Brigitta Bondy und Peter Zill, München
Die Einführung der Psychopharmaka in den 50er Jahren hat weltweit die psychiatrische Morbidität verringert und das Schicksal sowie die sozialen Folgen der Betroffenen erheblich verbessert. Obwohl zahlreiche Substanzen entwickelt wurden, die aufgrund ihrer unterschiedlichen Wirkungsspektren nahezu jeder Hypothese über zugrunde liegende pathogenetische Mechanismen gerecht werden, zeigten sich erhebliche interindividuelle Schwankungen beim Ansprechen auf die Behandlung oder das Auftreten von Nebenwirkungen. Wir gehen heute davon aus, dass etwa 20 bis 40% der Patienten nicht oder nicht ausreichend auf die Behandlung mit Antidepressiva oder Neuroleptika ansprechen. Außerdem treten bei bis zu 60% der „Responder“ Nebenwirkungen auf, ohne dass diese einer Überdosierung der Präparate mit erhöhten Serumkonzentrationen zugeordnet werden könnten [1]. In beiden Fällen ist der Nutzen der therapeutischen Interaktion nur gering und ein Wechsel hin zu einer anderen Behandlungsstrategie oft erforderlich. In der Praxis ist daher auch heute noch die „klinische“ Erfahrung von größerer Bedeutung als ein „gezieltes Auswählen“ der Substanzen anhand der unterschiedlichen Wirkungsmechanismen.
Das Ansprechen auf die Therapie ist ein komplexer Vorgang und wird durch eine Interaktion zahlreicher physiologischer Parameter und Umgebungsfaktoren beeinflusst (Abb. 1). Dazu gehören neben dem Alter, der Leber und Nierenfunktion der Patienten auch der Ernährungszustand, der Konsum von Tabak oder Alkohol. Seit langer Zeit ist aber auch bekannt, dass genetische Varianten sowohl die Wirkung der Pharmaka als auch das Auftreten unerwünschter Arzneimittelwirkungen (UAW) beeinflussen können. Dabei interessierten anfangs überwiegend pharmakokinetische Parameter wie Aufnahme, Verteilung, Aktivierung, Abbau oder Ausscheidung der Substanzen, denn damit verändert sich die Wirkstoffkonzentration am eigentlichen Zielort, was sich entweder in mangelnder Effektivität (bei zu geringen Konzentrationen) oder in toxischen Effekten (bei hohen Konzentrationen) äußert [2]. Heute, da die Angriffspunkte und Zielproteine der Psychopharmaka besser bekannt sind und neue Hypothesen entwickelt werden, rücken die pharmakodynamischen Aspekte, also die Interaktion der Substanzen mit den Zielproteinen, mehr in den Vordergrund. Das übergeordnete Ziel dieser Forschungsrichtung ist es, für jeden Patienten anhand des individuellen genetischen Musters die optimale Substanz zu finden und so eine angepasste, effektivere Therapie zu initiieren, die darüber hinaus noch mit geringeren Nebenwirkungen belastet ist. Die folgende Darstellung soll einen Überblick über die bisher vorliegenden Befunde sowie einen Ausblick auf zukünftige Ziele geben.
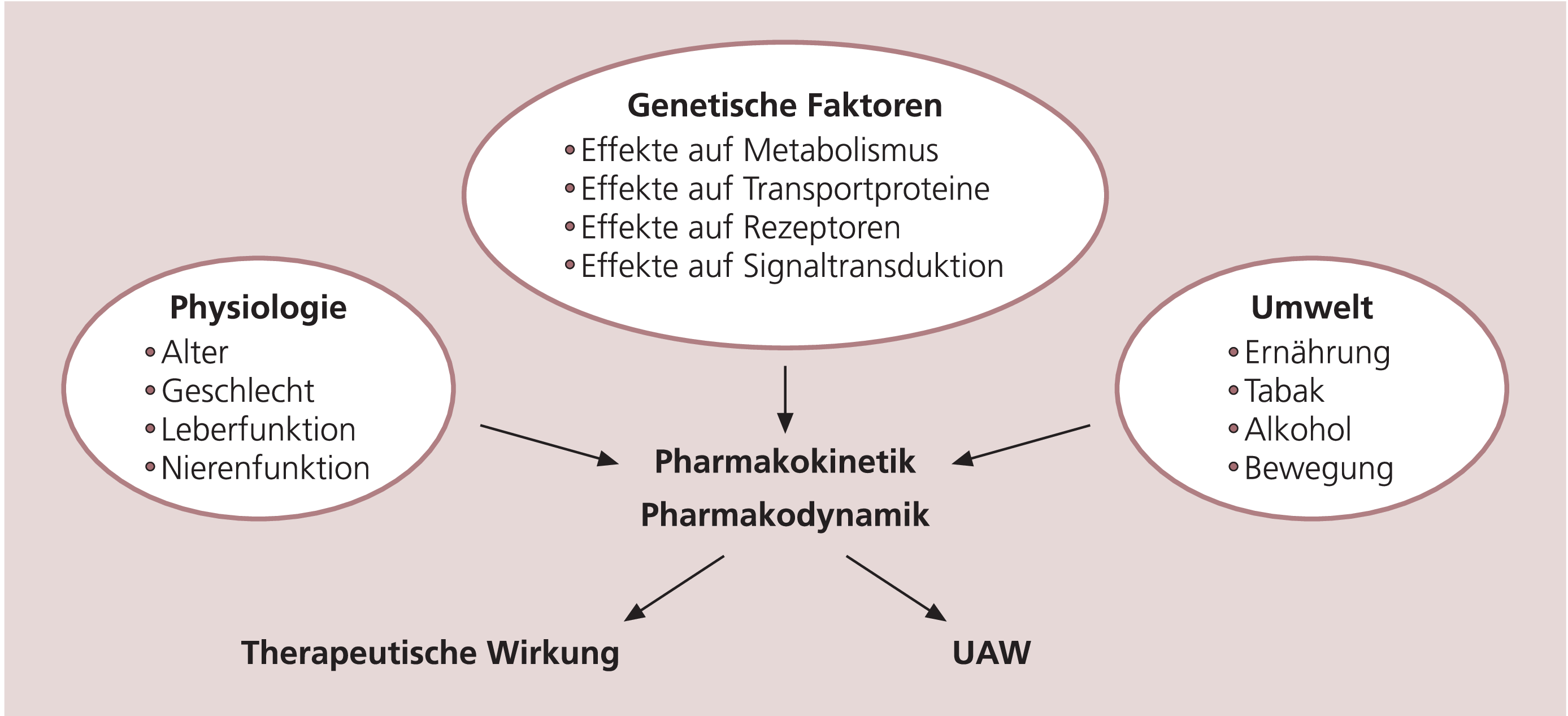
Abb. 1. Faktoren, die das Ansprechen auf eine Therapie beeinflussen
Kandidatengene der Pharmakodynamik
Psychopharmaka interagieren mit zahlreichen Mechanismen innerhalb des zentralen Nervensystems (ZNS), wie der Synthese von Neurotransmittern oder deren Speicherung und Freisetzung an den Synapsen, den Effekten abbauender Enzyme, den spezifischen prä- und postsynaptischen Rezeptoren sowie den Transportproteinen. Durch die Neurotransmitter-Rezeptor-Interaktion wird eine Reihe von Transduktionsmechanismen ausgelöst und das Signal intrazellulär weitervermittelt (Abb. 2). Entsprechend den früheren Hypothesen wurden vor allem Veränderungen in den Konzentrationen der Neurotransmitter als wichtiges Korrelat für den Wirkungsmechanismus betrachtet. Die neueren Konzepte stellen die Proteine jenseits der postsynaptischen Membran und die nachfolgende, veränderte Expression der Transkriptionsfaktoren und Proteine in den Mittelpunkt. Insbesondere die Familie der G-Proteine, denen durch die Kopplung zwischen Rezeptor und Neurotransmitter eine Schlüsselrolle in der Signaltransduktion zugeschrieben wird, ist von erheblichem Interesse, da durch diese die nachfolgende Kaskade von Reaktionen in Gang gesetzt wird. Aber auch andere Proteine der Signaltransduktion wie die Adenylatcyclase, Phosphodiesterasen, Kinasen, sowie das „cAMP regulatory element binding protein (CREB)“ sind potenzielle Zielproteine pharmakogenetischer Untersuchungen [3, 4]. Aus dieser Vielfalt der möglichen Reaktionen ergibt sich, dass Variationen in der DNS-Sequenz eines oder mehrerer dieser Kandidatengene zahlreiche Effekte nach sich ziehen können, welche sowohl die therapeutischen, also die gewünschten, wie auch die unerwünschten Wirkungen betreffen können.
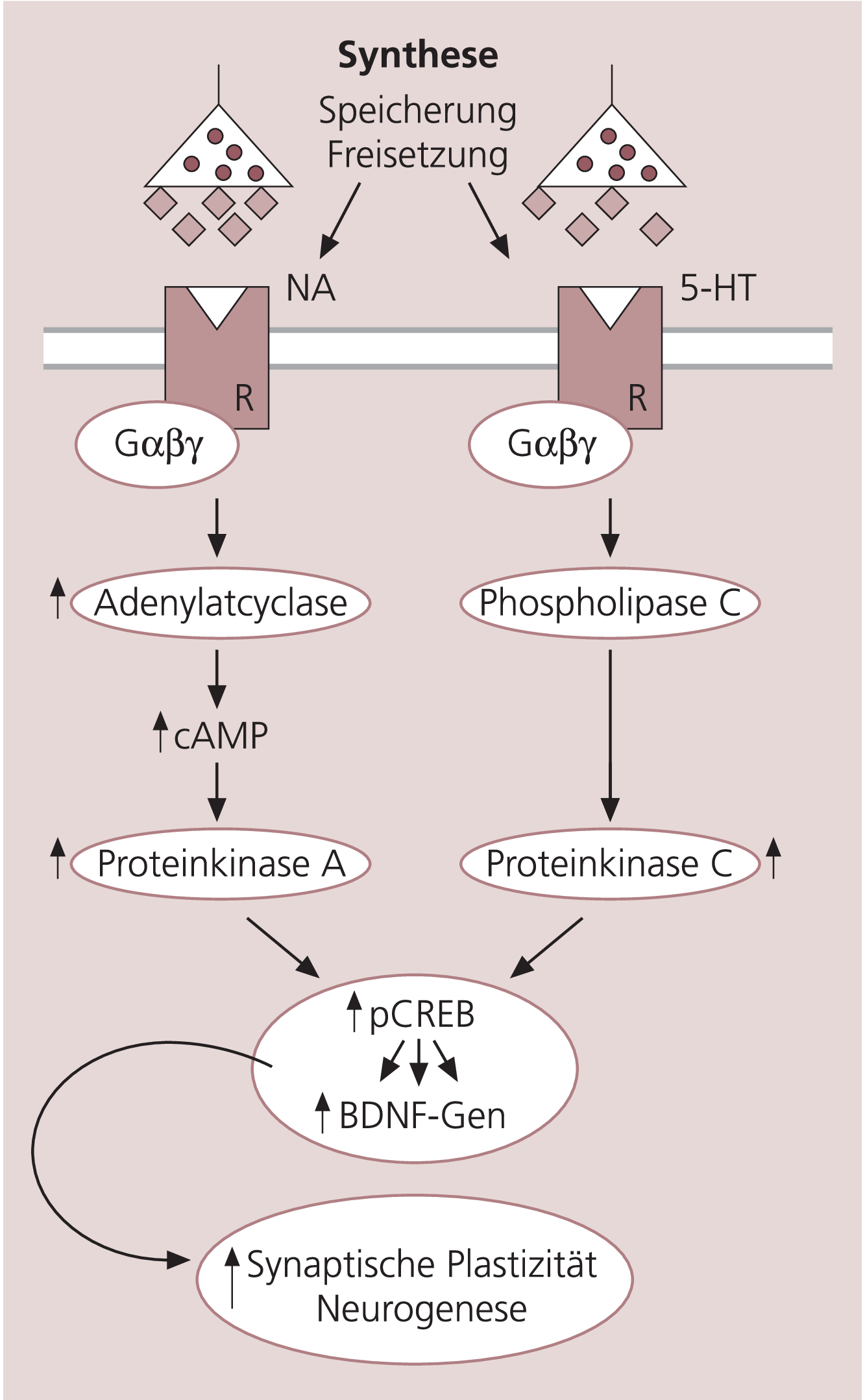
Abb. 2. Signaltransduktion mit potenziellen Kandidatengenen für die Wirkung von Psychopharmaka
NA = Noradrenalin, Gαβγ = G-Protein-αβγ-Komplex, cAMP = zyklisches Adenosinmonophosphat, CREB = cAMP regulatory element binding protein, pCREB = phosphoryliertes CREB, BDNF = Brain-derived neurotrophic factors
Pharmakogenetik der Schizophrenie
Die klassischen Hypothesen für die Ätiologie der Schizophrenie sowie für die Wirkung der Antipsychotika schlagen eine Beteiligung der dopaminergen und serotonergen Systeme vor. Die Mehrzahl der bisher vorliegenden pharmakogenetischen Untersuchungen wurde mit dem Prototyp der atypischen Neuroleptika, Clozapin, durchgeführt. Im Einklang mit dem pharmakodynamischen Spektrum der Substanz wurden vor allem Gene der serotonergen (5-HT2A, 5-HT2C, 5-HT6) und dopaminergen Rezeptoren (D2, D3 und D4) untersucht. Obwohl die Ergebnisse keinen eindeutigen Schluss zulassen (Tab. 1), wurde zumindest für einen Polymorphismus im Gen des 5-HT2A-Rezeptors, einen T102C-Austausch, ein Einfluss auf das Ansprechen auf Clozapin [5] und eventuell auch auf das Ansprechen auf klassische Neuroleptika [6] gezeigt.
Tab. 1. Pharmakogenetik der Dopamin (DA)- und Serotonin (5-HT)-Rezeptoren bei Schizophrenie und Ansprechrate auf Clozapin [mod. nach 38]
|
Rezeptor |
Gen |
Polymorphismus |
Ergebnis |
|
5-HT2A |
5-HT2A |
T102C |
Diskrepante Befunde, |
|
5-HT2A |
5-HT2A |
His45Tyr |
Diskrepante Befunde |
|
5-HT2C |
5-HT2C |
Cys23Ser |
Diskrepante Befunde, überwiegend negativ |
|
5-HT6 |
5-HT6 |
C267T |
Diskrepante Befunde |
|
D3 |
DRD3 |
Ser9Gly |
Diskrepante Befunde |
|
D4 |
DRD4 |
48 bp repeat |
Diskrepante Befunde, überwiegend negativ |
Unter den Dopamin-Rezeptoren gewann der D3-Subtyp an Bedeutung, vor allem wegen seiner überwiegenden Lokalisation in limbischen Strukturen des ZNS sowie wegen seiner Affinität für beide, klassische und atypische Neuroleptika. Der Ser9Gly-Polymorphismus des D3-Rezeptors, der auch zu funktionellen Änderung der Rezeptor-Eigenschaften führt, wurde bereits früh hinsichtlich einer möglichen Beteiligung für die Wirkung von klassischen Neuroleptika und Clozapin diskutiert. Die Ergebnisse sind allerdings ebenso diskrepant wie die mit dem D4-Rezeptor-Polymorphismus [18, 19], der wegen seiner Lokalisation im präfrontalen Kortex, einer Region, die mit den kognitiven Störungen Schizophrener in Beziehung gebracht wird, sowie wegen seiner besonderen Affinität gegenüber Clozapin in den Vordergrund des Interesses rückte [7].
Bei den D2-Rezeptoren, deren Blockade über viele Jahre als der entscheidende Wirkungsmechanismus klassischer Neuroleptika angesehen wurde, gibt es kaum Befunde, was sicherlich auch darin begründet ist, dass heute den Atypika in der Behandlung der Schizophrenie der Vorzug gegeben wird. Eigene Befunde zeigten, dass eine funktionell aktive Variante des D2-Gens einen Einfluss auf das individuelle Ansprechen auf Haloperidol haben kann [8].
Pharmakogenetik affektiver Störungen
Entsprechend den pathophysiologischen Mechanismen affektiver Störungen, die eine verminderte Funktion in der monoaminergen Neurotransmission postulieren, greifen Antidepressiva unterschiedlicher Klassen überwiegend in die serotonerge, noradrenerge und dopaminerge Neurotransmission ein. Vor allem der Serotonin-Transporter (5-HTT), das erste Zielprotein der weit verbreitet verordneten, selektiven Serotonin-Wiederaufnahmehemmer (SSRI) bietet sich als Kandidat für pharmakogenetische Untersuchungen an. Eine funktionelle Variante in der Promoterregion des 5-HTT-Gens resultiert in einem langen (L) oder kurzen (S) Allel. Das S-Allel reduziert die transkriptionelle Aktivität des 5-HTT-Promoters und führt letztendlich zu verminderter Expression des 5-HTT sowie verminderter Serotonin-Wiederaufnahme [9]. Zahlreiche Fall-Kontroll-Assoziationsstudien wurden in den letzten Jahren durchgeführt und haben gezeigt, dass Patienten, die zumindest ein L-Allel besitzen, besser und schneller auf die Behandlung mit SSRI wie Fluoxetin und Paroxetin ansprechen. Fasst man alle vorliegenden Studien zusammen, so zeigt sich ein erheblicher Einfluss des 5-HTT-Promoter-Polymorphismus in der Ansprechrate auf SSRI, allerdings nur bei der kaukasischen Population [10]. Für die asiatische Bevölkerung wurden gegensätzliche Befunde berichtet, was sich dahin gehend interpretieren lässt, dass in unterschiedlichen Populationen unterschiedliche Allele von Bedeutung sind, eventuell durch Interaktionen mit anderen Varianten in Genen, welchen in dieser ethnischen Gruppe eine besondere Bedeutung zukommt [10]. Weitere Untersuchungen zu den verschiedenen Serotonin-Rezeptor-Subtypen, des abbauenden Enzyms Monoaminoxidase, aber auch der Noradrenalin- und Dopamin-Transporter erbrachten keine überzeugenden Befunde (Tab. 2) [11].
Tab. 2. Relevante pharmakogenetische Befunde für Antidepressiva [mod. nach 11]
|
Gen |
Polymorphismus |
Ergebnis |
|
SERT |
L-Allel, S-Allel |
Response auf SSRI (Fluoxetin, Fluvoxamin, Citalopram, Paroxetin) |
|
5-HT2A |
T102C; C1420T; G1438A |
Keine Assoziation |
|
TPH1 |
A218C |
C/C assoziiert mit besserem Ansprechen auf Fluvoxamin, Paroxetin |
|
Gβ3 |
C825T |
T/T assoziiert mit besserem Ansprechen |
|
MAO-A |
30 bp repeat |
Keine Assoziation |
|
DRD2 |
Ser311Cys |
Keine Assoziation |
|
DRD3 |
Ser9Gly |
Keine Assoziation |
|
DRD4 |
48 bp repeats |
Keine Assoziation |
|
ACE |
ins/del |
Diskrepante Befunde |
|
BDNF |
Val166Met |
Keine Assoziation |
|
FKBP5 |
rs1360780 (C/T) |
TT assoziiert mit besserem Ansprechen |
SERT: Serotonin-Transporter; 5-HT2A: 5-HT2A-Rezeptor-Gen; TPH1: Tryptophan-Hydroxylase-1-Gen; Gβ3: G-Protein Beta-3-Untereinheit; MAO-A: Monoaminoxidase A; DRD2/3/4: Gene für die Dopamin-Rezeptoren D2/3/4; ACE: Angiotensin-Konversionsenzym; BDNF: Brain-derived Neurotrophic Factor; FKBP5: FK506 Binding Protein 5
Aktuelle Konzepte: Signaltransduktion, neuronale Plastizität, Stressresponse
Innerhalb des letzen Jahrzehnts haben sich unsere Kenntnisse bezüglich der pathogenetischen Mechanismen und der Effekte der Pharmaka wesentlich verändert. Es wurde deutlich, dass die durch die unterschiedlichen Wirkungsmechanismen erreichten Veränderungen der Neurotransmitter-Konzentrationen im synaptischen Spalt und die Interaktionen mit den entsprechenden Rezeptoren vor allem als initialer Schritt zu betrachten sind, durch den es über zahlreiche Aktivierungsschritte in der Synapse letztendlich zu substanziellen Veränderungen der Proteinexpression und damit zu langfristigen Veränderungen der neuronalen Funktion kommt. Damit lässt sich auch die Zeitspanne bis zum gewünschten therapeutischen Effekt sowohl für Antidepressiva als auch für Neuroleptika erklären.
Aufgrund der Schlüsselposition der G-Proteine wurde immer wieder darüber spekuliert, ob deren genetische Varianten einen Einfluss auf die Behandlung haben können. Bisher liegen erste Ergebnisse für eine Variante (GNB3 C825T) des Gens für die Beta-3-Untereinheit des G-Protein (Gβ3) vor und es zeigte sich, dass das T-Allel nicht nur mit einer gesteigerten Signaltransduktion assoziiert ist [12], sondern auch mit einem rascheren Ansprechen auf die Behandlung mit Antidepressiva [13–15], und zwar unabhängig von der jeweils verabreichten Substanz. Allerdings gibt es auch hier diskrepante Befunde, die darauf hinweisen, dass diesem Polymorphismus in unterschiedlichen Populationen eine unterschiedliche Bedeutung zukommt. Die weiteren Zielproteine der Signaltransduktion werden zurzeit untersucht, insbesondere wird überprüft, ob eine Veränderung der neuronalen Plastizität und Neuronogenese eine gemeinsame Endstrecke der antidepressiven Wirkung sein könnte [16].
Lange ist bekannt, dass im Vorfeld von Depressionen gehäuft krisenhafte Lebensereignisse auftreten, welche einen erheblichen Risikofaktor darstellen, später eine Depression zu entwickeln. Im Allgemeinen hat das Gehirn eine große Anpassungsfähigkeit an die Erfordernisse und Aktivitäten des täglichen Lebens. Besonders Glucocorticoide sind mit verantwortlich für die Adaptation des Gehirn auf Stress, da sie die elektrische Aktivität von Neuronen modulieren und für morphologische Veränderungen in Neuronenverbänden mit verantwortlich sind. Es gibt heute keinen Zweifel mehr, dass bei der Depression vor allem eine Kombination von genetisch bedingter Vulnerabilität sowie länger bestehender Stressbelastung mit den bekannten neuroendokrinen Veränderungen zu strukturellen und neurochemischen Veränderungen im Gehirn mit nachfolgendem Zelltod führen kann. Der genaue Mechanismus, über den Glucocorticoide zu einer Zellschädigung führen können, ist bisher nicht genau geklärt, es wird jedoch angenommen, dass es durch Downregulation des Transkriptionsfaktors CREB zur verminderten Produktion von neurotrophen Faktoren (wie BDNF) kommt, die für das Überleben und die Funktion von Nervenzellen von Bedeutung sind [12].
Zu den häufig replizierten Befunden in der Depressionsforschung gehört der Nachweis einer zentral und peripher gestörten Stresshormon-Regulation, die im Rahmen einer effektiven Behandlung wieder normalisiert wird [17]. Bekannt sind Hyperaktivität in der Hypothalamus-Hypophysen-Nebennierenrinden-(HPA-)Achse mit erhöhter Sekretion von Corticotropin-Releasing-Faktor (CRF) und nachfolgender Cortisol-Sekretion, verminderter Glucocorticoid-Rezeptorsensitivität mit gestörten Feedback-Mechanismen [18]. In diesem Zusammenhang sind Ergebnisse aus eigenen Untersuchungen von Bedeutung, die in zwei unabhängigen klinischen Studien einige Polymorphismen in Genen überprüften, die in der HPA-Achsenregulation und speziell für die Glucocorticoid-Rezeptorsensitivität eine wichtige Rolle spielen. Dabei gab es in beiden Gruppen genetische Varianten des FKBP-5-Gens, einem Cochaperon des Glucocorticoid-Rezeptors. Patienten mit der TT-Variante des Gens zeigten bereits nach einwöchiger Behandlung mit Antidepressiva eine signifikante Verbesserung ihrer Krankheitssymptome, während Patienten mit den CT- oder CC-Genotypen selbst nach fünf Wochen noch nicht diesen Grad an Besserung erreichten [19]. Zu bemerken ist hier vor allem auch, dass dieser Effekt nicht selektiv für eine Substanz beobachtet wurde, sondern jeweils bei SSRI, trizyklischen Antidepressiva und auch Mirtazapin-Monotherapie gleichermaßen auftrat.
Ebenfalls mögliche Störungen in der HPA-Achse betreffen unsere Ergebnisse hinsichtlich des Insertions/Deletions (I/D)-Polymorphismus im Angiotensin-Konversionsenzym-(ACE-)Gen. Da ACE neben seinen Blutdruck-regulierenden Wirkungen auch zahlreiche Effekte innerhalb des ZNS ausübt, wie die Beschleunigung des Abbaus von Substanz P [20] oder die Beteiligung in der stressinduzierten Aktivierung der HPA-Achse [21, 22], könnten Polymorphismen, die wie der I/D-Polymorphismus auch funktionell Einflüsse auf die ACE-Konzentration ausüben, bei der Depression eine Rolle spielen. Im Rahmen unserer Untersuchungen konnten wir zeigen, dass Patienten mit zumindest einem D-Allel schneller auf die unterschiedlichen Methoden wie medikamentöse Behandlung, Elektrokrampftherapie, transkranielle Magnetstimulation oder Schlafentzug ansprachen [23, 24], was sich auch in der Dauer des stationären Aufenthalts manifestierte. Diese Befunde zum ACE-Gen gewinnen an Bedeutung, weil es sich dabei um einen „missing link“ zwischen depressiven Störungen und Herz-Kreislauf-Erkrankungen handeln könnte [25] und weil neue Untersuchungen belegen, dass ACE die Charakteristika eines Signaltransduktionsmoleküls besitzt [26]. Dies bedeutet, dass über ACE die Signaltransduktion im ZNS und in der Peripherie beeinflusst werden kann.
Pharmakogenetik der UAW
Abgesehen davon, dass ein Teil der Patienten nicht oder nur ungenügend auf die Behandlung anspricht, ist die Behandlung, die sich in der Regel über einen längeren Zeitraum erstreckt, häufig von einer hohen Rate an Nebenwirkungen gekennzeichnet. Auch UAW werden wie die gewünschten Effekte über pharmakodynamische Prozesse induziert oder verstärkt und somit auch durch genetische Varianten beeinflusst. Obwohl sich diese pharmakogenetischen Untersuchungen erst in der Anfangsphase befinden, sind in naher Zukunft doch einige interessante Ergebnisse zu erwarten.
Spätdyskinesie
Eines der größten Probleme in der der Behandlung mit klassischen Antipsychotika ist das Auftreten extrapyramidal-motorischer Störungen und insbesondere der Spätdyskinesie. Da vor allem eine Überaktivität dopaminerger Transmission in den Basalganglien sowie eine Heraufregulierung von D2-Rezeptoren als Ursache der Spätdyskinesie diskutiert wird [27], hatten dopaminerge Gene bisher die höchste Priorität für pharmakogenetische Untersuchungen. Vor allem der D2-Rezeptor als lange diskutiertes primäres Zielprotein für klassische Antipsychotika wurde häufig untersucht, allerdings überwiegend mit negativem Befund (Tab. 3) [28]. Positiver sehen die Ergebnisse mit dem DRD3-Ser9Gly-Polymorphismus aus, da es hier in den letzten Jahren eine Reihe von unabhängigen Studien gab, die belegen konnten, dass der Austausch von Serin zu Glycin in Position 9 mit einem erhöhten Risiko verbunden ist, eine Spätdyskinesie zu entwickeln.
Tab. 3. Untersuchungen zur Assoziation zwischen genetischer Variante und UAW [mod. nach 28, 36]
|
UAW |
Gen |
Polymor- |
Ergebnis |
|
Spätdyskinesie |
DRD2 |
TaqI A und B |
Überwiegend negativ, |
|
Gewichtszunahme |
5-HT2C 5-HT2A |
Cys23Ser |
Ser-Allel assoziiert mit Gewichtszunahme, auch negative Befunde |
Metabolisches Syndrom
Auch wenn das Risiko einer Spätdyskinesie bei den so genannten Atypika deutlich reduziert ist, haben auch diese Substanzen gelegentlich schwerwiegende UAW, darunter vor allem Gewichtszunahme, Dyslipidämien, Glucose-Stoffwechselstörungen oder kardiovaskuläre Risiken wie QT-Zeitverlängerung [28].
Bei den metabolischen Wirkungen ist ein Zusammenhang mit schweren psychischen Störungen bereits um die Jahrhundertwende erkannt worden und die negativen Einflüsse von Phenothiazin auf die Glucosetoleranz wurden bereits in den 50er Jahren diskutiert. Dennoch schienen sich nach Einführung der Atypika diese Probleme zu häufen [29]. Vor allem eine häufige, deutliche Gewichtszunahme (>7% des Ausgangsgewichts) zählt zu den Problemen in der Behandlung mit nahezu allen neuen Substanzen, vor allem aber mit Clozapin, Olanzapin und Quetiapin, ebenso wie Störungen im Glucose- und Lipidstoffwechsel [29]. Die Tatsache, dass diese metabolischen Probleme durchaus nicht bei allen Patienten zu beobachten sind, lässt eine genetisch bedingte Vulnerabilität vermuten.
Die Regulierung von Hunger und Sättigung ist ein komplizierter Vorgang, in welchen zahlreiche zentrale und periphere Mechanismen involviert sind. Ferner ist bekannt, dass die Regulierung des Gewichts zu einem erheblichen Maße genetisch beeinflusst wird und heute sind bereits mehr als 300 Gene bekannt, die hier einen Einfluss haben [30]. Bisher wurde vor allem das serotonerge System berücksichtigt, da sowohl tierexperimentelle Studien als auch Untersuchungen am Menschen gezeigt haben, dass eine Erhöhung der 5-HT-Konzentration Sättigungsgefühl induziert und dass 5-HT2C-Agonisten die Nahrungsaufnahme vermindern [31]. Dies ist natürlich im Hinblick auf die 5-HT2-antagonistischen Wirkungen vieler Antipsychotika von erheblicher Bedeutung.
Die ersten Untersuchungen wurden mit einer strukturellen Variante des 5-HT2C-Rezeptorgens durchgeführt, die zu einer Cys23Ser-Substitution und zu einer Veränderung der Rezeptoreigenschaften führt, und es zeigte sich eine Assoziation der Mutante mit Gewichtszunahme unter Clozapin-Behandlung [32]. Neuere Studien belegen die Bedeutung des 5-HT2C-Gens, da eine Variante in der Promoterregion (–759C/T) sowohl mit Gewichtszunahme unter Olanzapin als auch Clozapin assoziiert werden kann, wenn sich dies auch nicht immer replizieren ließ [33–35].
Weitere Untersuchungen werden in Zukunft vor allem Proteine der Regulation des Lipid- und Glucosestoffwechsels betreffen sowie auch Mutationen im Leptin-Gen, das die Nahrungsaufnahme und den Energiehaushalt reguliert [36]. Bereits früher wurde eine Mutante in der DNS des Leptin-Gens mit extremer Adipositas assoziiert [37].
Zusammenfassung und Zukunftsperspektiven
Obwohl zahlreiche Studien gezeigt haben, dass Varianten in Genen sowohl die gewünschten als auch die unerwünschten therapeutischen Wirkungen beeinflussen können, sind wir noch weit davon entfernt für jeden Patienten eine „Therapie nach Maß“ wählen zu können. Die pharmakogenetischen Untersuchungen stecken noch in den Anfängen. Es gibt eine Reihe von Faktoren, welche diese Untersuchungen beeinflussen und bei weiteren Studien in Erwägung gezogen werden sollten.
Gerade die diskrepanten Befunde weisen darauf hin, dass das Ansprechen auf die Behandlung, ebenso wie die Störung selbst, ein komplexes Phänomen ist. Das bedeutet, eine Interaktion von mehreren Genen untereinander sowie mit anderen Faktoren, beispielsweise Umweltfaktoren, wird die Response beeinflussen. Höchstens in Einzelfällen wird man eine Mutation in einem Gen für die interindividuelle Variabilität verantwortlich machen können. Trotz dieser Probleme besteht Zuversicht, dass in nicht zu ferner Zeit für schwerwiegende UAW eine „Risiko-Kombination von genetischen Varianten“ identifiziert werden kann.
Danksagung
Einige der hier zitierten Arbeiten wurden vom Bundesministerium für Bildung und Forschung (BMBF) im Rahmen des Förderschwerpunktes Kompetenz-Netze in der Medizin gefördert.
Literatur
1. Kerwin R. Antipsychotic drugs. Medicine 2000;28:23–5.
2. Weinshilboum R. Inheritance and drug response. N Engl J Med 2003;348:529–37.
3. Johnson JA, Lima JJ. Drug receptor/effector polymorphisms and pharmacogenetics: current status and challenges. Pharmacogenetics 2003;13:525–34.
4. Young LT, Bakish D, Beaulieu S. The neurobiology of treatment response to antidepressants and mood stabilizing medications. J Psychiatry Neurosci 2002;27:260–5.
5. Basile VS, Masellis M, Potkin SG, Kennedy JL. Pharmacogenomics in schizophrenia: the quest for individualized therapy. Hum Mol Genet 2002;11:2517–30.
6. Mancama D, Arranz MJ, Kerwin RW. Genetic predictors of therapeutic response to clozapine: current status of research. CNS Drugs 2002;16:317–24.
7. Seeman P, Vantol HHM. Dopamine receptor pharmacology. Trends Pharmacol Sci 1994;15:264–70.
8. Schäfer M, Rujescu D, Giegling I, Guntermann A, et al. Association of short-term response to haloperidol treatment with a polymorphism in the dopamine D2 receptor gene. Am J Psychiatry 2001;158:802–4.
9. Heils A, Teufel A, Petri S, Stober G, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem 1996;66:2621–4.
10. Mancama D, Kerwin RW. Role of pharmacogenomics in individualising treatment with SSRIs. CNS Drugs 2003;17:143–51.
11. Serretti A, Artioli P. From molecular biology to pharmacogenetics: a review of the literature on antidepressant treatment and suggestions of possible candidate genes. Psychopharmacology (Berl) 2004;174:490–503.
12. Siffert W, Rosskopf D, Siffert G, Busch S, et al. Association of a human G-protein beta3 subunit variant with hypertension. Nat Genet 1998;18:45–8.
13. Zill P, Baghai TC, Zwanzger P, Schule C, et al. Evidence for an association between a G-protein beta3-gene variant with depression and response to antidepressant treatment. Neuroreport 2000;11:1893–7.
14. Serretti A, Lorenzi C, Cusin C, Zanardi R, et al. SSRIs antidepressant activity is influenced by G beta 3 variants. Eur Neuropsychopharmacol 2003;13:117–22.
15. Lee HJ, Cha JH, Ham BJ, Han CS, et al. Association between a G-protein beta 3 subunit gene polymorphism and the symptomatology and treatment responses of major depressive disorders. Pharmacogenomics J 2004;4:29–33.
16. Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry 1999;46:1181–91.
17. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev 1996;17:187–205.
18. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000;23:477–501.
19. Binder EB, Salyakina D, Lichtner P, Wochnik GM, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 2004;36:1319–25.
20. Nutt D. Substance-P antagonists: a new treatment for depression? Lancet 1998;352:1644–6.
21. Jeunemaitre X. [Genetic polymorphisms in the renin-angiotensin system]. Therapie 1998;53:271–7.
22. Jezova D, Ochedalski T, Kiss A, Aguilera G. Brain angiotensin II modulates sympathoadrenal and hypothalamic pituitary adrenocortical activation during stress. J Neuroendocrinology 1998;10:67–72.
23. Baghai T, Schüle C, Zwanzger P, Minov C, et al. Possible influence of the insertion/deletion polymorphism in the angiotensin I-converting enzyme gene on therapeutic outcome in affective disorders. Mol Psychiatry 2001;6:258–9.
24. Baghai TC, Schule C, Zwanzger P, Zill P, et al. Influence of a functional polymorphism within the angiotensin I-converting enzyme gene on partial sleep deprivation in patients with major depression. Neurosci Lett 2003;339:223–6.
25. Bondy B, Baghai TC, Zill P, Bottlender R, et al. Combined action of the ACE D- and the G-protein ß3-allele in major depression: a possible link to cardiovascular disorder? Mol Psychiatry 2002;7:1120–6.
26. Kohlstedt K, Brandes RP, Müller-Esterl W, Busse R, et al. Angiotensin-converting enzyme is involved in outside-in signaling in endothelial cells. Circ Res 2004;94:60–7.
27. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry 2000;61(Suppl 4):5–9.
28. Malhotra AK, Murphy GM Jr., Kennedy JL. Pharmacogenetics of psychotropic drug response. Am J Psychiatry 2004;161:780–96.
29. Mackin P, Watkinson HM, Young AH. Prevalence of obesity, glucose homeostasis disorders and metabolic syndrome in psychiatric patients taking typical or atypical antipsychotic drugs: a cross-sectional study. Diabetologia 2005;48:215–21.
30. Chagnon YC, Rankinen T, Snyder EE, Weisnagel SJ, et al. The human obesity gene map: the 2002 update. Obes Res 2003;11:313–67.
31. Malhotra AK. Candidate gene studies of antipsychotic drug efficacy and drug-induced weight gain. Neurotox Res 2004;6:51–6.
32. Reynolds GP, Zhang Z, Zhang X. Polymorphism of the promoter region of the serotonin 5-HT2C receptor gene and clozapine-induced weight gain. Am J Psychiatry 2003;160:677–9.
33. Basile VS, Masellis M, De Luca V, Meltzer HY, et al. 759C/T genetic variation of 5HT2C receptor and clozapine-induced weight gain. Lancet 2002;360:1790–1.
34. Ellingrod VL, Perry PJ, Ringold JC, Lund BC, et al. Weight gain associated with the -759C/T polymorphism of the 5HT2C receptor and olanzapine. Am J Med Genet B Neuropsychiatr Genet 2005;134:176-8.
35. Theisen FM, Hinney A, Bromel T, Heinzel-Gutenbrunner M, et al. Lack of association between the –759C/T polymorphism of the 5-HT2C receptor gene and clozapine-induced weight gain among German schizophrenic individuals. Psychiatr Genet 2004;14:139–42.
36. Templeman LA, Reynolds GP. Pharmacogenetics of antipsychotic side effects. Clin Neuropsychiatry 2004;1:108–16.
37. Clement K, Garner C, Hager J, Philippi A, et al. Indication for linkage of the human OB gene region with extreme obesity. Diabetes 1996;45:687–90.
38. Muller DJ, Shinkai T, De Luca V, Kennedy JL. Clinical implications of pharmacogenomics for tardive dyskinesia. Pharmacogenomics J 2004;4:77–87.
Prof. Dr. Brigitta Bondy, Peter Zill, Psychiatrische Klinik der Ludwig-Maximilians-Universität, Nussbaumstraße 7, 80336 München, E-Mail: brigitta.bondy@med.uni-muenchen.de
Pharmacodynamics and pharmacogenetics: Focus on adverse effects
Genetic factors are supposed to play a major role for the differences in response to treatment or the incidence of adverse drug effects in psychopharmacotherapy. The aim of pharmacogenetics is to elucidate this variability due to hereditary differences. According to the hypotheses on the mechanisms of drug action, several mutations in genes coding for neurotransmitter receptors, degrading enzymes, transport proteins or enzymes of the drug metabolizing system have been identified and investigated. Although there exist some controversy among the results, there is increasing evidence that several polymorphisms within the genes coding for serotonin and dopamine receptors and the serotonin transporter may be involved in drug action. Recent investigation is focusing the structures beyond the initial drug targets, as the G-proteins, which are initiating the signal transduction cascade and are thus the first step for alterations in gene expression.
Keywords: Pharmacogenetics, antidepressants, neuroleptics, polymorphism
Psychopharmakotherapie 2005; 12(04)