Jörg Czekalla, Neuss
Im Folgenden werden die wichtigsten Validitätskriterien der Evidence-based Medicine (EbM) erläutert, die den Umgang mit der Terminologie, die in EbM-Analysen von Publikationen angewandt wird, erleichtern sollen. Diese Erläuterungen können jedoch nur einen Überblick über diese umfangreiche Thematik geben. Für eine EbM-Analyse sind zusätzlich detaillierte Überprüfungen der angewandten statistischen Verfahren notwendig.
Aussagekraft und Verallgemeinerungsfähigkeit eines Therapieergebnisses können mit interner und externer Validität einer Studie beurteilt werden. Die interne Validität beschreibt die Gültigkeit der Aussage einer Studie und soll in der Studienplanung zum Beispiel durch die Struktur-, die Beobachtungs- und die Behandlungsgleichheit von Vergleichsgruppen gesichert werden.
Die externe Validität beschreibt die Gültigkeit der Aussagen für eine größere Zielpopulation. Ein hochwertiges Studiendesign soll die Interpretation erlauben, dass die beobachteten Studienergebnisse nur durch den zu untersuchenden Effekt erklärt werden können.
Bei der Validitätsprüfung richtet sich das Augenmerk insbesondere auf systematische Fehler oder Verzerrungen (Bias), wodurch die Studienergebnisse in eine bestimmte Richtung beeinflusst werden könnten, beispielsweise durch Studienabbrecher, inadäquate Randomisierung der Patienten und Behandlungsunterschiede.
Therapiestudien: Beurteilung des Studiendesigns
Randomisierte, kontrollierte und doppelblinde Studien gelten als der Goldstandard in der klinischen Forschung, da durch dieses Design ein möglicher Bias minimiert wird. Die Methodik muss jedoch in jedem Einzelfall kritisch geprüft werden. Wenn zum Beispiel in psychiatrischen Studien eine Intervention im Rahmen einer mittelfristig bis langfristig progredienten Erkrankung (z.B. Demenz) vorgenommen wird, sind parallele Behandlungsgruppen ideal. Im Rahmen akuter stationärer Behandlungen können auch so genannte Cross-over-Studien durchgeführt werden. In diesen Studien erhält jeder Patient in zufälliger Reihenfolge sowohl die zu prüfende Therapie als auch die Kontrolle, oft durch eine therapiefreie Wash-out-Phase unterbrochen. Der Vorteil besteht darin, dass bei gleicher statistischer Power, einen Therapieunterschied aufzudecken, weniger Patienten eingeschlossen werden müssen als in einer Studie mit parallelen Gruppen. Nachteilig kann jedoch – beeinflusst durch die Reihenfolge der Therapien – das Auftreten von Carry-over- und Perioden-Effekten (Übertragungs-Effekten, Wirkungsübertragungen) sein, welche die Ergebnisse beeinflussen können. Liegen derartige Effekte vor, so kann nur die erste Behandlungsperiode ausgewertet werden.
Randomisierung
Die randomisierte, kontrollierte Studie der Phase III (RCT) ist die Methode der Wahl zum Wirksamkeitsnachweis. Sie ist in der wissenschaftlich-medizinischen Forschung auf breiter Basis anerkannt und bietet günstige methodische Voraussetzungen für den Nachweis eines Kausalzusammenhangs. Die RCT kann zwei- oder mehrarmig, offen oder verblindet sein. Die Teilnehmer werden nach dem Zufallsprinzip der Interventionsgruppe (z.B. medikamentöse Behandlung) oder einer Kontrollgruppe (z.B. Plazebo oder bisherige Standardtherapie) zugeordnet. Im Idealfall geschieht dies zentral mit IT-gestützten Verfahren an einem vom Prüfzentrum getrennten Ort. Ziel ist hierbei, die gleichmäßige Verteilung bekannter und unbekannter prognostischer Faktoren in den Behandlungsgruppen zu garantieren.
Methoden, die dem Kliniker Einblick in das Verfahren gewähren oder Einfluss ermöglichen, sind nicht akzeptabel. Die Randomisierung verhindert die bewusste oder unbewusste Zuteilung von vermeintlich geeigneteren Patienten zu einer der vorgesehenen Behandlungen. Dies würde zu einer Verzerrung der Ergebnisse führen, da der Kliniker manche Patienten möglicherweise eher für eine Behandlung vorsieht als andere, beispielsweise Patienten mit einer schwereren Form der Erkrankung seltener der Plazebo-Gruppe oder der vermutlich weniger gut wirksamen Therapie zuordnet.
Die Zielsetzung eines randomisierten, kontrollierten Studiendesigns ist in der Regel die Evaluation eines einzigen Effekts (z.B. Medikamenten-Wirkung vs. Plazebo) in einer definierten Patientengruppe. In einem prospektiven Design werden Daten von Ereignissen erhoben, die nach der Entscheidung, eine Studie durchzuführen, erhoben werden.
Stratifizierung
Enthält die Grundgesamtheit von Studienpatienten unterschiedliche Teilgesamtheiten, dann werden geschichtete Zufallsstichproben (Strata) gewählt. Als Beispiel aus dem täglichen Leben für Schichtung soll der Vergleich mit einer Torte dienen: Sinnvolle und repräsentative Teilmenge einer Torte ist weder der Tortenboden, noch die Füllung, noch die Garnierung, sondern allenfalls ein Stück Torte, welches alle Schichten enthält.
Auf psychiatrische Patienten übertragen wäre folgender Sachverhalt vorstellbar: Zwei verschiedene Neuroleptika zur Behandlung der bipolaren Erkrankung sollen miteinander verglichen werden. Die Patientengruppe ist nach DSM-IV-Kriterien klassifiziert worden und enthält Patienten mit und ohne psychotische Symptomatik, solche in einer manischen oder gemischten Episode, Patienten mit beispielsweise unterschiedlicher Erkrankungsdauer, Vorbehandlung und Vorepisode sowie Rapid Cycler. Solche Faktoren müssen bei der Analyse berücksichtigt werden, sonst können die Resultate durch Vermengung dieser Faktoren unkontrolliert beeinflusst werden, da diese eine wesentliche Einflussgröße auf die Zielvariable Wirksamkeit darstellen.
Aus diesem Grund stratifiziert man die Patienten, um diese Risiken gleichmäßig auf die Therapiegruppen zu verteilen. Da die Gruppen etwa identisch sein sollten, können im Idealfall alle Unterschiede zum Studienendpunkt theoretisch auf die Intervention zurückgeführt werden. Jedoch kann selbst ein regelrechtes Randomisierungsverfahren nicht immer und absolut spätere Gruppenunterschiede ausschließen.
Geheimhaltung des Randomisierungsplans
Wenn die Randomisierung geheim gehalten wird, ist der Prüfarzt nicht in der Lage, zu kalkulieren, welche Therapie der nächst folgende Patient erhalten würde, und damit bewusst oder unbewusst das Behandlungsergebnis in eine bestimmte Richtung zu verzerren. Es wird meist nicht explizit im Text der Publikation erwähnt, ob der Randomisierungsplan geheim gehalten wurde, jedoch ist meist davon auszugehen, dass dies der Fall war, wenn die Randomisierung per Telefon oder mit einem System erfolgte, welches nicht im räumlichen Zusammenhang mit dem Prüfzentrum stand.
Doppelblindstudien
Bei Doppelblindstudien sind weder Prüfarzt noch Patient über die tatsächliche Therapie informiert. Mithilfe der Verblindung sollen Verzerrungen in der Beurteilung des Therapieeffekts und der Nebenwirkungen vermieden werden (Beobachtungsgleichheit). In Publikationen finden sich manchmal auch Hinweise, dass die Verblindung durch optisch und in der Menge nicht unterscheidbare Prüfmedikation gewährleistet werden sollte.
In psychiatrischen Studien werden die Schwere der Erkrankung und die psychopathologischen Symptome mit Ratingskalen gemessen (z.B. Y-MRS, HAMD-21, CGI-BP, PANSS). Da das Rating auf diesen Skalen wie kaum ein anderes Messinstrument subjektiven Einflüssen unterlegen sein kann, ist die Verblindung in diesen Studien sehr wichtig. Um die Beobachtungsgleichheit zusätzlich zu verbessern, wird häufig die Interrater-Reliabilität zwischen den einzelnen Beurteilern der jeweiligen Skala vor Studienbeginn anhand einer Messung der Korrelation jedes Beurteilers mit dem gruppenbezogenen medianen Score jedes Items durchgeführt. Beurteiler, die keine Korrelation von in der Regel mindestens 0,80 erreichen, dürfen in einer solchen Studie keine Patienten beurteilen.
Offene Studien
In offenen Studien (open-label) sind sowohl der Prüfarzt als auch der Patient über die tatsächliche Therapie informiert. Open-Label-Studien können beispielsweise dann nur durchgeführt werden, wenn die Darreichungsform (z.B. bestimmte Infusionen) oder notwendige Maßnahmen (z.B. Spiegelbestimmungen in einem Medikationsarm) die tatsächliche Medikation erkennen lassen. Um Beobachtungsgleichheit zu erreichen, ist es in solchen Studien daher besonders wichtig, objektive Zielparameter festzulegen, die das Risiko der verzerrten Beurteilung minimieren.
Statistik
Hypothese
Wissenschaftliche Therapieforschung erfordert eine medizinische Fragestellung mit entsprechender Hypothesenbildung zum therapeutischen Nutzen, die auf messbaren und im Allgemeinen quantifizierbaren klinischen Parametern beruht. Die Hypothesen müssen sachgerecht gestellt und mit dem vorhandenen Instrumentarium und den in die Untersuchung einzuschließenden Patienten schlüssig zu beantworten sein.
Die Entscheidung, ob ein Unterschied in der Wirkung zwischen zwei Prüfsubstanzen besteht, wird getroffen, indem Hypothesen mit statistischen Tests geprüft werden. Die Nullhypothese besagt, dass kein Unterschied in der Wirkung zweier Prüfsubstanzen besteht. Dieser Nullhypothese wird die Alternativhypothese (Research-Hypothese) gegenübergestellt, die die Behauptung aufstellt, dass ein Unterschied vorliege (siehe Kasten 1).
Kasten 1
Nullhypothese H0: μ = 0;
Alternativhypothese H1: μ ≠ 0
Nur die Research-Hypothese kann statistisch signifikant überprüft werden. Aus einer Nichtablehnung der Nullhypothese – und damit einem nicht-signifikanten Ergebnis – kann nicht auf die Gültigkeit der Nullhypothese geschlossen werden.
Statistische Tests
Hier stellt sich die Frage, ob die angewandten statistischen Verfahren zur Analyse wichtiger Zielkriterien zum einen beschrieben und darüber hinaus adäquat gewählt wurden.
Zur Beantwortung des letzteren Teils der Frage sei auf die Lehrbücher der Statistik verwiesen. An dieser Stelle sollen lediglich die wichtigsten Grundbegriffe des statistischen Testens definiert werden, da diese unabdingbar sind für die Interpretation von Studienergebnissen.
Die statistischen Tests (z.B. t-Test, χ2-Test, Wilcoxon-Test) verwenden zur Beurteilung der Hypothese eine Testgröße T, die aufgrund der Daten eine Entscheidung zwischen den beiden Hypothesen ermöglicht. Falls die Nullhypothese zutrifft (keine Veränderung unter Studienbedingungen), sind beispielsweise für die Prüfgröße T sehr kleine Werte (nahe bei Null) am wahrscheinlichsten. Große Betragswerte sind für T unwahrscheinlicher, falls die Nullhypothese zutrifft.
Der p-Wert (p=probability) gibt die Wahrscheinlichkeit an, mit der der aus den Daten berechnete Wert für T (oder ein größerer Wert) auftreten kann, falls die Nullhypothese zutrifft.
Signifikanzniveau: Ist das Signifikanzniveau festgelegt worden?
Seit etwa 60 Jahren wird nach R. A. Fisher üblicherweise eine Nullhypothese als abgelehnt betrachtet, falls der p-Wert kleiner als 0,01 bis 0,05 ist. Man spricht von einem signifikanten Testresultat. Ein p-Wert >0,05 spricht für ein nicht signifikantes Testergebnis.
Die Irrtumswahrscheinlichkeit, mit der eine wahre Nullhypothese fälschlicherweise abgelehnt wird – das heißt, auf einen eigentlich nicht vorhandenen Therapieeffekt geschlossen wird – soll möglichst klein sein, üblicherweise 5% (p<0,05). Dieses ist der Fehler 1. Art und wird mit α bezeichnet (Tab. 1). Man würde also irrtümlich annehmen, dass aufgrund der zufällig beobachteten Daten das Medikament eine Wirkung besitzt. Dieser Fehler kann auch als Konsumentenrisiko bezeichnet werden.
Tab. 1. Fehler bei der Testentscheidung
|
Unbekannte |
Entscheidung aufgrund des Versuchs und der daraus resultierenden Testentscheidung |
||
|
Nullhypothese |
Nullhypothese |
||
|
Nullhypothese |
Richtig |
α |
Richtige |
|
Nullhypothese |
Falsch |
Richtige |
β |
Umgekehrt kann auch eine tatsächlich vorliegende Wirkung des Medikaments aufgrund der vorliegenden Daten eventuell nicht aufgedeckt werden. Dies wird als Fehler 2. Art (β) bezeichnet. Dieser kann auch als Produzentenrisiko bezeichnet werden, denn dann könnte eine tatsächlich wirksame Substanz eventuell nicht auf den Markt gebracht werden.
95%-Konfidenzintervall: Wie präzise ist der berechnete Behandlungseffekt?
Das 95%-Konfidenzintervall (95%-KI) gibt an, innerhalb welcher Grenzen sich das wahre Ergebnis mit 95%iger Wahrscheinlichkeit befindet. Die Breite des Konfidenzintervalls hängt ab von
der Streuung der Messungen (breite Streuung – breites KI),
der Stichprobengröße (größere Stichprobe, schmaleres KI) und
der Höhe des Vertrauens (größere Wahrscheinlichkeit, breiteres KI).
Schließt dieses Intervall die Null ein, so bedeutet dies, dass die Nullhypothese μ=0 nicht abgelehnt werden kann. Es kann also kein Unterschied zwischen den Behandlungsgruppen aufgedeckt werden.
Umgekehrt gilt: Schließt das Intervall die Null nicht ein, so kann die Nullhypothese abgelehnt werden und damit die Alternativhypothese statistisch signifikant bestätigt werden. Dies bedeutet, dass auf einen Behandlungsunterschied geschlossen werden kann.
Statistische Signifikanz ist nicht unbedingt gleichbedeutend mit klinischer Relevanz. Als statistisch signifikant gilt, wenn die Nullhypothese abgelehnt werden kann, und als medizinisch relevant gilt, wenn der Effekt des Medikaments einen bestimmten Mindestwert W überschreitet. Dieser Wert W wird beispielsweise in psychiatrischen Studien vorher in Form von Ansprechkriterien festgelegt.
Die Abbildung 1 soll das Verständnis für die Unterscheidung von statistischer Signifikanz und klinischer Relevanz erleichtern. In dieser Grafik sind fiktive Ergebnisse von fünf vergleichenden Studien mit Median und 95%-Konfidenzintervallen dargestellt. Die verschiedenen Fälle A bis E zeigen die möglichen Situationen und Interpretationen, wenn das 95%-Konfidenzintervall zur Schätzung des Effekts verwendet wird.
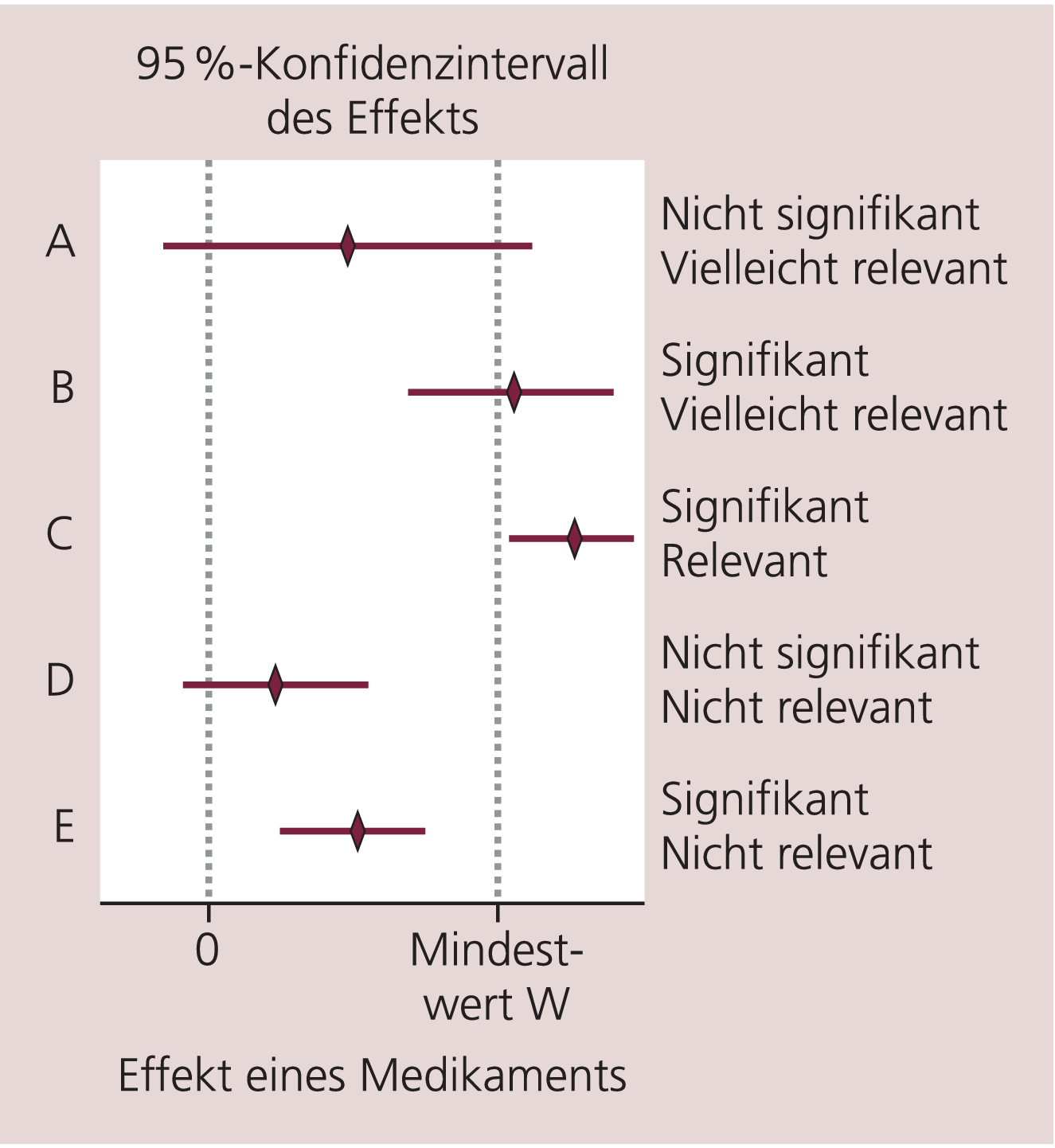
Abb. 1. Unterschied zwischen statistischer Signifikanz und medizinischer Relevanz
Zielkriterien
Sind klinisch sinnvolle Zielkriterien festgelegt und ist zwischen primären und sekundären Zielkriterien unterschieden worden?
Die Beurteilung eines Therapieerfolgs erfordert die Festlegung von eindeutigen und reproduzierbaren Zielgrößen. Ihre Auswahl hängt ab von der Relevanz für die jeweilige Erkrankung, Mechanismen der Therapie, Genauigkeit ihrer Bestimmung, Variabilität in der Studienpopulation, Verfügbarkeit effizienter und unverfälschter statistischer Auswerteverfahren sowie von der Verallgemeinerungsfähigkeit und Akzeptanz in der Fachwelt.
Fallzahlberechnung und Power
Bei der Planung einer Untersuchung muss der benötigte Stichprobenumfang (Fallzahl) vorher festlegt werden.
Die Studie hat mit der festgelegten Fallzahl eine bestimmte Power (meist 80%) (siehe Kasten 2), einen Therapieunterschied eines bestimmten Ausmaßes zu erkennen, und bezieht sich in der Regel auf das primäre Zielkriterium. Die Berechnung der benötigten Fallzahl erfolgt beispielsweise auf der Basis von Pilotstudien. Die Power beschreibt dabei die Wahrscheinlichkeit, eine richtige Alternativhypothese tatsächlich aufzudecken.
Kasten 2
Power = 1 – (Wahrscheinlichkeit Fehler 2. Art)
Beispiel: Eine Formulierung zur Fallzahlberechnung in einer psychiatrischen Studie kann folgendermaßen aussehen:
„… Auf der Basis vorangegangener Studien wurde berechnet, dass eine Fallzahl von 160 Patienten (80/Gruppe) erforderlich ist, um eine Effektgröße von 0,48 auf der ADHD-Rating-Scale-IV mit einer Power von 82% aufzudecken“ [3].
Patienten
Einschluss charakteristischer Patienten: Wurden sinnvolle Ein- und Ausschlusskriterien vor Beginn der Studie festgelegt? Ist das Patientenkollektiv repräsentativ? Oft nicht vorhanden, aber ein Gütezeichen, ist die Information darüber, wie viele Patienten vor der Randomisierung gescreent wurden und aus welchen Gründen nicht alle tatsächlich eingeschlossen worden sind. Die nicht eingeschlossene Zielpopulation sollte sich im Idealfall nicht von den eingeschlossenen Studienpatienten unterscheiden.
Baseline-Homogenität: Waren die Behandlungsgruppen zu Studienbeginn in wichtigen Einflussgrößen (Alter, Geschlecht, Krankheitsstadium) gleich strukturiert? Informationen hierüber findet man typischerweise in Tabelle 1 einer Publikation, die die Patientencharakteristika enthält.
Datenerhebung und -auswertung
Follow-up: War der Beobachtungszeitraum ausreichend lang, um das erwartete Ereignis beobachten zu können, und wurden ausreichend viele Patienten über die gesamte Studiendauer beobachtet?
Im Idealfall beenden alle randomisierten Patienten die Studie und können in die Auswertung einbezogen werden. Realistisch ist jedoch, dass es in jeder Studie Patienten gibt, die die Studie nicht beenden. Häufige Gründe sind beispielsweise, dass nach der Randomisierung festgestellt wird, dass
der Patient die Einschlusskriterien nicht erfüllt,
vermutete Nebenwirkungen (adverse events) auftreten,
der Patient die Motivation verliert,
medizinische Gründe den Ausschlag geben (Schwangerschaft, Begleiterkrankungen) oder
die Nachkontrolle unmöglich wird, weil der Patient wegzieht oder verstirbt.
Wenn die Anzahl der Drop-outs zu hoch ist, kann dies die Ergebnisse der Studie und die daraus hergeleiteten Schlussfolgerungen verzerren. Die vorher für den Nachweis eines Effekts berechnete Fallzahl wird somit nicht mehr gewährleistet. In der Praxis hat sich gezeigt, dass Studien in bestimmten Therapiebereichen (z.B. kardiovaskuläre Studien) mit einem Follow-up <80% der Patienten nicht als valide betrachtet werden können. Fachzeitschriften wie „Evidence-based Medicine“ und „ACP Journal Club“ publizieren keine Studien mit einem Follow-up unterhalb dieser Grenze. Für psychiatrische Studien müssen aber gegebenenfalls andere Kriterien gelten.
Intention-to-treat-Prinzip: Wurden alle Patienten in den Gruppen analysiert, in die sie randomisiert worden sind?
Wenn diejenigen Patienten, die die Studie abgebrochen haben, einfach ignoriert werden, führt dies meist zu einer systematischen Verzerrung – fast immer zu Gunsten der Intervention. Deshalb gehört es zum Standard, die Ergebnisse von Vergleichsstudien auf der Basis der Gesamtzahl der ursprünglich zu Behandelnden (intention to treat) zu analysieren. Das bedeutet, dass Daten von allen Patienten, die ursprünglich in eine Gruppe randomisiert wurden, zu untersuchen sind; sowohl die Daten von denen, die die Studie abgebrochen haben, die ihre Medikation nicht genommen haben, die aus irgendwelchen Gründen in die andere Gruppe kamen, als auch derjenigen, die ordnungsgemäß die Studie beendet haben. Der Grund für dieses Vorgehen liegt darin, dass nur dann Strukturgleichheit in den Behandlungsgruppen gewährleistet werden kann, wenn diese bei der Analyse beibehalten werden.
Per-protocol-Analyse: Im Gegensatz dazu steht die Analyse „per protocol“, bei der nur die Patienten analysiert werden, für die keine schweren Protokollverstöße vorliegen (prüfplankonforme Patienten).
Behandlungsgleichheit: Wurden alle Gruppen gleich behandelt, abgesehen von der zu prüfenden Therapie?
Beobachtungsgleichheit: Wurden alle Gruppen auf gleiche Weise beurteilt und erfolgte die Datenerhebung in gleichen Zeitintervallen?
Berechnung des Behandlungseffekts
Wie groß ist der Behandlungseffekt?
Wurden klinisch sinnvolle Ansprechkriterien festgelegt? Kann mithilfe der angegebenen Daten die absolute Risikoreduktion (ARR) und die Number needed to treat (NNT) berechnet werden?
Dies sind nützliche Größen, um den Behandlungseffekt einer Intervention einschätzen zu können. In nur wenigen Publikationen sind diese Größen bereits berechnet, in den meisten Fällen muss diese Kalkulation selbst durchgeführt werden.
ARR: Absolute Risikoreduktion; Maß zur Einschätzung des Behandlungseffekts;
Berechnung: Differenz der Risiken in der Kontrollgruppe (CER = control event rate) und der experimentellen Gruppe (EER = experimental event rate) (ARR = CER–EER)
NNT: Number needed to treat; ebenfalls ein Maß zur Einschätzung des Behandlungseffekts; die NNT gibt die Patientenzahl an, die für die Zeitdauer der Studie behandelt werden muss, um die erwünschte Wirkung zu erreichen; Berechnung: Kehrwert der ARR (NNT = 1/ARR)
Beispiel: In der folgenden 4-armigen randomisierten, kontrollierten Studie [4] wurden im Rahmen einer 8-wöchigen Therapie drei verschiedene Dosisstufen eines ADHD-Präparats mit Plazebo verglichen.
Das Ansprechen auf die Therapie war definiert als Reduktion des ADHD-RS-Parent:Inv-Gesamtscores ≥25% von der Ausgangssituation zum Endpunkt (ADHD RS = Attention Deficit Hyperactivity Disorder Rating Scale).
Die Ansprechraten in den vier Behandlungsgruppen betrugen:
46,5% bei Patienten, die das Präparat in niedriger Dosis erhielten,
56,0% bei Patienten, die das Präparat in mittlerer Dosis erhielten,
56,1% bei Patienten, die die das Präparat in hoher Dosis erhielten, und
30,1% in der Plazebo-Gruppe.
In dieser beispielhaft angeführten Studie zeigen sich statistisch signifikante absolute Risikoreduktionen in den Behandlungsgruppen mit der mittleren und der hohen Dosierung im Vergleich zu Plazebo (Tab. 2). Die 95%-Konfidenzintervalle beinhalten nicht die Null, so dass die Nullhypothese μ=0 abgelehnt werden kann. Die Erfüllung dieser Hypothese würde bedeuten, dass kein Unterschied zwischen den Behandlungsgruppen besteht. Somit gilt die Alternativhypothese µ≠0. Dies bedeutet, dass auf einen Behandlungsunterschied geschlossen werden kann.
Tab. 2. Ansprechen auf eine medikamentöse ADHD-Therapie: Beispiel zur Verdeutlichung (Ansprechraten in vier Behandlungsgruppen, gemessen als Reduktion im ADHD-RS-Parent:Inv-Gesamtscore ≥25% von der Ausgangssituation zum Endpunkt) [4]
|
Behandlungsgruppen im Vergleich |
ARR |
95%-KI |
NNT |
|
Präparat in niedriger Dosis vs. Plazebo |
16,4 |
–3,3 bis 36,0 |
6,1 |
|
Präparat in mittlerer Dosis vs. Plazebo |
25,8 |
10,1 bis 41,5 |
3,9 |
|
Präparat in hoher Dosis vs. Plazebo |
26,0 |
10,2 bis 41,8 |
3,8 |
ARR=Absolute Risikoreduktion; NNT=Number needed to treat; 95%-KI=95%-Konfidenzintervall
Die NNT für die beiden Dosisstufen liegt bei nur 3,9 und 3,8 Patienten, welche über die Zeitdauer dieser Studie behandelt werden müssen, um ein Ansprechen zu erreichen. Diese Zahl ist beeindruckend niedrig, wenn man sich die NNT aus anderen Therapiebereichen mit erheblich längerer Zeitdauer zum Vergleich heranzieht.
Zum Beispiel bedarf es einer NNT von 50 bisher gesunden Personen mit Hypercholesterolämie, die über den Zeitraum von 5,2 Jahren mit Lovastatin behandelt werden müssen, um einen Myokardinfarkt, instabile Angina pectoris oder plötzlichen Herztod zu vermeiden [2].
Das drastischste Beispiel ist dem Bereich der antihypertensiven Therapie entnommen: 128 Hypertoniker müssen über 5,5 Jahre mit antihypertensiver Medikation behandelt werden, um ein Ereignis (Tod, Schlaganfall oder Myokardinfarkt) zu verhindern [1].
Bewertung der wissenschaftlichen Aussagekraft nach Evidenzklassen
Die vorangegangenen Ausführungen bezogen sich auf randomisierte, kontrollierte Studien, welche der zweithöchsten Evidenzklasse I b entsprechen (Tab. 3). Die höchste Evidenzklasse I a wird repräsentiert durch die Metaanalyse. Den Abschluss der Evidenzklassen bilden die sogenannte Expertenmeinung (Klasse IV) und der Fallbericht (Klasse V).
Tab. 3. Evidenzklassen (FDA 5/98: Guidance for Industry, WHO and AHCPR 1994)
|
Evidenzklassen |
|
|
Ia |
Evidenz aufgrund von Metaanalysen randomisierter kontrollierter Studien in systematischen Übersichtsarbeiten |
|
Ib |
Evidenz aufgrund von mindestens einer randomisierten, kontrollierten Studie |
|
IIa |
Evidenz aufgrund mindestens einer gut angelegten, kontrollierten Studie ohne Randomisierung |
|
IIb |
Evidenz aufgrund mindestens einer gut angelegten, quasi-experimentellen Studie |
|
III |
Evidenz aufgrund gut angelegter, nicht-experimenteller, deskriptiver Studien (z. B. Fall-Kontroll-Studien) |
|
IV |
Evidenz aufgrund von Berichten/Meinungen von Expertenkreisen, Konsensuskonferenzen und/oder klinischer Erfahrung anerkannter Autoritäten ohne transparenten Beleg |
|
V |
Fallbericht |
Die Metaanalyse (Evidenzlevel Ia)
Definition
Einen systematischen und quantitativen Review sowie die Zusammenfassung von Einzelstudien zu einem Gesamtergebnis bezeichnet man als Metaanalyse. Die Ziele einer Metaanalyse lassen sich wie folgt darstellen:
Systematischer Review aller Erkenntnisse aus klinischen Studien
Bereitstellung einer quantitativen Zusammenfassung der Einzelresultate klinischer Studien
Kombination der Einzelresultate über die Studien und deren Gesamtinterpretation
Arten der Metaanalyse
Es wird unterschieden zwischen:
Metaanalyse auf der Grundlage publizierter Daten (Metaanalyse von Veröffentlichungen)
Metaanalyse auf der Grundlage von Übersichtstabellen und statistischen Maßzahlen der einzelnen Studien (Metaanalyse von Übersichtsdaten)
Metaanalyse auf der Grundlage von Individualdaten
Protokoll und Ziel
Für eine qualitativ hochwertige Metaanalyse ist ein vorab festgelegtes Protokoll mit genauer Angabe der Fragestellung und des entsprechenden Zielkriteriums unerlässlich.
Nachdem die Einschlusskriterien detailliert im Protokoll festgelegt worden sind, ist die Identifizierung geeigneter Studien für den Einschluss in die geplante Metaanalyse der nächste Schritt. Dabei genügt es in der Regel nicht, relevante randomisierte Studien in bibliographischen Archiven wie Medline zu suchen. Vielmehr sollten alle relevanten Studien (publiziert und nicht publiziert) nach Möglichkeit gefunden werden. Voraussetzung ist, auch Kongressbände, Register mit laufenden Studien (current controlled trials, clinical trials.gov) sowie die sogenannte „graue“ Literatur (z.B. Dissertationen, interne Berichte, Veröffentlichungen der Pharmaindustrie, Zeitschriften ohne Gutachtersystem) zu screenen. Gespräche mit Experten können zusätzlich wertvolle Informationen über unpublizierte Studien ergeben. Es muss in diesem Rahmen auch eine Entscheidung über den Einschluss fremdsprachiger Studien (nicht englisch oder deutsch) getroffen werden.
Eine gute Metaanalyse enthält daher auch eine Liste mit nicht berücksichtigten Studien unter Angabe des jeweiligen Grundes.
Auswahl valider, homogener und vergleichbarer Studien
Ein qualitativer Review der eingeschlossenen Studien über ihre methodische Qualität ist ein notwendiger Schritt für die spätere Gesamtinterpretation der Ergebnisse. Dabei sollten unter anderem Design und Durchführung der Primärstudien auf systematische Verzerrungen untersucht werden.
Fehlende Randomisierung, nicht ausreichender Follow-up der eingeschlossenen Patienten oder beispielsweise fehlende Verblindung können zu einer Verzerrung der Individualstudien und damit im Endeffekt schließlich auch der gesamten Metaanalyse führen. Nicht immer sind diese Informationen aus den Publikationen ersichtlich. Es sollte dann die Entscheidung getroffen werden, solche Studien wegen mangelnder Dokumentation eventuell aus der Metaanalyse auszuschließen. Eine Metaanalyse kann qualitativ nur so wertvoll sein wie die einzelnen eingeschlossenen Individualstudien. Liegt eine Verzerrung in den Individualstudien vor, so wird in der Regel diese Verzerrung auch in der Metaanalyse vorliegen.
Methodik der Evaluation des primären Zielkriteriums aus den einzelnen Studien
Eine Metaanalyse kann mit individuellen Patientendaten, aber auch anhand der Ergebnisse der statistischen Analyse der Primärstudien durchgeführt werden.
Nachdem geeignete Studien für die Metaanalyse ausgewählt wurden, müssen die den einzelnen Studien gemeinsamen statistischen Maßzahlen identifiziert und kombiniert werden. Dabei werden zum Beispiel für einzelne Daten, wie Ansprechraten, die Risikodifferenz, das relative Risiko oder das Odds-Ratio berechnet. Für stetige Daten (z.B. Summenscores, Gewichtsveränderungen) werden in der Regel Mittelwertsdifferenzen untersucht.
Ergebnisdarstellung/Publikation
Standards für die Qualität der Berichterstattung wurden von einer Expertenkonferenz 1999 im QUOROM Statement (Quality of reporting of metaanalyses) festgelegt [5]. Das Statement besteht aus einer Checkliste und einem Flussdiagramm. Die Checkliste enthält Empfehlungen zur adäquaten Darstellung der Ergebnisse einer Metaanalyse, unterteilt nach den Abschnitten Abstract, Einleitung, Methoden, Ergebnisse und Diskussion. Sie ist in Abschnitte gegliedert, die sich auf Literatursuche, Studienauswahl, Validitätsbewertung, Datenextraktion, Studiencharakteristika, quantitative Datensynthese sowie den Studienablaufplan (trial flow) beziehen. Das Flussdiagramm gibt die Anzahl der gefundenen, ein- und ausgeschlossenen RCT sowie die Gründe für den Ausschluss an. (www.consort-statement.org/QUOROM.pdf)
Diskussion des Publikationsbias
Eine Metaanalyse ist wie jede andere Studie anfällig für Verzerrungen (Bias). Ein wichtiger zusätzlicher Bias ist der Publikationsbias. Hierbei handelt es sich um die systematische Überschätzung des Gesamteffekts aufgrund der fehlenden Veröffentlichung von Studien mit nicht-signifikanten Resultaten. Diese Studien werden häufig nicht publiziert, wenn das Ergebnis unspektakulär ist oder kein Interesse an der Veröffentlichung besteht. Wenn nur Primärstudien eingeschlossen werden, die einen signifikanten Effekt oder eine Tendenz dazu aufweisen, findet man in der Metaanalyse möglicherweise einen relevanten signifikanten Effekt. Der Einfluss der nicht aufgenommenen Studien sollte daher diskutiert werden. Dabei versucht man abzuschätzen, wie viele nicht gefundene Studien mit nicht-signifikantem Ergebnis oder welche Fallzahlen es bräuchte, damit das Ergebnis der Metaanalyse nicht mehr bestehen bleibt (Robustheit der Metaanalyse).
Diskussion der externen Validität
Wurden die Resultate angemessen interpretiert und wurde im breiteren Zusammenhang der klinischen Problematik diskutiert, inwiefern sich die Ergebnisse der Metaanalyse auf eine größere Zielpopulation übertragen lassen? Zum Beispiel können nur Rückschlüsse auf die Verträglichkeit einer Therapie bei älteren Patienten >60 Jahre gezogen werden, wenn nur dieses Kollektiv in allen Primärstudien eingeschlossen wurde.
Zusammenfassung
Evidence-based Medicine ist nicht auf randomisierte, kontrollierte Studien und Metaanalysen begrenzt. Sie beinhaltet die Suche nach der jeweils besten wissenschaftlichen Evidenz, um klinische Fragestellungen beantworten zu können. Um etwas über die Genauigkeit eines therapeutischen Verfahrens zu erfahren, benötigt man auch gut durchgeführte Querschnitts- und Beobachtungsstudien von Patienten, bei denen die Erkrankung klinisch repräsentativ behandelt wird – hier sind kontrollierte Studien deutlich limitiert. Für eine prognostische Fragestellung, wie insbesondere für die Langzeittherapie oder Rehabilitation, werden methodisch angepasste Follow-up-Studien von Patienten benötigt, die in einem einheitlichen, frühen Stadium ihrer Krankheit in die Studie aufgenommen wurden.
Obwohl randomisierte, kontrollierte klinische Studien als „Goldstandard“ für den Wirksamkeitsnachweis einer Therapie (siehe Zulassungsverfahren) dienen, sind für manche Fragestellungen kontrollierte Studien also keinesfalls optimal bzw. ist keine kontrollierte Studie für die besondere Situation unseres Patienten durchgeführt worden, muss die nächstbeste externe Evidenz gefunden und berücksichtigt werden. Hier kann es sich vor allem um naturalistische Studien (auch Anwendungsbeobachtungen) handeln.
Literatur
1. Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985;291:97–104.
2. Down JR, Clearfield M, Weis S, Whitney E, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. JAMA 1998;279:1615–22.
3. Michelson et al. Once-daily atomoxetine treatment for children and adolescents with attention deficit hyperactivity disorder: A randomized, placebo-controlled study. Am J Psychiatry 2002;159:1896–1901.
4. Michelson et al. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics 2001;108:1–9.
5. Moher D, Cook DJ, Eastwood S, Olkin I, et al. for the QUOROM Group. Improving the quality of reports of meta-analyses of randomized controlled trials: the QUOROM statement. Lancet 1999;354:1896–1900.
6. Sackett DL, Straus SE, Richardson WS, Rosenberg W, et al. Evidence-based Medicine. How to practice and teach EBM. New York: Churchill Livingstone, 1997.
7. Thompson SG. Why sources of heterogeneity in meta-analysis should be investigated. In: Chalmers I, Altman DG (eds.). Systematic reviews. London: BMJ Publishing, 1995:64–74.
Dr. med. Jörg Czekalla, ECPM, Psychiater; FMH Pharmazeutische Medizin, Executive Director, Therapeutic Area CNS, Medical & Scientific Affairs, Janssen-Cilag GmbH, Raiffeisenstr. 8, 41470 Neuss, E-Mail:
Critical appraisal of clinical studies and meta-analyses – An educational article on the most important validity criteria according to Evidence-based Medicine (EbM)
The Oxford Dictionary of Current English defines „evidence“ as „available facts, circumstances, etc. indicating whether a thing is true or not true.“ In conjunction with clinical expertise, which can just be acquired by clinical practice, evidence-based medicine (EbM) is considered as a lifelong learning process which is targeted on adjustment on fast varying medical knowledge by continuous problem-focused learning. Thus, there is an increasing need for medical education due to the systematic assessment of clinical studies by mental health care specialists.
The associated methodology has been developed in the eighties by the Cochrane Collaboration in Oxford, whose intention is to compile systematic reviews regarding assessment of therapies as well as to update and publish them. The critical appraisal of clinical studies and meta-analyses is based on validity criteria with regard to study design, biostatistics, study population and interpretation according to Sackett et al., and will be summarized. According to the methods of evidence-based medicine the estimation of clinical benefit by calculation of absolute risk reduction (ARR) und number needed to treat (NNT) will be described with a focus on psychiatric studies and examples. Meanwhile efforts are made by psychiatrists to make decisions on best evidence available at present in combination with clinical expertise.
Keywords: Psychiatry, evidence-based medicine, clinical studies, critical appraisal, meta-analysis, clinical benefit assessment, absolute risk reduction (ARR), number needed to treat (NNT)
Psychopharmakotherapie 2006; 13(06)