Christoph Hiemke, Christian Hampel, Mainz, und Harald Weigmann, Ingelheim
Als der Psychiater Roland Kuhn 1956 die antidepressive Wirksamkeit von Imipramin (z.B. Tofranil®) entdeckte, wussten weder er noch der Pharmakologe Robert Domenjoz, der Imipramin zur Prüfung zur Verfügung gestellt hatte, über welche molekularen Strukturen die Substanz wirkt. Erst später wurde herausgefunden, dass Substanzen vom Imipramin-Typ, die trizyklischen Antidepressiva, die Wiederaufnahme der Monoamine Noradrenalin und/oder Serotonin hemmen, aber auch an zahlreiche Neurotransmitter-Rezeptoren binden. Rezeptorinteraktionen wurden als Ursache für Nebenwirkungen identifiziert, beispielsweise Mundtrockenheit, Obstipation, Akkommodations- und Miktionsstörungen durch anticholinerge Effekte und Sedierung durch antihistaminerge Wirkung. Serotonin und Noradrenalin wurden als depressionsrelevante Neurotransmitter und ihre Transporter als primäre molekulare Angriffsorte der antidepressiven Wirkung erkannt. Um nebenwirkungsärmere Substanzen als die trizyklischen Antidepressiva zu finden, suchte man konsequenterweise nach Serotonin- und Noradrenalin-selektiven Wiederaufnahmehemmern, die keine Rezeptoren blockieren. Die meisten der seitdem eingeführten Antidepressiva sind selektive Serotonin-Wiederaufnahmehemmer (SSRI). Sie wurden Antidepressiva der ersten Wahl [16], allerdings nicht wegen einer verbesserten therapeutischen Effizienz, sondern wegen besserer Verträglichkeit und größerer Sicherheit [29]. Trotz der damit verbundenen Fortschritte der antidepressiven Therapie sprechen etwa 30% der Patienten auf die antidepressive Monotherapie nicht oder unzureichend an. Daher werden häufig Therapien angewandt, beispielsweise Kombinationstherapien, für die es keine oder nur schwache Evidenz für eine Wirksamkeit gibt [1]. Eine aus pharmakologischer Sicht plausible Augmentationsstrategie ist die gleichzeitige Aktivierung der beiden depressionsrelevanten Neurotransmittersysteme Serotonin und Noradrenalin [16]. Erste Studien zeigen, dass bei einer Kombination von Fluoxetin (z.B. Fluctin®), einem SSRI, und Desipramin, einem selektiven Noradrenalin-Wiederaufnahmehemmer, die Wahrscheinlichkeit einer Remission größer ist als mit einer Fluoxetin- oder Desipramin-Monotherapie [26]. Für die Annahme, dass durch eine gleichzeitige Blockierung von Serotonin- und Noradrenalin-Transportern die antidepressive Wirkung verstärkt werden kann, sprechen auch Befunde mit Venlafaxin [37]. In niedriger Dosis hemmt Venlafaxin ausschließlich die Wiederaufnahme von Serotonin, während in Dosen von 225 mg zusätzlich die von Noradrenalin gehemmt wird [12]. Entsprechend konnte durch Erhöhung der Venlafaxin-Dosis von 75 auf 225 mg die Wirkung gesteigert werden [17, 32]. Duloxetin bietet angesichts seiner etwas potenteren und ausbalancierteren Hemmung der Serotonin- und Noradrenalin-Wiederaufnahme [15] gegenüber Venlafaxin eine vielversprechende Option im Rahmen der Depressions-Behandlung, aber auch für andere Indikationen.
Chemische Eigenschaften
Duloxetin ist ein Thiophenpropanderivat mit sekundärer Aminogruppe, an das über eine Etherbindung Naphthalin gekoppelt ist (chemische Bezeichnung: (+)-(S)-N-Methyl-γ-(napthyloxy)-2-thiophenpropylamin; relative Molekülmasse 297,4 g/mol, Summenformel C18H19NOS, Strukturformel s. Abb. 1). Duloxetin liegt als Hydrochlorid vor (1 mg Salz entspricht 0,89 mg Base).
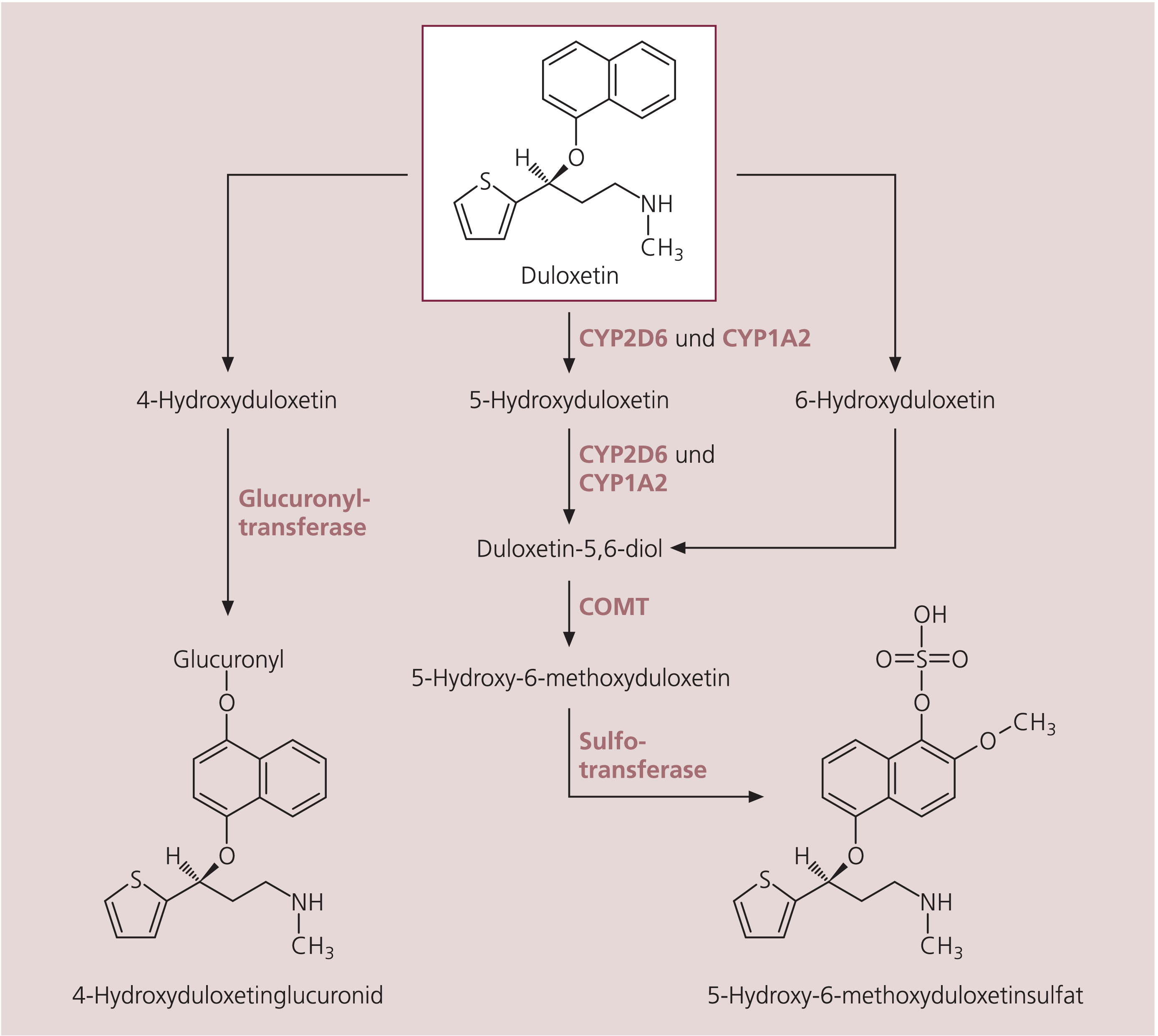
Abb. 1. Chemische Struktur und Metabolisierung von Duloxetin. An der Metabolisierung sind die Cytochrom-P450-Isoenzyme CYP2D6 und CYP1A2 sowie Catechol-O-methyltransferase (COMT) und zwei andere Transferasen beteiligt [nach 19, 20]
Präklinische Pharmakologie
An Membranen aus Zelllinien, die humane Monoamin-Transporter exprimieren, wurde die Bindung von Duloxetin und anderen Antidepressiva an Noradrenalin- und Serotonin-Transporter gemessen. Ein objektives Maß für die Bindungsaffinität ist die Gleichgewichtshemmkonstante Ki. Dieser Wert beschreibt die Konzentration, die erforderlich ist, um die Hälfte der spezifischen Bindungsstellen zu besetzen. Verglichen mit anderen Antidepressiva (Tab. 1) ist Duloxetin in vitro der Inhibitor mit der höchsten Affinität zu beiden Wiederaufnahmetransportern [2, 38]. Seine Affinität zu Noradrenalin-Transportern ist höher als die der Nordrenalin-selektiven Substanzen Reboxetin (Edronax® und Solvex®) und Nortriptylin (Nortrilen®) und höher als die des ebenfalls dual wirksamen Antidepressivums Venlafaxin. Aus dem Verhältnis der Ki-Werte lässt sich die Selektivität für die Interferenz mit einem der beiden Transporter ablesen. Duloxetin ist demnach mit einem Wert von 9 ähnlich wie Amitriptylin (z.B. Saroten®) einzuordnen.
Tab. 1. In-vitro-Affinität von Duloxetin und anderen Antidepressiva zu humanen Monoamin-Transportern und -Rezeptoren und Hemmung der Wiederaufnahme von Serotonin und Noradrenalin in Synaptosomen
|
Transporteraffinitäten |
Verhältnis |
Rezeptoraffinitäten |
Wiederaufnahmehemmung |
||||||||
|
SERT |
NAT |
NA:5-HT |
Alpha1 |
mACh |
H1 |
5-HT2A |
5-HT |
NA |
|||
|
Duloxetin |
0,8 |
7,5 |
9 |
>1000 |
>1000 |
>1000 |
504 |
4,6 |
16 |
||
|
Serotonin- und Noradrenalin-Wiederaufnahmehemmer |
|||||||||||
|
Amitriptylin |
4,3 |
35 |
8,1 |
27 |
15 |
1 |
12 |
8,4 |
13,9 |
||
|
Clomipramin |
0,28 |
38 |
136 |
38 |
37 |
31 |
64 |
1 |
16 |
||
|
Venlafaxin |
82 |
2480 |
30 |
>1000 |
>1000 |
>1000 |
>1000 |
77 |
538 |
||
|
Selektive Noradrenalin-Wiederaufnahmehemmer |
|||||||||||
|
Nortriptylin |
154 |
2,2 |
0,01 |
59 |
149 |
10 |
26 |
154 |
2,2 |
||
|
Reboxetin |
1070 |
8 |
0,01 |
>1000 |
>1000 |
>1000 |
>1000 |
1070 |
8 |
||
|
Selektive Serotonin-Wiederaufnahmehemmer |
|||||||||||
|
Citalopram |
1,6 |
4070 |
3392 |
>1000 |
>1000 |
>1000 |
283 |
1,8 |
6100 |
||
|
Escitalopram |
1,1 |
6514 |
2606 |
>1000 |
>1000 |
>1000 |
>1000 |
– |
– |
||
|
Fluoxetin |
0,81 |
240 |
296 |
>1000 |
>1000 |
>1000 |
>1000 |
20 |
1230 |
||
|
Fluvoxamin |
2,2 |
1300 |
591 |
>1000 |
>1000 |
>1000 |
280 |
3,8 |
620 |
||
|
Paroxetin |
0,13 |
40 |
308 |
>1000 |
107 |
>1000 |
>1000 |
0,7 |
33 |
||
|
Sertralin |
0,29 |
420 |
1448 |
370 |
625 |
>1000 |
>1000 |
3,4 |
220 |
||
Angegeben sind die Gleichgewichtshemmkonstanten (Ki) als Maß für die Bindungsaffinität bzw. die Hemmung der Aufnahme der Mononamine in Synaptosomen. Sie entsprechen den Konzentrationen, die notwendig sind, die Bindung an Serotonin- und Noradrenalin-Transporter (SERT und NAT) sowie Alpha1-Adrenozeptoren (Alpha1), muscarinische Acetylcholin-Rezeptoren (mACh), Histamin-H1-Rezeptoren (H1) und Serotonin2A-Rezeptoren (5-HT2A) bzw. die synaptosomale Aufnahme von Serotonin (5-HT) und Noradrenalin (NA) zu 50% zu hemmen. Ein Maß für die Selektivität der Antidepressiva ist das Verhältnis der Hemmung der Bindung an SERT zu NAT [nach 2, 3, 25, 30, 38, 41].
Im Unterschied zu Amitriptylin oder anderen trizyklischen Antidepressiva besitzt Duloxetin keine klinisch relevante Bindungsaffinität zu Neurotransmitter-Rezeptoren (Tab. 1), was auf gute Verträglichkeit und Sicherheit der Substanz hinweist.
Eine Möglichkeit zur Beurteilung der Potenz der Hemmung von Monoamin-Transportern ist die Messung des Transports von radioaktiv markierten Monoaminen in isolierte Nervenendigungen, so genannte Synaptosomen. An Synaptosomen aus dem Gehirn von Ratten fand man für Duloxetin eine potente Hemmung der Aufnahme von Serotonin und Noradrenalin. [41]. Duloxetin-Konzentrationen, die notwendig waren, die Aufnahme von Noradrenalin zu 50% zu hemmen, betrugen 4,6±1,1 nmol/l für Serotonin und 16±3 nmol/l für Noradrenalin. Duloxetin ist demnach potenter als Venlafaxin, für welches als Hemmkonzentrationen 77±2 nmol/l für die Serotonin- und 538±43 nmol/l für die Noradrenalin-Wiederaufnahme angegeben wurden [41]. Aus dem Verhältnis der Wiederaufnahmehemmung von Serotonin zu Noradrenalin lässt sich die Selektivität einer Substanz ablesen. Diese Werte sprechen für eine weniger serotonerg betonte Hemmung von Duloxetin als von Venlafaxin.
Die duale Wirkung von Duloxetin – Hemmung der Wiederaufnahme von Serotonin und Noradrenalin – ist auch in vivo belegt. Durch Mikrodialysestudien an Ratten [18] wurde nachgewiesen, dass Duloxetin ähnlich wie Clomipramin (z.B. Anafranil®) und Venlafaxin die Konzentrationen von sowohl Serotonin als auch Noradrenalin in der Extrazellulärflüssigkeit steigert (Abb. 2).
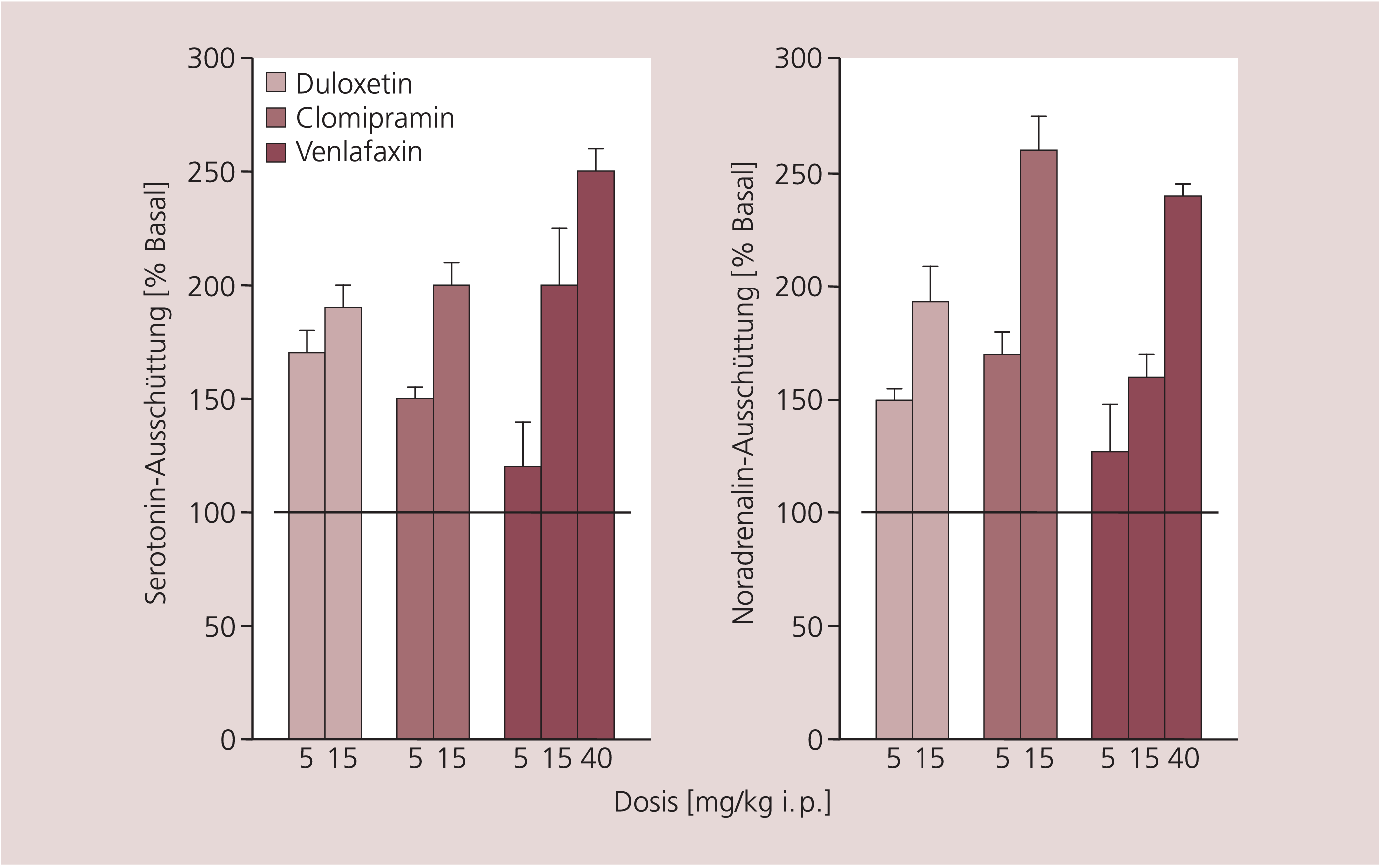
Abb. 2. Veränderung der extrazellulären Konzentrationen von Serotonin und Noradrenalin im frontalen Kortex der Ratte durch Duloxetin, Clomipramin und Venlafaxin. Dargestellt sind die Effekte einer Akutgabe der Antidepressiva (i.p.) mit anschließender (0,5 bis 4 h) Messung der Konzentrationen der Neurotransmitter mit Hilfe der Mikrodialysetechnik [nach 18].
Pharmakokinetik
Duloxetin wird nach peroraler Einnahme aus der magensaftresistenten Kapsel resorbiert. Die pharmakokinetischen Kenndaten sind in Tabelle 2 zusammengefasst. Die maximale Plasmakonzentration (Cmax) wird 6 Stunden nach Einnahme erreicht. Die gleichzeitige Einnahme von Duloxetin mit der Nahrung verzögert die Zeit bis zum Erreichen des Konzentrationsmaximums um annähernd 4 auf 10 Stunden [33, 36]. Die absolute orale Bioverfügbarkeit von Duloxetin liegt im Mittel bei 50% [33]. Duloxetin wird zu etwa 96% an humane Plasmaproteine gebunden (Albumin und saures Alpha-1-Glykoprotein) und verteilt sich gut im Gewebe.
Tab. 2. Pharmakokinetische Kenndaten von Duloxetin
|
Bioverfügbarkeit, oral |
50% (Bereich 32–80%) |
|
tmax |
6 Stunden |
|
t1/2 |
12 Stunden (Bereich 9–19 Stunden) |
|
Steady State erreicht nach |
3 Tagen |
|
Plasmaclearance |
0,9 l·h–1·kg–1 |
|
Verteilungsvolumen |
17 l/kg |
|
Plasmaeiweißbindung |
96% |
|
Dosis:Plasmakonzentration |
Lineare Beziehung |
|
Mittlere Plasmakonzentration |
20 bis 80 ng/ml bei 60 mg/d |
|
Metabolisierung |
Durch CYP2D6 und CYP1A2 und Transferasen |
|
Keine pharmakologisch aktiven Metaboliten in klinisch relevanten Konzentrationen |
|
|
Interaktionspotenzial |
Mäßiger Inhibitor von CYP2D6 |
|
Ausscheidung |
Etwa 70% renal, etwa 20% über die Fäzes |
tmax=Zeit bis zum Erreichen der maximalen Plasmakonzentration (nach oraler Gabe) t1/2=Eliminationshalbwertszeit im Plasma CYP2D6 und CYP1A2=Isoenzyme der Cytochrom-P450-Enzymfamilie (CYP) Zusammengestellt nach [8, 20, 25, 33–36]
Duloxetin wird umfangreich verstoffwechselt (Abb. 1). Die am Metabolismus beteiligten Enzyme sind die Cytochrom-P450-Isoenzyme (CYP) CYP2D6 und CYP1A2 und Catechol-O-methyltransferase (COMT). Hauptmetaboliten sind zwei pharmakologisch inaktive Konjugate, das Glucuronsäure-Konjugat von 4-Hydroxyduloxetin und das Schwefelsäure-Konjugat von 5-Hydroxy-6-methoxyduloxetin. Die Eliminationshalbwertszeit beträgt nach oraler Einnahme im Mittel 12 Stunden. Die Plasmaclearance ist bei Frauen und älteren Personen geringfügig vermindert und bei Rauchern um etwa das 1,5fache erhöht [35]. Dies erfordert in Einzelfällen eine Dosisanpassung. Die höhere Clearance von Duloxetin bei Rauchern ist vermutlich auf eine Induktion von CYP1A2 zurückzuführen. Die Duloxetin-Metaboliten werden zu etwa 70% über den Urin und zu etwa 20% mit den Fäzes ausgeschieden [20]. Angesichts der hepatischen Metabolisierung und überwiegend renalen Ausscheidung von Duloxetin darf die Substanz nicht bei Patienten mit einer Lebererkrankung, die zu einer Lebefunktioneinschränkung führt, oder bei schweren Nierenfunktionseinschränkungen (Creatinin-Clearance unter 30 ml/min) eingesetzt werden. Bei einer Tagesdosis von 60 mg liegen im Steady State die Plasmakonzentrationen von Duloxetin zwischen 20 und 80 ng/ml [25]. Duloxetin ist sowohl Substrat als auch Inhibitor von CYP2D6 [34]. Bei Kombination von Duloxetin mit Medikamenten, die Substrate oder Inhibitoren von CYP2D6 oder CYP1A2 sind, ist auf mögliche Wechselwirkungen zu achten.
Klinische Wirkungen
Depressions-Behandlung
Für die Behandlung von Episoden einer Major Depression ist die empfohlene Dosis 60 mg/d, die unabhängig von den Mahlzeiten eingenommen werden kann. Für Dosierungen oberhalb von 60 mg/d gibt es keine Hinweise auf zusätzliche Wirkungsvorteile. Bei fehlender oder unzureichender Wirkung kann bis 120 mg gesteigert werden.
Neben einer Reihe von Studien zur Wirksamkeit von Duloxetin in verschiedenen Dosierungen [13] wurden zwei Plazebo-kontrollierte Studien in der empfohlenen Dosierung von 60 mg/d durchgeführt [5, 6]. Diese zeigten Remissionsraten nach 9 Wochen unter Duloxetin von 44 bzw. 43% versus 16 bzw. 28% unter Plazebo. Eine gepoolte Analyse von sechs randomisierten doppelblinden Vergleichsstudien zeigte eine Überlegenheit von Duloxetin im Vergleich zu den SSRI: Während die Remissionsraten unter SSRI nach 8 Wochen im Durchschnitt bei 38% lagen, lag sie unter Duloxetin bei 43% [9].
Die Depression umfasst häufig auch Angstsymptomatik. Die Wirkung von Duloxetin auf diese Problematik wurde auch geprüft. Die Angstsymptomatik nahm bereits nach 2 Wochen signifikant gegenüber Plazebo ab [7].
Physiologischerweise unterdrücken vom Hirnstamm in das Rückenmark absteigende serotonerge und noradrenerge Projektionen aus der Peripherie eingehende Schmerzafferenzen. Im Zuge des angenommenen Defizits an monoaminerger Neurotransmission bei der Depression kommt es möglicherweise zu einer Dysfunktion, die zu schmerzhaften physischen Symptomen führt, für die sich keine organische Ursache am Ort der Symptome findet. Angesichts der pathophysiologischen Konzepte bietet Duloxetin nicht nur bei depressiven Patienten, sondern auch bei Vorliegen schmerzhafter Beschwerden eine Behandlungsoption. In einer Phase-III-Studie untersuchten Detke et al. [4] auch die Veränderung körperlicher Beschwerden, insbesondere von Schmerzen, im Rahmen der Depression im Therapieverlauf. Im Vergleich zu den Ausgangswerten zeigte die 9-wöchige Studie eine statistisch signifikante Verbesserung der Schmerzen unter Duloxetin.
Therapie der Stress-induzierten Harninkontinenz
Harninkontinenz ist ein unwillkürlicher Urinverlust. Die durchschnittliche Harninkontinenz-Prävalenz bei Frauen beträgt 28% [23]. Die drei häufigsten Formen der Harninkontinenz sind Belastungsinkontinenz, Dranginkontinenz und eine Kombination aus Belastungs- und Dranginkontinenzsymptomen [11]. Die Belastungsinkontinenz entsteht auf dem Boden einer Sphinkter- und/oder Beckenbodenschwäche, während die Ursache der Dranginkontinenz in einer Blasenüberaktivität (Detrusorinstabilität oder -hypersensitivität) bei völlig intaktem Verschlussapparat liegt. Etwa 49% der inkontinenten Frauen weisen Symptome einer Belastungsinkontinenz auf; 22% haben Symptome einer Dranginkontinenz und 29 % haben eine Mischinkontinenz [23]. Bis vor kurzem war die Pharmakotherapie der Belastungsinkontinenz auf den Einsatz weniger peripher an Blase und Sphinkter wirksamer Substanzen wie Sympathomimetika (Midodrin – z.B. Gutron®) oder Estrogene in Mono- oder Kombinationstherapie beschränkt. Häufiger jedoch wurde wegen des ausgeprägten Wunschs der belastungsinkontinenten Patientin nach einer medikamentösen Behandlung zur Vermeidung einer sonst nötigen Operation auf ein bestenfalls kapazitätssteigerndes Antimuscarinikum zurückgegriffen, das allerdings keinen stimulierenden Effekt auf den Sphinkter- und Beckenbodenapparat entfaltet. Durch die Experimente von Thor et al. [39] an anästhesierten Katzen wurde die Möglichkeit einer zentralnervösen Steuerung und Beeinflussbarkeit des Miktionsreflexes über serotonerge und noradrenerge Mechanismen als therapeutische Option erkannt. Es wurde eine sphinktertonisierende und detrusorstabilisierende Wirkung von Duloxetin nachgewiesen [39]. Da die Miktion trotz kontinuierlicher Duloxetin-Wirkung mit stetig erhöhter Serotonin- und Noradrenalin-Konzentration im spinalen Kerngebiet des sphinkterinnervierenden Nervus pudendus (Nucleus onuf) völlig unbeeinträchtigt und ohne Detrusor-Sphinkter-Dyskoordination ablief, wird die Duloxetin-Wirkung mit einer indirekten Modulierung des zentralen Schlüsselneurotransmitters Glutamat erklärt, welcher in der Speicherphase der Blase im Nucleus onuf ausgeschüttet und im Moment der Miktion zurückgehalten wird [21, 31].
Duloxetin wurde als Medikament für die Behandlung einer Belastungsinkontinenz klinisch geprüft. Drei Phase-III-Studien mit jeweils vergleichbaren Studiendesigns und Patientenpopulationen geben Aufschluss über die klinische Wirksamkeit und Verträglichkeit von Duloxetin bei Frauen mit Belastungsinkontinenz [6, 22, 40]. Sie belegten alle die Wirksamkeit von Duloxetin (2×40 mg täglich) gegenüber Plazebo. Die Wirksamkeit definierte sich jeweils über eine statistisch signifikante Reduktion der Inkontinenz-Episoden-Frequenz (IEF) und eine Zunahme der Inkontinenz-bezogenen Lebensqualität. Eine Metaanalyse der Dosisfindungsstudie [14] und der drei Phase-III-Studien [6, 28, 40] zeigte, dass Patientinnen bei einer durchschnittlichen Baseline-IEF von 16,9/Woche unter der Einnahme von Duloxetin eine mediane Reduktion der IEF um 52% erreichten.
Duloxetin wurde für die Behandlung der Belastungsinkontinenz zugelassen. Die empfohlene Dosis beträgt 2×40 mg täglich, die unabhängig von den Mahlzeiten eingenommen werden kann. Nach 2 bis 4 Wochen müssen Nutzen und Verträglichkeit der Behandlung bei den Patientinnen überprüft werden. Wenn die Patientinnen länger als 4 Wochen noch beeinträchtigende Nebenwirkungen wahrnehmen, kann die Dosis auf 2×20 mg täglich reduziert werden. Generell empfiehlt sich ein begleitendes Beckenbodentraining.
Sicherheit, Verträglichkeit und Arzneimittelinteraktionen
Duloxetin ist ein gut verträgliches Medikament. Es induziert keine klinisch relevanten Veränderungen von Herzfrequenz, Blutdruck, EKG, Leberenzymen und anderen Laborwerten [15]. Mittlere Änderungen des systolischen und diastolischen Blutdrucks betrugen etwa 1,5 mmHg. Die mit Duloxetin behandelten Patienten wiesen eine Erhöhung der Herzfrequenz um etwa 1,5 Schläge pro Minute auf, während die Herzfrequenz der mit Plazebo behandelten Patienten um etwa 0,5 Schläge pro Minute abnahm. Duloxetin verlängerte das QTc-Intervall nicht [27]. Selten trat unter der Behandlung mit Duloxetin ein Hypertonus auf (0,8 bzw. 0% vs. 0% unter Plazebo) [4, 5].
Bei schweren Leber- oder Nierenfunktionseinschränkungen (Creatinin-Clearance unter 30 ml/min) ist die Gabe von Duloxetin kontraindiziert [8].
Bei Frauen mit Belastungsinkontinenz induziert Duloxetin keine Veränderungen der Stimmungslage [14].
Das Nebenwirkungsprofil ist ähnlich wie das von SSRI [10]. Ungefähr 10% der 1139 Patienten, die Duloxetin wegen einer Depression in den Plazebo-kontrollierten Studien erhielten, brachen die Behandlung wegen Nebenwirkungen ab, unter Plazebo waren es 4% der 777 Patienten. Nausea (Duloxetin 1,4%, Plazebo 0,1%) bildete den häufigsten Abbruchgrund. In vier Plazebo-kontrollierten klinischen Studien wurde über jeweils 12 Wochen die Sicherheit von Duloxetin untersucht [4, 5, 10]. Insgesamt erhielten 958 Patientinnen Duloxetin und 955 Patientinnen Plazebo. Die am häufigsten berichteten Nebenwirkungen waren Übelkeit, Mundtrockenheit, Müdigkeit, Schlaflosigkeit und Obstipation. Über Übelkeit klagten 23,2%; sie war überwiegend, d.h. bei annähernd 80% der Patientinnen, leicht bis mäßig ausgeprägt. In den meisten Fällen (81%) verschwand die Übelkeit innerhalb von 1 bis 4 Behandlungswochen [14].
Auch mit sexuellen Funktionsstörungen, die unter SSRI häufig beobachtet werden [24], ist zu rechnen [15, 27]. Sie scheinen seltener aufzutreten als unter Paroxetin (z.B. Seroxat®). Die Datenlage ist allerdings ergänzungsbedürftig.
Abruptes Absetzen von Duloxetin sollte wie bei Venlafaxin und SSRI vermieden werden. Als Absetzeffekte können Unruhe, Ängstlichkeit und Übelkeit auftreten.
Pharmakodynamische Interaktionen
Da Duloxetin eine Serotonin stimulierende Substanz ist, ist wie bei allen SSRI bei Kombination mit Medikamenten, die Serotonin potenzieren, wegen des Risikos eines Serotoninsyndroms Vorsicht geboten. Die Kombination mit irreversiblen Monoaminoxidaseinhibitoren ist kontraindiziert [8].
Pharmakokinetische Interaktionen
Duloxetin ist sowohl Substrat als auch Inhibitor von CYP2D6 [34]. Bei gleichzeitiger Gabe kann Duloxetin die Eliminationshalbwertszeit der CYP2D6-Substrate Desipramin (trizyklisches Antidepressivum) und Tolterodin (Detrusitol®, Anticholinergikum zur Behandlung der Dranginkontinenz) erhöhen. In einer Studie an gesunden Probanden, die Desipramin bei konstanten Plasmaspiegeln von Duloxetin erhielten, war die maximale Plasmakonzentration von Desipramin 1,7fach erhöht [34]. Die Verabreichung von Duloxetin mit CYP2D6-Inhibitoren (z.B. SSRI) sollte mit Vorsicht erfolgen, da beispielsweise Paroxetin (20 mg/d) die Plasmaclearance von Duloxetin um 37% senkt [8].
Die Pharmakokinetik des CYP1A2-Substrates Theophyllin (z.B. Uniphyllin®), einem Bronchodilatator zur Behandlung von Asthma bronchiale und weiteren Atemwegserkrankungen bzw. deren Symptomen, wird durch Duloxetin nicht wesentlich beeinflusst. Demnach inhibiert Duloxetin kaum das Isoenzym CYP1A2. Der CYP1A2-Inhibitor Fluvoxamin (z.B. Fevarin®), ein SSRI, erhöht bereits in niedrigen Konzentrationen die Plasmakonzentration und verlängert die Eliminationshalbwertszeit von Duloxetin. Daher sollte Duloxetin nicht gemeinsam mit CYP1A2-Inhibitoren wie Fluvoxamin verabreicht werden [8].
Fazit
Duloxetin ist ein dual wirksamer Hemmer der neuronalen Wiederaufnahme von Serotonin und Noradrenalin. Es besitzt keine klinisch relevante Affinität für histaminerge, dopaminerge, cholinerge und adrenerge Rezeptoren. In der Depressions-Behandlung erklärt die hohe Affinität und duale Wirkung die in Studien gezeigte Überlegenheit gegenüber Plazebo und SSRI. Außerdem erstreckt sich die Wirkung auf eine deutliche Reduktion von Angst- und Schmerzsymptomen, die vielfach mit Depressionen assoziiert sind. Duloxetin ist weiterhin der erste zur Behandlung der Belastungsinkontinenz bei Frauen zugelassene Arzneistoff. Neben einer guten Wirksamkeit zeichnet sich Duloxetin durch eine gute Verträglichkeit aus.
Literatur
1. Bauer M, Whybrow P, Angst J, Versani M, et al. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment for Unipolar Depressive Disorders. Part 1: Acute and continuation treatment of major depressive disorder. World Biol Psychiatry 2002;3:5–43.
2. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, Shaw JL, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporter in vitro and in vivo, human serotonin receptor subtypes and other neuronal receptors. Neuropsychopharmacology 2001;25:871–80.
3. Cusack B, Nelson A, Richelson E. Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology 1994;114:559–65.
4. Detke MJ, Lu Y, Goldstein DJ, McNamara RK, et al. Duloxetine once daily dosing versus placebo in the acute treatment of major depression. J Psychiatr Res 2002;36: 383–90.
5. Detke MJ, Lu Y, Goldstein DJ, Hayes JR, et al. Duloxetine, 60 mg once daily, for major depressive disorder: a randomized double-blind placebo-controlled trial. J Clin Psychiatry 2002;62:308–15.
6. Dmochowski RR, Miklos JR, Norton PA, Zinner NR, et al. Duloxetine versus placebo for the treatment of North American women with stress urinary incontinence. J Urol 2003;170:1259–63.
7. Dunner DL, Goldstein DJ, Mallinckrodt C, Lu Y, et al. Duloxetine in treatment of anxiety symptoms associated with depression. Depress Anxiety 2003;18:53–61.
8. Eli Lilly and Boehringer Ingelheim – Yentreve Summary of Product Characteristics, 2003. Data on file.
9. Goldstein DJ, Lu Y, Detke MJ, Lee TC, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389–99.
10. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack MA. Duloxetine in the treatment of major depressive disorder: a double-blind placebo-controlled trial. J Clin Psychiatry 2002;62:225–31.
11. Hampel C, Wienhold D, Beken N, et al. Prevalence and natural history of female incontinence. Eur Urol 1997;32(Suppl 2):3–12.
12. Harvey AT, Rudolph RL, Preskorn SH. Evidence of the dual mechanisms of action of venlafaxine. Arch Gen Psychiatry 2000;57:503–9.
13. Hirschfeld RM, Vornik LA. Newer antidepressants: review of efficacy and safety of escitalopram and duloxetine. J Clin Psychiatry 2004;65(Suppl 4):46–52.
14. Hurley DJ, Baygani S, Simmon SA, et al. Duloxetine for stress urinary incontinence: a meta-analysis of safety. Int J Gynaecol Obstet 2003;83(Suppl 3):95(abs tp82).
15. Karpa KD, Cavanaugh JE, Lakoski JM. Duloxetine pharmacology: profile of a dual monoamine modulator. CNS Drug Review 2002;8:361–76.
16. Kent JM. SNaRIs, NaSSAs, and NaRIs: new agents for the treatment of depression. Lancet 2000;355:911–8.
17. Khan A, Upton GV, Rudolph RL, Entsuah R, et al. The use of venlafaxine in the treatment of major depression and major depression associated with anxiety: a dose-response study. Venlafaxine Investigator Study Group. J Clin Psychopharmacol 1998;18:19–25.
18. Koch S, Hemrick-Luecke SK, Thompson LK, Evans DC, et al. Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology 2003;45:935–44.
19. Kuo F, Gillespie TA, Kulanthaivel P, Lantz RJ, et al. Synthesis and biological activity of some known and putative duloxetine metabolites. Bioorg Med Chem Lett 2004;14:3481–6.
20. Lantz RJ, Gillespie TA, Rash TJ, Kuo F, et al. Metabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjects. Drug Metab Dispos 2003;31:1142–50.
21. Michel MC, Peters SL. Role of serotonin and noradrenaline in stress urinary incontinence. BJU Int 2004;94(Suppl 1):23–30.
22. Millard R, Morre K, Rencken R, Yalcin I, et al. Duloxetine versus placebo in the treatment of stress urinary incontinence: a four-continent randomized clinical trial. BJU Int 2004;93:311–8.
23. Minassian VA, Drutz HP, Al-Badr A. Urinary incontinence as a worldwide problem. Int J Gynecol Obstet 2003;82:327–38.
24. Montgomery SA, Baldwin DS, Riley A. Antidepressant medications: a review of the evidence for drug-induced sexual dysfunction. J Affect Disord 2002;69:119–40.
25. Müller WE, Haen E, Fritze J, Rüther E, et al. Selektive Serotonin- und Noradrenalin-Wiederaufnahmehemmer (SSNRI). Antidepressiva mit dualem Wirkungsmechanismus. Psychopharmakotherapie 2004;11:71–5.
26. Nelson JC, Mazure CM, Jatlow PI, Bowewers MB Jr, et al. Combining norepinephrine and serotonin reuptake inhibition mechanisms for treatment of depression: a double-blind, randomized study. Biol Psychiatry 2004;55:296–300.
27. Nemeroff CB, Schatzberg AF, Goldstein DJ, Detke MJ, et al. Duloxetine for the treatment of major depressive disorder. Psychopharmacol Bull 2002;36:106–32.
28. Norton PA, Zinner NR, Yalcin I, Bump RC. Duloxetine Urinary Incontinence Study Group. Duloxetine versus placebo in the treatment of stress urinary incontinence. Am J Obstet Gynecol 2002;187:40–8.
29. Oeljeschläger B, Müller-Oerlinghausen B. Wege zur Optimierung der individuellen antidepressiven Therapie. Dtsch Ärzteblatt 2004;101:1337–40.
30. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Therap 1997;283:1305–22.
31. Rajaofetra N, Massagia JG, Marlier L, Poulat P, et al. Serotonergic, noradrenergic and peptidergic innervation of Onuf’s nucleus of normal and transected spinal cord of baboons (Papio papio). J Comp Neurol 1992:318:1–17.
32. Rudolph RL, Fabre LF, Feighner JP, Rickels K, et al. A randomized, placebo-controlled, dose-response trial of venlafaxine hydrochloride in the treatment of major depression. J Clin Psychiatry 1998;59:116–22.
33. Sharma A, Goldberg MJ, Cerimele BJ. Pharmacokinetics and safety of duloxetine, a dual-serotonin and norepinephrine reuptake inhibitor. J Clin Pharmacol 2000;40:161–7.
34. Skinner MH, Kuan HY, Pan A, Sathirakul K, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170–7.
35. Skinner MH, Kuan H-Y, Skerjanec A, Seger ME, et al. Effect of age on the pharmacokinetics of duloxetine in women. Br J Clin Pharmacology 2003;57:54–61.
36. Skinner MH, Skerjanec A, Seger M, et al. The effect of food and bedtime administration on duloxetine pharmacokinetics. Int J Clin Pharmacol Ther 2000;67:129.
37. Stahl SM, Entsuah R, Rudolph RL. Comparative efficacy between venlafaxine and SSRIs: a pooled analysis of patients with depression. Biol Psychiatry 2002;52:1166–74.
38. Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 1997;340:249–58.
39. Thor KB, Katofiasc MA. Effects of duloxetine, a combined serotonin and norepinephrine reuptake inhibitor, on central neural control of lower urinary tract function in the chloralose-anesthetized female cat. J Pharmacol Exp Ther 1995;274:1014–24.
40. Van Kerreboreck P, Abrams P, Lange R, Slack M, et al. Duloxetine vs. placebo in the treatment of European and Canadian women with stress urinary incontinence. Br J Obstet Gynaeceol 2004;111:249–57.
41. Wong DT, Bymaster FP. Dual serotonin and noradrenaline uptake inhibitor class of antidepressants potential for greater efficacy or just hype? Prog Drug Res 2002;58:169–222.
Prof. Dr. rer. nat. Christoph Hiemke, Psychiatrische Klinik der Universität Mainz, Untere Zahlbacher Str. 8, 55101 Mainz, E-Mail: hiemke@mail.uni-mainz.de
Dr. med. Christian Hampel, Urologische Klinik der Universität Mainz, Langenbeckstr. 1, 55101 Mainz, E-Mail: hampel@urologie.klinik.uni-mainz.de
Dr. med. Dr. rer. nat. Harald Weigmann, Boehringer Ingelheim Pharma GmbH & Co KG, Abt. Medizinische Wissenschaft, Binger Str. 173, 55216 Ingelheim, E-Mail: Harald.Weigmann@ ing.boehringer-ingelheim.com
Pharmacology of duloxetine. A therapeutic option for different indications.
Duloxetine is a thiophenpropane derivative with a secondary amine function that inhibits both, noradrenaline and serotonin transporters. Given orally, it is well absorbed. Due to extensive first pass metabolism, however, its mean bioavailability is only 50% (range 32 to 80%). Maximal plasma concentrations are observed after 6 hours. In plasma duloxetine is bound to proteins by about 96%. Duloxetine is extensively metabolized to numerous metabolites which do not contribute to the pharmacological actions of the drug. Major enzymes involved in phase I metabolism are cytochrome P450 isoenzymes (CYP) CYP2D6 and CYP1A2. Duloxetine metabolites are excreted by about 70% via urine and about 20% via feces. Its elimination half life is about 12 hours. Under daily doses of 60 mg, expected steady state plasma concentrations range between 20 and 80 ng/ml. Duloxetine is a weak inhibitor of CYP2D6 which is so far not known to be of clinical relevance. Considering its in vitro binding affinities to human serotonin and noradrenaline transporters, duloxetine is equipotent to escitalopram plus desipramine. Duloxetine does not bind to neurotransmitter receptors. Clinical studies have shown antidepressant efficacy. Moreover, duloxetine is effective in the treatment of stress urinary incontinence. Therapeutically effective daily doses are 60 and 80 mg for the treatment of depression and urinary incontinence, respectively. Tolerability and safety of duloxetine are similar to those of selective serotonin reuptake inhibitors. Duloxetine is thus a new therapeutic option that acts on two distinct molecular unities and can be used for differential indications.
Keywords: Duloxetine, pharmacology, pharmacokinetics, depression, urinary incontinence
Psychopharmakotherapie 2006; 13(01)