ansprüchen an Patientensicherheit und Wissenschaftlichkeit genügen und von den Kommissionen positiv beurteilt werden können. Einige solcher Gespräche haben, von GSK initiiert, bereits stattgefunden und zu hilfreichen Ergebnissen geführt [1, 2]. Nach Ansicht der konsultierten Expertenrunde sind Plazebo-kontrollierte Studien wissenschaftlich notwendig, ethisch und machbar [1] und sie sind notwendig für die Entwicklung wirksamerer und verträglicherer Antidepressiva [2].
Mit solchen Gesprächen würde gleichzeitig auch eine der Forderungen, insbesondere von Universitätskliniken, am Design der Prüfpläne mitzuarbeiten, prinzipiell erfüllt.
Ein weiterer Wunsch, nur Patienten einzuschließen, die nicht schwer depressiv sind (schwere Depression: HAMD ≥ 24), ist zwar verständlich, birgt aber auch Probleme. Ein Antidepressivum sollte nämlich besonders bei schwerkranken Patienten seine Wirksamkeit beweisen. Weiterhin ist beispielsweise das Wirksamkeitskriterium „Response“ (Reduktion des Depressions-Scores um wenigstens 50%) bei leicht Kranken einfacher zu erreichen als bei schwer Kranken [9]. Das bedeutet, dass bei leicht kranken Patienten die Ansprechrate unter Plazebo deutlich ansteigt und dass es statistisch umso schwieriger wird, die Wirksamkeit der Prüfsubstanz zu zeigen, je weniger krank die Patienten sind. Die Krankheit sollte daher den Schweregrad einer behandlungsbedürftigen Depression haben (HAMD-17-Score in der Regel ≥ 18). Darüber hinaus sollten aber die Patienten nicht nach dem Krankheitsschweregrad allein selektiert werden und die Studie sollte zudem die alltägliche Behandlungsrealität reflektieren: So sollte sowohl eine Dosiserhöhung bei geringer Wirksamkeit als auch eine Dosissenkung bei Unverträglichkeit möglich und die erlaubte Begleitmedikation nicht allzu stark eingeschränkt sein.
Eine Herausforderung, die in der vorliegenden Umfrage häufig als Grund für die Ablehnung Plazebo-kontrollierter Studien angegeben wurde, ist die Schwierigkeit der Patientenrekrutierung. Patienten setzen eine Plazebo-Behandlung grundsätzlich mit einer Nicht-Behandlung gleich, sofern sie nicht über deren Wesen aufgeklärt werden. Natürlich muss es dem Patienten nach umfangreicher Erklärung (der Vorzüge und der Risiken) der Studie freistehen, ohne nachteilige Folgen eine Teilnahme abzulehnen. Dies tun die Patienten bei Plazebo-kontrollierten Studien häufiger als bei anderen Vergleichsstudien. Tatsächlich ist die Screening-Ausfallrate in Plazebo-kontrollierten Studien erheblich, so dass erheblich mehr Patienten gescreent werden müssen als beispielsweise bei einem Vergleich zweier Antidepressiva.
Die wesentlichen Unbekannten bei der Planung Plazebo-kontrollierter Antidepressiva-Studien sind das Interesse der Prüfärzte, die Zustimmung der Ethikkommissionen und die Bereitschaft der Patienten zur Teilnahme. Die vorliegende Umfrage zur Machbarkeit solcher Studien in Europa hat nur das Interesse von Prüfärzten erfragt. Das Ergebnis war ermutigend und selbst auf einer konservativen Basis (den Patientenzahlen der interessierten Ärzte, die ein positives Votum erwarten) erscheinen diese Studien realisierbar.
Von weiterem Interesse wäre, inwieweit die Ergebnisse dieser Umfrage auf die Machbarkeit Plazebo-kontrollierter Studien bei anderen psychischen Störungen, wie Angsterkrankungen oder der Schizophrenie, übertragbar ist. Während uns zu Angststörungen oder der Zwangsstörung keine Machbarkeitsumfragen bekannt sind, zeigte eine Erhebung zur Machbarkeit Plazebo-kontrollierter Akutstudien mit Antipsychotika [5], dass die Bereitschaft forschender Psychiater, an solchen Studien teilzunehmen, vergleichsweise gering ist. Während in der vorliegenden Umfrage mehr als die Hälfte der Antwortenden (55–60%) an der Teilnahme Plazebo-kontrollierter Antidepressiva-Studien interessiert war, konnten sich nur 30% der Antwortenden eine Teilnahme an Plazebo-kontrollierten Antipsychotika-Studien vorstellen. Ein Drittel der Prüfärzte, die bereits an einer Plazebo-kontrollierten Schizophrenie-Studie teilgenommen hatten, war nicht mehr bereit, sich an einer weiteren zu beteiligen. Die Frage, ob die unterschiedlichen Ergebnisse beider Umfragen tatsächlich eine unterschiedliche Teilnahmebereitschaft bei Depressions- und Schizophrenie-Studien aufzeigen, ist dennoch nicht eindeutig zu beantworten, da das vorliegende Umfrageergebnis trotz relativ hoher Rücklaufquoten (34%) nicht repräsentativ ist. Der Vergleich der Ergebnisse beider Umfragen lässt jedoch darauf schließen, dass Plazebo-kontrollierte Antidepressiva-Studien eher machbar sind als Plazebo-kontrollierte Antipsychotika-Studien.
Literatur
1. Adam D, Kasper S, Möller H-J, Singer EA. Placebo-controlled trials in major depression are necessary and ethically justifiable. Eur Arch Psychiatry Clin Neurosci 2005;255:258–60.
2. Baldwin D, Broich K, Fritze J, Kasper S, et al. Placebo-controlled studies in depression: necessary, ethical and feasible. Eur Arch Psychiatry Clin Neurosci 2003;253:22–8.
3. EMEA – Note for guidance on clinical investigation of medicinal products in the treatment of depression. London, 2002.
4. FDA – Guidelines for the evaluation of antidepressant drugs. Rockville, 1977.
5. Fleischhacker WW, Burns T for the European group for research in schizophrenia. Feasibility of placebo-controlled clinical trials of antipsychotic compounds in Europe. Psychopharmacology 2002;162:82–4.
6. Helmchen H. Ethisch nicht zu vertreten! Der Neurologe und Psychiater 2003;5:11–2.
7. Khan A, Khan S, Brown WA. Are placebo controls necessary to test new antidepressants and anxiolytics? Int J Neuropsychopharmacol 2002;5:193–7.
8. Khan A, Warner HA, Brown WA. Symptom reduction and suicide risk in patients treated with placebo in antidepressant trials. Arch Gen Psychiatry 2000;57:311–7.
9. Szegedi A, Wetzel H, Angersbach D, Philipp M, et al. Response to treatment in minor and major depression: results of a double-blind comparative study with paroxetine and maprotiline. J Affect Disord 1997;45:167–78.
10. Walsh BT, Seidman SN, Sysko R, Could M. Placebo response in studies of major depression. JAMA 2002;287:1840–7.
11. World Medical Association: Declaration of Helsinki – Ethical principles for medical research involving human subjects. Edinburgh, 2000.
Priv.-Doz. Dr. Dieter Angersbach, Dr. Dr. Norbert Banik, Prof. Dr. Siegfried Schön, GlaxoSmithKline GmbH & Co. KG, Theresienhöhe 11, 80339 München, E-Mail: dieter.angersbach@gsk.com
PPT – Leserforum
Zum Beitrag „Escitalopram bei berufstätigen Menschen – Ergebnisse einer Anwendungsbeobachtung an 2378 Patienten“ (Psychopharmakotherapie 2006;13:142–6):
Ein „Hauptzielparameter“ in dieser Anwendungsbeobachtung (AWB) waren die Krankenstandstage in den drei Monaten vor und während der Behandlung mit Escitalopram. Von ursprünglich 2378 Patienten wurden die Krankenstandstage bei 2299 Patienten analysiert (Abb. 3). Diese reduzierten sich durchschnittlich von 11 auf 5,4 Tage.
Der Rückgang der Krankentage könnte auch durch saisonale Effekte (Winter/Frühjahr) verursacht sein, was nicht diskutiert wird.
Des Weiteren waren über die Hälfte der Patienten (55,6%) zu Studienbeginn nicht vorbehandelt. Wollte man die Studieneffekte (Krankenstandstage, klinischer Verlauf) allein auf die Medikation reduzieren, so zeigte sich, dass durch die antidepressive Therapie eine Besserung der Zielparameter eintrat. Ein Vorteil von Escitalopram wird jedoch nicht gezeigt; eine Vergleichsmedikation ist nicht gegeben. Eine Auswertung der vorbehandelten Patienten allein wäre möglich gewesen, zumindest hätte dies diskutiert werden müssen.
Auch diese AWB liefert keinen weiteren Hinweis auf die Überlegenheit einer Escitalopram-Therapie gegenüber Citalopram, wie dies im Editorial suggeriert wird. Vielmehr wird deutlich, dass durch die AWB 2287 (2378 x 96,2%) Patienten neu auf Escitalopram eingestellt wurden (was sicher auch ein Ziel einer AWB ist). Die Therapie der Patienten entspricht bei 10 mg/d 297310 Euro je Quartal (Preise Deutschland 01.08.06). Bei Anwendung eines Citalopram-Generikums ließen sich über 50% einsparen. Dies sollte in einer Publikation, die auf gesundheitsökonomische Aspekte eingeht, und in einem kritischen Journal berücksichtigt werden.
Dr. Holger Neye, Kassenärztliche Vereinigung Niedersachsen, Berliner Allee 22, 30175 Hannover
Stellungnahme der Autoren:
Wir danken Dr. Neye für sein Schreiben, in dem er einige wichtige Fragen aufwirft, die wir gerne beantworten möchten:
Der Rückgang der Krankenstandstage ist nicht durch saisonale Effekte erklärbar: Von 1469 Patienten war das genaue Datum des Einschlusses in die Studie erhebbar. Unter Zuhilfenahme von klimatologischen Daten konnte für jeden dieser Patienten die durchschnittliche tägliche Sonnenscheindauer im Intervall von drei Monaten vor und nach dem Studieneinschluss berechnet werden. Tatsächlich war hier ein deutlicher und statistisch signifikanter Unterschied fassbar (t=68,021, df=1468, p<0,001): In den drei Monaten vor der Studie betrug die mittlere tägliche Sonnenscheindauer 5,9 Stunden versus 2,8 Stunden in den drei Monaten unter Therapie mit Escitalopram. Dies lässt die Schlussfolgerung zu, dass einige unserer Patienten während der AWB mit Escitalopram eine zusätzliche saisonale Verschlechterung ihrer depressiven Erkrankung erfahren haben und unsere Ergebnisse den wahren Unterschied in den Krankenstandstagen vor und nach der Therapie mit Escitalopram eher unterschätzen.
Diese AWB war als offene Studie ohne Komparatorsubstanz konzipiert und natürlich nicht auf einen direkten Vergleich mit anderen Therapien ausgelegt. Demzufolge können wir aus der vorliegenden Arbeit auch nicht auf Unterschiede zwischen Escitalopram und anderen Antidepressiva schließen. Im Editorial [5] wird nicht auf unsere Untersuchung, sondern auf Metaanalysen [1, 3, 4] Bezug genommen, die eine Überlegenheit von Escitalopram gegenüber anderen Antidepressiva, insbesondere Citalopram, in Bezug auf die Wirkung und die Wirkungslatenz belegen. Weiters lassen pharmakoökonomische Studien [2, 6] den Schluss zu, dass Escitalopram verglichen mit Citalopram (selbst bei fehlenden Akquisitionskosten für Citalopram) eine kostenökonomische Therapie darstellt.
In unserer Stichprobe liegen von 807 Patienten Informationen über eine Vorbehandlung mit Antidepressiva vor: Die Gruppe der Patienten, die schon vor der Anwendungsbeobachtung mit Antidepressiva vorbehandelt worden war, hatte durchschnittlich mehr Krankenstandstage (15,1±19,9 Tage) als die Patienten ohne Vortherapien (10,5±17,1 Tage; t=–3,337, df=665,173, p=0,001). In beiden Gruppen kam es während der dreimonatigen Therapie mit Escitalopram zu einer signifikanten Reduktion der Krankenstandstage (Patienten mit antidepressiver Vorbehandlung: 7,6±16,0 Tage; t=7,995, df=317, p<0,001; Patienten ohne Vortherapien: 6,5±15,9 Tage; t=4,562, df=390, p<0,001). Ein statistisch signifikanter Unterschied zwischen diesen beiden Gruppen unter Therapie mit Escitalopram war nicht mehr fassbar (t=–0,878, df=734, p=0,380).
Nachdem 68,7% der mit Antidepressiva vorbehandelten Patienten vor der Behandlung mit Escitalopram eine Therapie mit einem anderen SSRI (z.B. Citalopram) erhalten hatten und meist aufgrund von insuffizienter Wirkung auf Escitalopram umgestellt wurden, ist die deutliche Reduktion der Krankenstandstage in dieser Subgruppe besonders eindrucksvoll. Eine weitere Therapie mit einem generischen Citalopram-Präparat wäre für viele dieser Patienten wahrscheinlich weder klinisch zielführend noch vom pharmakoökonomischen Standpunkt sinnvoll gewesen.
Literatur
1. Auquier P, Robitail S, Llorca P-M, Rive B. Comparison of escitalopram and citalopram efficacy: a meta-analysis. Int J Psychiatr Clin Pract 2003;7:259–68.
2. Hemels ME, Kasper S, Walter E, Einarson TR. Cost-effectiveness of escitalopram versus citalopram in the treatment of severe depression. Ann Pharmacother 2004;38:954–60.
3. Kasper S, Spadone C, Verpillat P, Angst J. Onset of action of escitalopram compared with other antidepressants: results of a pooled analysis. Int Clin Psychopharmacol 2006;21:105–10.
4. Kennedy SH, Andersen HF, Lam RW. Efficacy of escitalopram in the treatment of major depressive disorder compared with conventional selective serotonin reuptake inhibitors and venlafaxine XR: a meta-analysis. J Psychiatry Neurosci 2006;31:122–31.
5. Müller WE. Alte und neue Antidepressiva im Fokus. Psychopharmakotherapie 2006;13:129.
6. Wade AG, Toumi I, Hemels ME. A pharmacoeconomic evaluation of escitalopram versus citalopram in the treatment of severe depression in the United Kingdom. Clin Ther 2005;27:486–96.
Doz. Dr. Dietmar Winkler, Dr. Edda Pjrek, Dr. Nikolas Klein, O. Univ. Prof. Dr. Dr.h.c. Siegfried Kasper, Klinische Abteilung für Allgemeine Psychiatrie, Medizinische Universität Wien, Währinger Gürtel 18–20, 1090 Wien, Österreich, E-Mail: dietmar.winkler@meduniwien.ac.at
Zum Beitrag „Dopaminagonisten in der Behandlung des Restless-Legs-Syndroms“ (Psychopharmakotherapie 2006;13:191–6):
In der genannten Arbeit heißt es: „Zunächst sollte eine entsprechende Eisen-Substitution vorgenommen werden, da sie die RLS-Symptome in einigen Fällen verbessern kann.“ Als Quelle wird eine Arbeit von Davis et al., J Neurol 2000;43:70–5 angegeben.
Dabei handelt es sich um eine randomisierte, doppelblinde Studie über 12 Wochen. Zwischen Verum und Plazebo fanden sich keine signifikanten Differenzen, weder in den primären noch in den sekundären Outcome-Parametern. Der einzige Satz unter der Rubrik „Conclusions“ im Abstract dieser Arbeit lautet wörtlich: „Eisen scheint keine effektive empirische Behandlung für das Restless-Legs-Syndrom zu sein.“ (Iron sulfate does not appear to be an effective empiric treatment for restless legs syndrome.)
Dies ist nicht die Art der Literaturaufbereitung, die ein praktisch tätiger Arzt von einem Übersichtsreferat erwartet.
Dr. med. Matthias Mindach, Humboldstr. 5, 15230 Frankfurt (Oder), E-Mail: Mindach@ibel.de
Stellungnahme der Autorin:
Da Fokus meines Beitrags die Behandlung des Restless-Legs-Syndroms (RLS) mit Dopaminagonisten war, konnte ich auf die Bedeutung des Eisens beim RLS nur sehr verkürzt eingehen und bitte zu entschuldigen, dass entsprechend auch nur eine, in diesem Fall nicht glücklich gewählte, Literaturstelle zitiert werden konnte.
Als wichtigster Parameter eines sekundären RLS hat sich Ferritin gezeigt, das hierbei niedrig normal oder erniedrigt gemessen wird (s. auch Leitlinien der Deutschen Gesellschaft für Neurologie und Allen et al. 2003). Zahlreiche Untersuchungen (kranielle Kernspintomographie [cMRT], transkranielle Dopplersonographie [TCD], Liquordiagnostik, Autopsien, Korrelationen von niedrigen Ferritinwerten mit dem Ausmaß der Symptome) konnten zeigen, dass Eisen in der Pathophysiologie des RLS eine bedeutende Rolle spielt (z.B. Earley et al. 2006, Clardy et al. 2006, Schmidauer et al. 2005, Mizuno et al. 2005, Ulfberg and Nystrom 2004, Sun et al. 1998). Entsprechend wird eine Eisen-Dopamin-Verbindung als ein zentraler Faktor in der Pathophysiologie des RLS angesehen (Allen 2004).
In der Tat gibt es leider nur sehr wenige Studien zu einer Therapie des RLS mit Eisen. Earley und Mitarbeiter haben 2004 Ergebnisse einer offenen Studie mit intravenöser Eisen-Substitution beim RLS veröffentlicht, von der ein Großteil der Behandelten profitierte. Weitere randomisierte, doppelblinde Studien stehen bislang noch aus. Bekannt ist allerdings, dass ein erniedrigtes Ferritin <50 µg/l mit einer Zunahme der RLS-Symptome einhergehen kann (O’Keeffe 1994, Sun et al. 1998, Allen et al. 2003), entsprechend sollte dann eine Eisen-Substitution erfolgen, da in vielen Fällen die Beschwerden des RLS dadurch gelindert werden.
Priv.-Doz. Dr. med. Svenja Happe, Abteilung Klinische Neurophysiologie, Klinikum Bremen-Ost, Züricher Str. 40, 28325 Bremen, E-Mail: svenja.happe@klinikum-bremen-ost.de
Placebo-controlled antidepressant studies in Europe – a feasibility survey
Despite the availability of numerous effective antidepressants, psychiatrists agree that in everyday clinical practice there is a large need of still more potent substances. From a scientific point of view, placebo-controlled, double-blind studies are inevitable for the marketing approval of new drugs and constitute a regulatory requirement. In a “Note of Clarification” on the declaration of Helsinki, the World Medical Association (WMA) clarified that on certain conditions, which in our opinion also apply to antidepressants, placebo-controlled studies are admissible from an ethical point of view, even if effective drugs are available. In order to explore the feasibility of such studies in Europe, a survey was conducted in more than 900 psychiatric centers (psychiatrists established in private practices, smaller hospitals and university hospitals) in 21 European countries to examine their interest and potential for participating in predefined trial designs. Just over half of the 331 centers answering the survey were interested in participating, but no more than every other of these centers expected the respective ethics committee to provide an approval for such a study. It is envisaged that these centers could screen about 800 depressive patients for a two-armed phase II study and about 1,200 for a three-armed phase III study within one year. The numerous centers which were uncertain about their ethics committee’s opinion might possess a large potential for contributing additional patients.
Keywords: Antidepressants, clinical studies, placebo control, feasibility, center recruitment
Psychopharmakotherapie 2006; 13(06)