Kai-Uwe Kühn und Boris B. Quednow, Bonn, Michael Riedel, München, Olaf Krampe, Marburg, und Wolfgang Maier, Bonn
Das bizyklische Isobenzofuran-Derivat Citalopram ist ein selektiver Serotonin-Wiederaufnahmehemmer (SSRI), der seit 1996 in Deutschland zur Behandlung depressiver Erkrankungen zugelassen ist [9]. Citalopram ist ein sehr selektiver SSRI, es hemmt die Serotonin-Wiederaufnahme 3400-mal stärker als die von Noradrenalin und 22000-mal stärker als die von Dopamin [16, 17]. Darüber hinaus hat Citalopram nur eine geringe oder keine Affinität zu Acetylcholin-, Histamin-, Noradrenalin-, Dopamin-, GABA- oder Opiat-Rezeptoren [16]. Die Metaboliten des Citaloprams zeigen eine vergleichbare Selektivität, aber eine schwächere Inhibition der Serotonin-Wiederaufnahme [15] und scheinen kaum zu den klinischen Effekten beizutragen [17]. Citalopram besitzt eine hohe Bioverfügbarkeit und im Gegensatz zu den meisten anderen SSRI oder tri- und tetrazyklischen Antidepressiva weist es eine lineare Pharmakokinetik und eine geringe Plasma-Eiweißbindung auf [1, 4]. Aufgrund der langen Plasma-Halbwertszeit von 35 Stunden kann die Substanz einmal täglich verabreicht werden. Im Unterschied zu Paroxetin (z. B. Seroxat®) oder Fluoxetin (z.B. Fluctin®) hemmen Citalopram oder seine Metaboliten die eigene Metabolisierung nicht, so dass die Kumulationsgefahr nicht erhöht ist [1]. Zusätzlich zeigt Citalopram nur eine geringe Aktivität am Cytochrom-P450-Isoenzym-System und weist ein geringes Risiko für Arzneimittelinteraktionen auf [4]. Diese pharmakokinetischen und -dynamischen Eigenschaften tragen dazu bei, dass Citalopram auch bei Patienten mit einer Polypharmakotherapie – wie beispielsweise somatisch multimorbiden oder älteren Patienten – eingesetzt werden kann. Es sollte darauf hingewiesen werden, dass es bei einer gleichzeitigen Einnahme mit Substanzen, die ebenfalls die serotonerge Neurotransmission steigern (z.B. Lithiumsalze und andere serotonerge Antidepressiva, insbesondere MAO-Hemmer), zu pharmakodynamischen Arzneimittelinteraktionen kommen kann, wobei im Extremfall die Entwicklung eines Serotonin-Syndroms droht.
In kontrollierten Studien war Citalopram in der Behandlung der Depression Plazebo signifikant überlegen und zeigte eine vergleichbare Wirksamkeit wie andere SSRI und trizyklische oder tetrazyklische Antidepressiva [17, 25]. Darüber hinaus bestätigen zahlreiche Studien die Wirksamkeit von Citalopram bei Angst- und Zwangserkrankungen [3, 23, 24].
Doch obwohl zahlreiche kontrollierte klinische Studien die Wirksamkeit und gute Verträglichkeit von Citalopram in der Behandlung der Depression bestätigen konnten, ist generell die Vergleichbarkeit solcher Studien mit dem Einsatz einer Substanz im klinischen Alltag immer wieder in Frage gestellt worden [6, 19, 20]. Es konnte bereits gezeigt werden, dass die Patientenpopulation in klinischen Studien mit Antidepressiva von der Alltagspopulation der Patienten stark abweicht [28]. Anwendungsbeobachtungen (AWB) bieten die Möglichkeit, die Wirksamkeit und Sicherheit einer Substanz unter Alltagsbedingungen zu beobachten [6, 18, 19, 26]. AWB sind Beobachtungsstudien, die dazu bestimmt sind, Erkenntnisse bei der Anwendung verkehrsfähiger Arzneimittel zu sammeln. Ihr besonderes Charakteristikum ist die weitestgehende Nichtbeeinflussung des behandelnden Arztes in Bezug auf Indikationsstellung sowie Wahl und Durchführung der Therapie im Einzelfall. Ziel ist die Beobachtung von Behandlungsmaßnahmen in der routinemäßigen Anwendung durch Arzt und Patient. Eine AWB kann ohne Vergleichsgruppe, beispielsweise arzneimittelorientiert, oder mit zwei oder mehr zu vergleichenden Gruppen, beispielsweise indikationsorientiert, angelegt sein. Sie wird mit Handelsware durchgeführt. AWB bergen eine Reihe möglicher Interessenkonflikte im Spannungsfeld zwischen Datenschutz, Schutz des Patienten, Schutz und Haftung des Arztes und Interesse des Sponsors [BfArM, 12.11.1998]. Allerdings können ausschließlich AWB aufgrund ihres Charakters eine umfassende Aussage über Arzneimittelinteraktionen in der klinischen Praxis ermöglichen. Dieses Dilemma zeigt sowohl die Existenzberechtigung als auch die größte Schwäche von Anwendungsbeobachtungen auf [10, 14].
Arzneimittelinteraktionen können durch erhöhte Toxizität oder unwirksame Arzneimittelspiegel den Krankheitsverlauf negativ beeinflussen [10]. Da Arzneimittelinteraktionen nicht zwangsläufig während der Medikamentenentwicklung umfassend untersucht werden und aufgrund der Tatsache, dass sich Probanden in klinischen Studien zum Teil erheblich von einem Großteil der Patienten unterscheiden, sind Anwendungsbeobachtungen ein essenzielles Werkzeug in der Gewährleistung und Überwachung der Arzneimittelsicherheit [14, FDA Oct 1996 www.fda.gov/medwatchwww.fda.gov/medwatch].
Ziel der vorliegenden AWB war es, die Wirksamkeit und Sicherheit von Citalopram in der Behandlung der Depression unter Alltagsbedingungen in der Praxis niedergelassener Psychiater, Neurologen und Nervenärzte zu untersuchen.
Patienten und Methoden
Es nahmen 206 niedergelassene Fachärzte für Neurologie, Psychiatrie oder Nervenheilkunde verteilt über das gesamte Bundesgebiet teil. Die teilnehmenden Ärzte wurden für jeden eingeschlossenen Patienten honoriert. Im Zeitraum von April bis Oktober 2002 wurden von den teilnehmenden Ärzten prospektiv alle Patienten mit der Diagnose einer depressiven Erkrankung nach ICD-10 dokumentiert, die neu auf das Citalopram-Generikum Serital® eingestellt wurden. Es wurden Patienten mit neu diagnostizierter unipolarer Depression oder Patienten mit einer bestehenden unipolaren depressiven Erkrankung ohne aktuelle antidepressive Therapie in die Anwendungsbeobachtung eingeschlossen. Abgesehen von den Einschränkungen, die in der Fachinformation zu Citalopram genannt werden, bestanden keine weiteren Kriterien für den Ausschluss von Patienten von der AWB. Der Beobachtungszeitraum betrug maximal zwölf Wochen. Die Datenerhebungen fanden zum Einschluss, nach 4 bis 6 sowie nach 8 bis 12 Wochen statt. Die AWB wurde von der Temmler Pharma GmbH & Co. KG finanziert und durchgeführt.
Die 610 untersuchten Patienten waren im Mittel 49,8 ± 14,1 (Standardabweichung) Jahre alt, durchschnittlich groß (169,8 ± 7,8 cm) und normalgewichtig (72,7 ± 11,6 kg). Etwas mehr als zwei Drittel der teilnehmenden Patienten waren Frauen (67,7 %). Bei 55,6 % der Patienten wurde eine depressive Störung erstmals diagnostiziert. Die einzelnen Diagnosen sind in Tabelle 1 dargestellt. Die aktuelle depressive Episode bestand im Mittel seit 83 ± 124,0 Tagen (Median: 54). Bei Patienten mit einer bereits diagnostizierten Depression war die erste depressive Episode erstmals vor 7,3 ± 7,2 Jahren (Median: 5,0) aufgetreten und im Mittel gingen 4,3 ± 3,2 depressive Phasen (Median: 4,0) der aktuellen Episode voraus.
Tab. 1. Diagnose nach ICD-10
|
ICD-10-Kategorie |
Patienten |
||
|
n |
[%] |
||
|
F32.0 |
Leichte depressive Episode |
32 |
5,2 |
|
F32.1 |
Mittelgradige depressive Episode |
206 |
33,8 |
|
F32.2 |
Schwere depressive Episode ohne psychotische Symptome |
96 |
15,7 |
|
F32.3 |
Schwere depressive Episode mit psychotischen Symptomen |
10 |
1,6 |
|
F32.8 |
Sonstige depressive Episoden |
14 |
2,3 |
|
F32.9 |
Depressive Episode, nicht näher bezeichnet |
38 |
6,2 |
|
F33.0 |
Rezidivierende depressive Störung, gegenwärtig leichte Episode |
15 |
2,5 |
|
F33.1 |
Rezidivierende depressive Störung, gegenwärtig mittelgradige Episode |
124 |
20,3 |
|
F33.2 |
Rezidivierende depressive Störung, gegenwärtig schwere Episode |
50 |
8,2 |
|
F33.3 |
Rezidivierende depressive Störung, gegenwärtig schwere Episode mit psychotischen Symptomen |
12 |
2,0 |
|
F33.4 |
Rezidivierende depressive Störung, gegenwärtig remittiert |
2 |
0,3 |
|
F33.8 |
Sonstige rezidivierende depressive Störung |
5 |
0,8 |
|
F33.9 |
Rezidivierende depressive Störung, nicht näher bezeichnet |
5 |
0,8 |
|
F34.1 |
Dysthymia |
1 |
0,2 |
|
Gesamt |
610 |
100,0 |
|
Befunderhebung
Alle Untersucher waren Fachärzte für Psychiatrie, Neurologie oder Nervenheilkunde. Die Patienten wurden bei Einschluss, nach 4 bis 6 sowie nach 8 bis 12 Wochen untersucht. Als robustes Maß für klinische Wirksamkeit wurde die „Clinical Global Impression Scale“ (CGI) verwendet [2, 21]. Die Schwere der depressiven Erkrankung wurde mit der Hamilton-Depressions-Skala (HAMD), 17-Item-Version, gemessen [11, 12]. HAMD und CGI wurden bei Einschluss sowie nach 8 bis 12 Wochen erhoben. Des Weiteren wurden die Vorbehandlung bei einer bestehenden Depression, die Begleiterkrankungen sowie die Begleitmedikation anamnestisch erhoben. Die teilnehmenden Ärzte sollten für die Patienten, die während einer früheren depressiven Episode bereits eine medikamentöse Therapie erhalten hatten, den Grund für die Neueinstellung auf Serital® angeben. Folgende Antwortmöglichkeiten waren vorgegeben:
Bessere Wirkung erhofft
Bessere Verträglichkeit erhofft
Therapieresistenz
Geringere Kosten
Die unerwünschten Ereignisse wurden mit Hilfe des WHO Adverse Reaction Dictionary (German Version) nach Organsystem und Hauptbegriff kodiert.
Die teilnehmenden Patienten wurden bei der Zwischenuntersuchung und am Ende der Studie gebeten die Wirksamkeit und Verträglichkeit der Citalopram-Behandlung auf einer vierstufigen Skala (sehr gut, gut, mäßig, schlecht) zu beurteilen.
Die Interrater-Reliabilität der CGI-Skala (Item 1) wie auch der HAMD erwies sich in früheren Untersuchungen als sehr robust: sie lag in der CGI-Skala für Ärzte bei 0,66 [7] und für die HAMD zwischen 0,52 und 0,95 [13]. Beide Instrumente eignen sich daher gut für den Einsatz in einer AWB. Die Selbstrating-Skalen zur Wirksamkeit und Verträglichkeit sind auf ihre Interrater-Reliabilität bislang nicht untersucht.
Statistik
Um Therapieeffekte über den Beobachtungszeitraum zu überprüfen, wurden mit den Daten der HAMD und des CGI-Wilcoxon-Tests gerechnet (Einschluss vs. Endpunkt des Beobachtungszeitraums). Um Einflussfaktoren wie Geschlecht, Alter, Erkrankungsdauer zu untersuchen, wurden Varianzanalysen (ANOVA) mit Messwiederholung und Pearsons-Produkt-Moment-Korrelationen eingesetzt. Häufigkeitsdaten wie Response- und Remissionsraten wurden anhand von χ2-Tests analysiert. Das Signifikanzniveau wurde bei p<0,05 (zweiseitig) festgesetzt.
Studienziele und Zielparameter
Ziel der AWB war die Untersuchung von Wirksamkeit und Verträglichkeit von Citalopram in der ambulanten Therapie depressiver Patienten. Als primäre Zielkriterien wurde hierfür die Änderung der Werte im HAMD und im CGI gegenüber dem Ausgangswert gewählt. Sekundäre Zielkriterien waren die Erfassung von unerwünschten Arzneimittelwirkungen (UAW) und der Begleitmedikation.
Ergebnisse
Psychopharmakologische Vor- und Begleitbehandlung
Bei 42,5 % der teilnehmenden Patienten wurde die depressive Erkrankung in der Vorgeschichte bereits pharmakologisch behandelt. In Tabelle 2 ist die Art und Häufigkeit der Vorbehandlung dargestellt.
Tab. 2. Pharmakologische Vorbehandlung der depressiven Erkrankung (Mehrfachnennungen möglich)
|
Substanz oder Substanzgruppe |
n |
|
Trizyklische Antidepressiva |
103 |
|
SSRI |
67 |
|
– Davon Citalopram |
19 |
|
Johanniskraut-Präparate |
27 |
|
Mirtazapin |
17 |
|
Venlafaxin |
16 |
|
Opipramol |
14 |
|
MAO-Hemmer |
8 |
|
Reboxetin |
7 |
|
Typische Neuroleptika |
7 |
|
Nefazodon/Trazodon |
6 |
|
Tetrazyklische Antidepressiva |
4 |
|
Benzodiazepine |
3 |
|
Atypische Neuroleptika |
1 |
|
Betablocker |
1 |
|
Homöopathika |
1 |
|
Lithiumsalze |
1 |
|
Pflanzliche Sedativa |
1 |
|
Gesamt |
284 |
Darüber hinaus erhielten ein Drittel (33,3 %) aller Patienten auch während der AWB eine psychotrope Begleitmedikation. Am häufigsten wurden Hypnotika/Sedativa (10,3 % aller Patienten), Antidepressiva (10,0 % aller Patienten), Tranquillanzien/Anxiolytika (9,7 % aller Patienten) und Neuroleptika (7,4 % aller Patienten) als psychotrope Begleitmedikation verabreicht. Von den 61 Patienten mit einer antidepressiven Begleitmedikation, erhielten 43 (70,5 %) trizyklische Antidepressiva, 6 (9,8 %) Opipramol, 4 (6,6 %) Lithiumsalze, 2 (3,3 %) SSRI, 2 (3,3 %) Johanniskraut-Präparate, 2 (3,3 %) tetrazyklische Antidepressiva, 1 (1,6 %) Mirtazapin und 1 (1,6 %) Trazodon.
Begleiterkrankungen und nicht-psychotrope Begleitmedikation
Insgesamt litten 230 Patienten (37,7 %) unter Begleiterkrankungen. 375 Erkrankungen wurden dokumentiert, wobei die meisten Krankheiten aus dem Bereich Erkrankungen des Kreislaufsystems (16,1 % der Patienten) zu verzeichnen waren. Die am häufigsten genannten Krankheiten waren essenzielle Hypertonie (11,1 % der Patienten), Rückenschmerzen (3,0 % der Patienten) und Diabetes mellitus (2,5 % der Patienten). Tabelle 3 gibt die Häufigkeit der ICD-10-Diagnosen an, bezogen auf die Patientenzahl. Neben der Dokumentation von Begleiterkrankungen und nicht-psychotropen Begleitmedikamenten sollte der Arzt beurteilen, ob es sich um einen multimorbiden Patienten handelt. Bei 42 Patienten (6,9 %) wurde diese Frage mit „ja“ beantwortet; allerdings fehlte bei 112 Patienten (18,4 %) die Angabe zur Multimorbidität. Insgesamt nahmen 28,9 % der Patienten begleitend nicht-psychotrope Medikamente ein. 10,7 % der Patienten nahmen mehr als ein nicht-psychotropes Medikament begleitend ein. Tabelle 4 zeigt die Häufigkeiten der nicht-psychotropen Begleitmedikation für die einzelnen Hauptgruppen nach Rote Liste, die mindestens 4-mal genannt wurden.
Tab. 3. Begleiterkrankungen nach ICD-10-Kategorien (ngesamt = 610, *identische Codes pro Patient werden einfach gezählt)
|
ICD-10-Kategorie |
Patienten |
||
|
n* |
[%] |
||
|
IX |
Krankheiten des Kreislaufsystems (I00–I99) |
98 |
16,1 |
|
IV |
Endokrine, Ernährungs- und Stoffwechselkrankheiten (E00–E90) |
52 |
8,5 |
|
XIII |
Krankheiten des Muskel-Skelett-Systems und des Bindegewebes (M00–M99) |
47 |
7,7 |
|
VI |
Krankheiten des Nervensystems (G00–G99) |
35 |
5,7 |
|
V |
Psychische und Verhaltensstörungen (F00–F99) |
22 |
3,6 |
|
XI |
Krankheiten des Verdauungssystems (K00–K93) |
22 |
3,6 |
|
XXI |
Faktoren, die den Gesundheitszustand beeinflussen und zur Inanspruchnahme des Gesundheitswesens führen (Z00–Z99) |
15 |
2,5 |
|
XVIII |
Symptome und abnorme klinische und Laborbefunde, die anderenorts nicht klassifiziert sind (R00–R99) |
9 |
1,5 |
|
X |
Krankheiten des Atmungssystems (J00–J99) |
8 |
1,3 |
|
XIX |
Verletzungen, Vergiftungen und bestimmte andere Folgen äußerer Ursachen (S00–T98) |
5 |
0,8 |
|
I |
Bestimmte infektiöse und parasitäre Krankheiten (A00–B99) |
4 |
0,7 |
|
II |
Neubildungen (C00–D48) |
4 |
0,7 |
|
VII |
Krankheiten des Auges und der Augenanhangsgebilde (H00–H59) |
4 |
0,7 |
|
VIII |
Krankheiten des Ohres und des Warzenfortsatzes (H60–H95) |
3 |
0,5 |
|
XII |
Krankheiten der Haut und der Unterhaut (L00–L99) |
2 |
0,3 |
|
XIV |
Krankheiten des Urogenitalsystems (N00–N99) |
2 |
0,3 |
|
III |
Krankheiten des Blutes und der blutbildenden Organe sowie bestimmte Störungen mit Beteiligung des Immunsystems (D50–D89) |
1 |
0,2 |
|
Andere |
24 |
3,9 |
|
Tab. 4. Begleittherapien der Patienten nach Hauptgruppe [Rote Liste] (ngesamt = 610, Präparate mit gleicher Hauptgruppe pro Patient einfach gezählt)
|
Hauptgruppe |
Patienten |
||
|
n |
[%] |
||
|
27 |
Betarezeptoren-, Calciumkanalblocker und Hemmstoffe des Renin-Angiotensin-Systems |
57 |
9,3 |
|
05 |
Analgetika/Antirheumatika |
31 |
5,1 |
|
74 |
Schilddrüsentherapeutika |
25 |
4,1 |
|
60 |
Magen-Darm-Mittel |
15 |
2,5 |
|
12 |
Antidiabetika |
13 |
2,1 |
|
17 |
Antihypertonika |
13 |
2,1 |
|
36 |
Diuretika |
13 |
2,1 |
|
15 |
Antiepileptika |
12 |
2,0 |
|
55 |
Koronarmittel |
10 |
1,6 |
|
70 |
Parkinsonmittel und andere Mittel gegen extrapyramidale Störungen |
7 |
1,1 |
|
76 |
Sexualhormone und ihre Hemmstoffe |
7 |
1,1 |
|
11 |
Antidementiva (Nootropika) |
6 |
1,0 |
|
28 |
Broncholytika/Antiasthmatika |
6 |
1,0 |
|
66 |
Neuropathiepräparate und andere neurotrope Mittel |
6 |
1,0 |
|
44 |
Gichtmittel |
5 |
0,8 |
|
31 |
Corticoide (Interna) |
4 |
0,7 |
|
53 |
Kardiaka |
4 |
0,7 |
|
58 |
Lipidsenker |
4 |
0,7 |
|
62 |
Mineralstoffpräparate |
4 |
0,7 |
|
79 |
Thrombozytenaggregationshemmer |
4 |
0,7 |
|
Sonstige (mit Häufigkeit < 4) |
25 |
4,1 |
|
|
Nicht kodierbare Begleitmedikamente |
2 |
0,3 |
|
Behandlungsdauer, Dosierung und Compliance
Zwischen Aufnahme- und Zwischenuntersuchung lagen im Mittel 36,5 ± 13,2 Tage (Median: 35 Tage), der mittlere zeitliche Abstand zwischen Aufnahme- und Abschlussuntersuchung betrug 80,6 ± 20,3 Tage (Median: 77 Tage). Das vorgeschriebene Zeitfenster wurde bei der Zwischenuntersuchung von 69,0 % und bei der Abschlussuntersuchung von 53,3 % der Patienten eingehalten.
Bei der Aufnahmeuntersuchung erhielten 83,0 % aller Patienten die Dosis von 20 mg Serital®. In 8,2 % der Fälle wurde die doppelte Dosis und in 7,7 % der Fälle wurde die halbe Dosis verabreicht. 0,7 % erhielten eine Anfangsdosis zwischen 20 und 40 mg und 0,5 % erhielten eine Anfangsdosis von > 40 mg. Die mittlere Tagesdosis betrug bei der Aufnahmeuntersuchung 21,2 ± 7,1 mg und stieg bis zur Zwischenuntersuchung auf 26,5 ± 10,2 mg und bis zur Abschlussuntersuchung auf 27,6 ± 11,4 mg an. Bei 31,6 % der Patienten ergab sich im Verlauf der Anwendungsbeobachtung eine Dosiserhöhung (bei der Mehrheit dieser Patienten [65,8 %] kam es zu einer Verdopplung der Dosis von 20 auf 40 mg), in 2,0 % der Fälle wurde die Tagesdosis reduziert. Bei 7,2 % wurde Serital® im Laufe der Anwendungsbeobachtung ganz abgesetzt. Bei 45,5 % der Patienten, die das Prüfpräparat absetzten, wurde ein unerwünschtes Ereignis als Grund für die Umstellung angegeben. Weitere Abbruchgründe sind in Tabelle 5 aufgezählt. Die Compliance war insgesamt sehr hoch: 88,8 % der Patienten nahmen zur Zwischenuntersuchung mehr als 80 % der verordneten Tabletten ein. Zur Abschlussuntersuchung stieg der Anteil der Patienten auf 92 %.
Tab. 5. Gründe für den Abbruch der Behandlung mit Serital® (*Mehrfachangaben möglich)
|
Abbruchgrund |
n |
Anteil bezogen auf Therapieabbrecher (n = 44) [%]* |
Anteil bezogen auf alle Patienten (n = 610) [%] |
|
Mangelnde Compliance |
14 |
31,8 |
2,3 |
|
Unzureichende Wirkung |
13 |
29,5 |
2,1 |
|
Interkurrente Erkrankung |
0 |
0,0 |
0,0 |
|
Unerwünschtes Ereignis |
20 |
45,5 |
3,3 |
|
Sonstiges |
7 |
15,9 |
1,1 |
Psychopathologie
Die Begründung der teilnehmenden Ärzte für eine medikamentöse Neueinstellung auf Serital® (bezogen auf die Patienten, die zu einem früheren Zeitpunkt eine pharmakologische Behandlung erhalten hatten) sind in Tabelle 6 angegeben.
Tab. 6. Gründe für eine Neueinstellung auf Serital® (Mehrfachnennungen möglich)
|
Grund |
n |
Anteil bezogen auf Patienten mit Angaben zum Grund (n = 265) [%] |
Anteil bezogen auf alle Patienten (n = 610) [%] |
|
Bessere Wirkung erhofft |
212 |
80,0 |
34,8 |
|
Bessere Verträglichkeit erhofft |
123 |
46,4 |
20,2 |
|
Therapieresistenz |
55 |
20,8 |
9,0 |
|
Geringere Kosten |
35 |
13,2 |
5,7 |
|
Sonstige |
12 |
4,5 |
2,0 |
Insgesamt zeigte sich in Wilcoxon-Tests sowohl in der HAMD (Z = –20,87; n = 597; p<0,001) als auch im Item 1 der CGI (Z = –19,74; n = 589; p<0,001) eine signifikante Verbesserung der depressiven Symptomatik zwischen Einschluss und Abschlussuntersuchung (Abb. 1). Bei 96,3 % der Patienten besserte sich der Schweregrad der Depression gemessen mit der HAMD, bei 1,8 % der Patienten blieb er unverändert, während sich bei ebenso vielen Patienten das psychopathologische Zustandsbild verschlechterte. Bei 74,8 % der Patienten ergab sich eine Abnahme des HAMD-Gesamtscores um mindestens 50 % des Ausgangswertes, wobei über die Hälfte der behandelten Patienten (65,4 %) während des Beobachtungszeitraums voll remittierte (HAMD bei Abschluss ≤ 7). Gemessen mit der CGI (Item 1) besserte sich der Schweregrad der Erkrankung bei 86,9 % der Patienten.
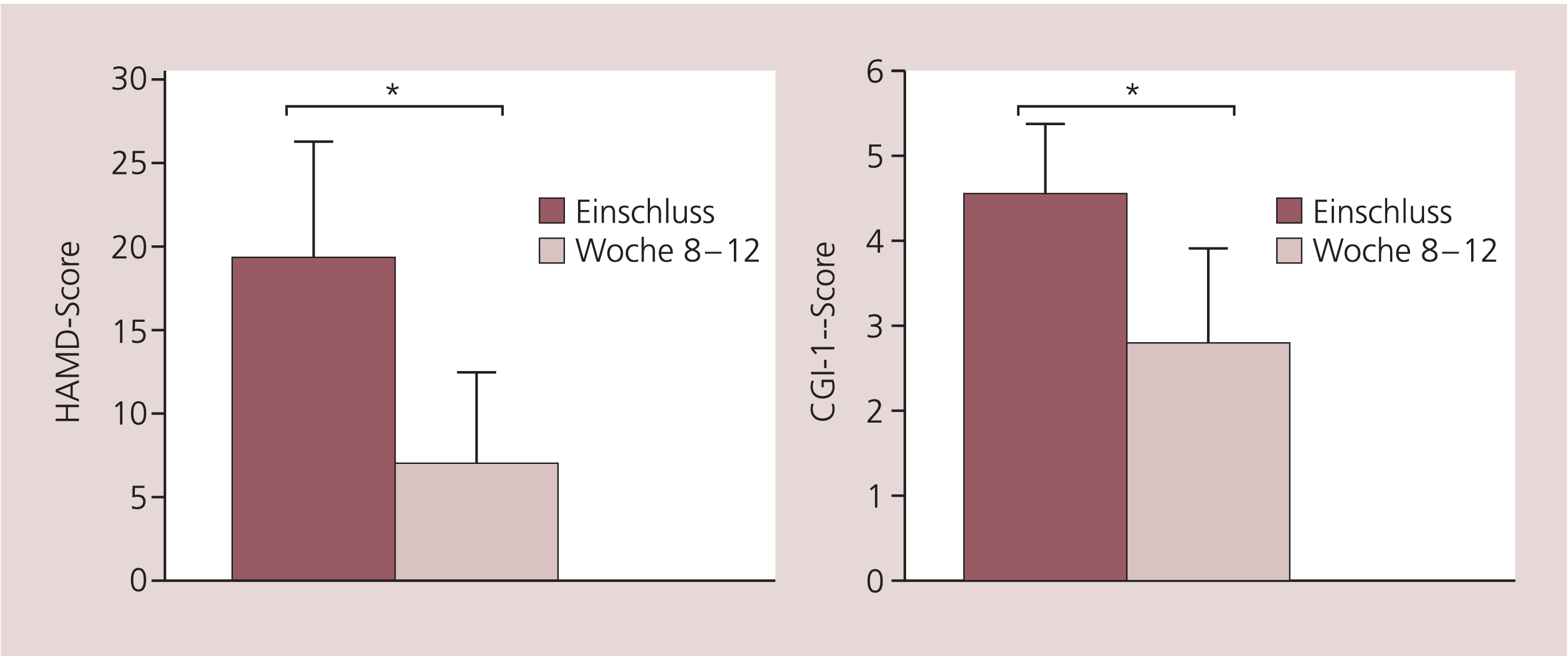
Abb. 1. Wirksamkeit von Serital®: Schwere der depressiven Erkrankung bei Einschluss und bei der Abschlussuntersuchung nach 8 bis 12 Wochen gemessen mit der Hamilton-Depressions-Skala (HAMD) und der „Clinical Global Impression Scale“ (CGI); * p<0,001
Bei 11,5 % der Patienten blieb der Schweregrad der Erkrankung unverändert und bei neun Patienten (1,5 %) verschlechterte sich der Schweregrad. Die Gesamtbeurteilung der Zustandsänderung in Item 2 der CGI in der Abschlussuntersuchung ergab ein vergleichbares Bild. Die therapeutische Wirksamkeit (CGI-Item 3) in der Abschlussuntersuchung wurde von 52,1 % der Ärzte als „sehr gut“, von 35,5 % als „mäßig“ und von 8,3 % als „gering“ beurteilt, während 4,2 % den Zustand der Patienten als „unverändert oder verschlechtert“ einstuften.
Unerwünschte Ereignisse und Verträglichkeit
Bei 47 Patienten (7,7 %) traten insgesamt 80 unerwünschte Ereignisse (UE) auf. Am häufigsten betrafen die UE den Verdauungstrakt (3,8 %) und das psychische Befinden (2,3 %). Die Häufigkeitsverteilung der UE nach Organsystem ist in Tabelle 7 aufgeführt. Bei drei UE wurde ein Kausalzusammenhang mit der Prüfsubstanz als gesichert bezeichnet, bei 36 UE als wahrscheinlich und bei weiteren 25 als möglich. Bei einem Patienten wurde der Ausgang der UE als bleibender Schaden beschrieben (Gewichtszunahme).
Tab. 7. Häufigkeitsverteilung der unerwünschten Ereignisse nach Organsystem (WHO Adverse Reaction Dictionary, ngesamt = 610, *Identische Codes pro Patient werden einfach gezählt)
|
Organsystem |
Patienten |
||
|
n* |
[%] |
||
|
0600 |
Verdauungstrakt |
23 |
3,8 |
|
0500 |
Psychiatrische Störungen |
14 |
2,3 |
|
0410 |
Zentrales und peripheres Nervensystem |
10 |
1,6 |
|
1810 |
Generalisierte Störungen |
5 |
0,8 |
|
0100 |
Haut und Hautanhangsgebilde |
4 |
0,7 |
|
0800 |
Stoffwechselstörungen |
4 |
0,7 |
|
1010 |
Herz-Kreislauf-System, Allgemein |
2 |
0,3 |
|
1030 |
Herzrhythmusstörungen |
1 |
0,2 |
|
Nicht kodierbar |
1 |
0,2 |
|
Es traten bei drei Patienten insgesamt vier schwere unerwünschte Ereignisse (SUE) auf: Kreislaufversagen und Schwindel, aggressive Reaktion, Zustand verschlechtert. Diese Nebenwirkungen remittierten vollständig. Im Urteil der untersuchenden Ärzte bestand kein Zusammenhang mit der Prüfsubstanz.
Zur Zwischenuntersuchung beurteilten 88,2 % der Patienten die Verträglichkeit von Serital® als gut oder sehr gut, zur Abschlussuntersuchung stieg der Anteil auf 91,6 % (Tab. 8).
Tab. 8. Verträglichkeitsbeurteilung durch den Patienten
|
Verträglichkeit |
Zwischenuntersuchung |
Abschlussuntersuchung |
||
|
n |
[%] |
n |
[%] |
|
|
Sehr gut |
208 |
34,1 |
299 |
49,0 |
|
Gut |
330 |
54,1 |
260 |
42,6 |
|
Mäßig |
38 |
6,2 |
25 |
4,1 |
|
Schlecht |
17 |
2,8 |
14 |
2,3 |
|
Keine Angaben |
17 |
2,8 |
12 |
2,0 |
|
Gesamt |
610 |
100,0 |
610 |
100,0 |
Korrelationen
Das Alter der Patienten war signifikant negativ mit der Verminderung des CGI-Scores (Item 1) (r = –0,16; p<0,0001) wie auch mit der Verminderung des HAMD-Scores (r = –0,11; p<0,01) über den Behandlungszeitraum korreliert. Das bedeutet, dass jüngere Patienten besser auf die Behandlung ansprachen. Bei den weiteren Subgruppenanalysen wurde daher – soweit möglich – das Alter als Kovariable mit einbezogen. Das Geschlecht zeigte in keinem der Wirksamkeitsmaße einen Einfluss.
Die Wirksamkeits- und Verträglichkeitsbeurteilung durch die Patienten bei der Abschlussuntersuchung war signifikant und zum Teil sehr hoch mit der Wirksamkeits- und Verträglichkeitsbeurteilung durch die Ärzte korreliert (vs. CGI-Item 2, 3.1 und 3.2: r = 0,28–0,76; p<0,001; vs. CGI-Item 1 und HAMD-Reduktion: r = –0,31 bis –0,69; p<0,001).
Einflussfaktoren auf den Behandlungsverlauf
Die ANOVA mit Messwiederholung von CGI (Item 1) und HAMD zeigte, dass Patienten mit einem Rezidiv gegenüber Patienten mit einer Erstdiagnose einer Depression signifikant schwerer erkrankt waren (Zwischensubjekteffekte: CGI: F1,585 = 13,8; p<0,001; HAMD: F1,593 = 13,5; p<0,001). Ersterkrankte profitierten aber über den Behandlungszeitraum im Item 1 (Interaktion Zeit x Gruppe: F1,585 = 5,24; p<0,05) sowie zur Abschlussuntersuchung im Item 2 (F1,571 = 6,83; p<0,01) und 3.1 (F1,574 = 10,8; p<0,001) des CGI auch signifikant stärker von der Behandlung. Zudem war die Responserate bei den Ersterkrankten (80 % vs. 72 %) signifikant höher (χ2 = 5,11; p<0,05) und die ersterkrankten Patienten beurteilten auch die Wirksamkeit bei der Zwischen- (F1,587 = 5,05; p<0,05) und Abschlussuntersuchung (F1,594 = 5,15; p<0,05) signifikant besser.
Patienten mit einer psychotropen Begleitmedikation waren ebenfalls signifikant schwerer erkrankt (Zwischensubjekteffekte: CGI-Item 1: F1,585 = 26,0; p<0,001; HAMD: F1,593 = 22,6; p<0,001) und zeigten im HAMD eine signifikant stärkere Besserung (Interaktion Zeit x Gruppe: F1,593 = 13,9; p<0,001) als Patienten ohne psychotrope Begleitmedikation.
Ebenso erwiesen sich Patienten mit einer Begleiterkrankung insgesamt als schwerer depressiv (Zwischensubjekteffekte: CGI-Item 1: F1,585 = 5,22; p<0,05; HAMD: F1,593 = 5,40; p<0,05) und zeigten in der ANOVA mit Messwiederholung nach Kontrolle für das Alter keine bessere oder schlechtere Wirksamkeit der Prüfsubstanz. Die signifikant besseren Ansprech- (81 % vs. 69 %; χ2 = 11,2; p<0,001) und Remissionsraten (73 % vs. 57 %; χ2 = 14,7; p<0,001) der Patienten ohne Begleiterkrankung könnten daher auf den signifikanten Altersunterschied beider Gruppen (t607 = 8,82; p<0,001) zurückzuführen sein. Allerdings beurteilten die Patienten ohne Begleiterkrankung die Wirksamkeit bei der Zwischen- (F1,587 = 5,07; p<0,05) und Abschlussuntersuchung (F1,594 = 11,7; p<0,001) sowie die Verträglichkeit bei der Abschlussuntersuchung (F1,594 = 7,51; p<0,01) auch nach Kontrolle für das Alter signifikant besser. Die Beurteilung der Wirksamkeit anhand der Items 2 (F1,571 = 6,58; p<0,01) und 3.1 (F1,574 = 3,88; p<0,05) sowie der unerwünschten Wirkungen anhand des Items 3.2 (F1,569 = 3,95; p<0,05) des CGI durch die Ärzte fiel bei der Abschlussuntersuchung auch nach Kontrolle für das Alter signifikant besser aus für die Gruppe der Patienten ohne Begleiterkrankung.
Diskussion
Bei der vorliegenden Untersuchung handelt es sich um die erste Anwendungsbeobachtung mit Citalopram bei depressiven Patienten in der Facharztpraxis. Die Anwendungsbeobachtung wurde mit dem Generikum Serital® durchgeführt.
Generika sehen sich mitunter dem Vorwurf ausgesetzt, weniger effektiv oder weniger sicher als das jeweilige Originalpräparat zu sein [8]. Insbesondere die Umstellung von einem Originalpräparat auf ein Generikum wird kritisch diskutiert und es wurde bereits, beispielsweise bei bestimmten Antikonvulsiva in der Behandlung der Epilepsie, von einer Umstellung auf ein Generikum abgeraten [27]. Vor allem der Einsatz verschiedener Generika in der Therapie eines Patienten kann zu erheblichen klinischen Problemen führen [8]. Der Gesetzgeber verlangt vor der Zulassung von Generika unter anderem den Nachweis der Bioäquivalenz (§21, §105 AMG). Ein Präparat wird dann als bioäquivalent angesehen, wenn die Plasmakonzentration des Wirkstoffs im Vergleich zum Originalprodukt innerhalb bestimmter, international akzeptierter Grenzen verläuft. Der klinische Nachweis der vergleichbaren Wirksamkeit und Verträglichkeit unter Praxisbedingungen muss für die Zulassung eines Generikums nicht erbracht werden, wenn die Bioäquivalenz so in vitro nachgewiesen werden konnte (Bekanntmachung über die Zulassung nach §21 Arzneimittelgesetz [Bioverfügbarkeit/Bioäquivalenz] vom 18. Dezember 2002; BAnz. Nr. 58 vom 25.03.2003, S. 5296). Hilfs- und Füllstoffe eines Generikums müssen jedoch nicht mit denen des Originalpräparats identisch sein. Diese Problematik betrifft jedoch nicht allein Generika, sondern auch so genannte „Parallelimporte“ [5].
In der vorliegenden Arbeit konnte für das Citalopram-Generikum Serital® eine geringe Rate an UAW und eine gute Wirksamkeit in der Behandlung der Depression auch unter Alltagsbedingungen – bei einer hohen Rate internistischer Begleiterkrankungen (37,7 % der Patienten) sowie Patienten mit internistischer (28,9 %) und psychiatrischer (33,3 %) Komedikation – gezeigt werden. Frühere klinische und kontrollierte Studien mit Citalopram in der Behandlung der Depression zeigten eine vergleichbar gute Wirksamkeit und Verträglichkeit [17, 25]. Die Studie von Zimmerman et al. [28] zur mangelnden Repräsentativität von Patientenpopulationen in klinischen Studien bestätigt die Notwendigkeit, die Wirksamkeit und Sicherheit einer Substanz auch unter Alltagsbedingungen, wie sie in einer AWB eher erreicht werden können, zu überprüfen. Die vorliegende AWB bestätigte damit einerseits auch unter Alltagsbedingungen die geringe Neigung des Citaloprams zu Arzneimittelinteraktionen. Andererseits deuten die Daten darauf hin, dass sich das Generikum in seiner klinischen Wirksamkeit und Verträglichkeit nicht vom Originalpräparat unterscheidet [22]. Darüber hinaus zeigte sich, dass insbesondere jüngere Patienten sowie – auch nach Kontrolle für diesen Alterseffekt – Ersterkrankte, Personen ohne Begleiterkrankungen und Patienten mit psychotroper Begleitmedikation signifikant besser auf die Behandlung mit Citalopram ansprachen.
Auffällig ist auch die übereinstimmende Fremd- und Selbstbeurteilung durch Ärzte und Patienten. Das Urteil der behandelnden Ärzte abgebildet in der Reduktion des CGI (Item 1) und der HAMD über den Behandlungszeitraum sowie das Wirksamkeits- und Sicherheitsurteil im CGI (Item 2, 3.1 und 3.2) war am Ende der Studie mit der subjektiven Beurteilung der Wirksamkeit und Verträglichkeit durch den Patienten signifikant korreliert. Dieser Befund tritt der häufig geäußerten Kritik entgegen, dass die teilnehmenden Ärzte in AWB ausschließlich ihre eigene Arbeit beurteilen und die Ergebnisse dadurch verzerren würden [20].
Das hier geprüfte Citalopram-Generikum zeigte in der Behandlung der Depression auch unter Alltagsbedingungen eine vergleichbar gute Wirksamkeit und eine vergleichbare gute Verträglichkeit wie das Originalpräparat in kontrollierten klinischen Studien. Häufig geäußerte Befürchtungen zur Unterlegenheit von Generika werden zumindest in diesem Fall durch die vorliegenden Daten nicht gestützt.
Literatur
1. Baumann P, Larsen F. The pharmacokinetics of citalopram. Rev Contemp Pharmacother 1995;6:287–95.
2. Beneke M, Rasmus W. „Clinical global impressions” (EDCEU): Some critical comments. Pharmacopsychiatry 1992;25:172–6.
3. Boerner RJ, Möller HJ. The importance of new antidepressants in the treatment of anxiety/depressive disorders. Pharmacopsychiatry 1999;32:119–26.
4. Brøsen K, Naranjo CA. Review of pharmacokinetic and pharmacodynamic interaction studies with citalopram. Eur Neuropsychopharmacol 2001;11:275–83.
5. Buchberger D. Generika – Bioäquivalenz und Bioverfügbarkeit am Beispiel Isosorbiddinitrat. Pharmazeutische Rundschau 1998;2:12.
6. Buchberger R, Wagner W. Fluvoxamine: safety profile in extensive post-marketing surveillance. Pharmacopsychiatry 2002;35:101–8.
7. Dahlke F, Lohaus A, Gutzmann H. Reliability and clinical concepts underlying global judgments in dementia: implications for clinical research. Psychopharmacol Bull 1992;28:425–32.
8. Drewe J. Kostenersparnis, eine trügerische Wahrheit? As de Coeur News 2002;15:10–1.
9. El-Armouche A, Zolk O, Eschenhagen T. Citalopram. Dtsch Med Wochenschr 2003;128:2253–6.
10. Gex-Fabry M, Balant LP. Therapeutic drug monitoring databases for postmarketing surveillance of drug-drug interactions. Drug Saf 2001;24:947–59.
11. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960;23:56–62.
12. Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 1967;6:278–96.
13. Hedlund JL, Vieweg BW. The Hamilton rating scale for depression: a comprehensive review. J Operational Psychiatry 1979;10:149–65.
14. Hennessy S. Postmarketing drug surveillance: an epidemiologic approach. Clin Ther 1998;20(Suppl):C32–9.
15. Hyttel J. Citalopram – pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Prog Neuropsychopharmacol Biol Psychiatry 1982;6:277–95.
16. Hyttel J, Arndt J, Sanchez C. The pharmacology of citalopram. Rev Contemp Pharmacother 1995;6:271–85.
17. Keller MB. Citalopram therapy for depression: a review of 10 years of European experience and data from U.S. clinical trials. J Clin Psychiatry 2000;61:896–908.
18. Laux G, Baier D. Quality-monitoring of psychotropic drug therapy in post-marketing surveillance. Results of a drug utilization observation (DUO) study on moclobemide. Pharmacopsychiatry 1997;30:21–7.
19. Linden M. Phase IV research and drug utilization observation studies. Pharmacopsychiatry 1997;30:1–3.
20. Linden M, Baier D, Gothe H, Kohnen R. What happens to patients after the end of a clinical trial? Systematic follow-up observational study of an open moclobemide trial in major depression. Pharmacopsychiatry 1997;30:35–43.
21. National Institute of Mental Health (NIMH). 028 CGI. Clinical global impressions. In: Guy W (Hrsg.). ECDEU assessment manual for psychopharmacology. Revised edition. Rockville Maryland: NIMH, 1976:217–22.
22. Parker NG, Brown CS. Citalopram in the treatment of depression. Ann Pharmacother 2000;34:761–71.
23. Pato MT. Beyond depression: citalopram for obsessive-compulsive disorder. Int Clin Psychopharmacol 1999;14(Suppl 2):19–26.
24. Pollock BG. Citalopram: a comprehensive review. Expert Opin Pharmacother 2001;2:681–98.
25. Tan JY, Levin GM. Citalopram in the treatment of depression and other potential uses in psychiatry. Pharmacotherapy 1999;19:675–89.
26. Thase ME. How should efficacy be evaluated in randomized clinical trials of treatments for depression? J Clin Psychiatry 1999;60(Suppl 4):23–31.
27. Wolf P. Gefahrvoller Wechsel. Ärztliche Praxis 1999;11:14–5.
28. Zimmerman M, Mattia JI, Posternak MA. Are subjects in pharmacological treatment trials of depression representative of patients in routine clinical practice? Am J Psychiatry 2002;159:469–73.
Priv.-Doz. Dr. med. Kai-Uwe Kühn, Dr. rer.nat. Boris B. Quednow, Prof. Dr. med. Wolfgang Maier, Klinik und Poliklinik für Psychiatrie und Psychotherapie, Universitätsklinikum Bonn, Sigmund-Freud-Straße 25, 53105 Bonn, E-Mail: k.u.kuehn@uni-bonn.de
Dr. med. Michael Riedel, Ludwig-Maximilians-Universität München, Psychiatrische Klinik und Poliklinik, Nussbaumstr. 7, 80336 München
Dr. med. Olaf Krampe, Temmler Pharma & Co. KG, Temmlerstr. 2, 35039 Marburg
Safety and efficacy of citalopram in the treatment of depressive disorders
Background: The objective of this post-marketing surveillance study was to document the efficacy and tolerability of the selective serotonin reuptake inhibitor citalopram in patients with depression under routine clinical conditions. Post-marketing surveillance studies permit to assess a substance in a much larger patient population and under real-life conditions than is possible in clinical trials and are therefore a useful tool to detect drug interactions and rare adverse drug reactions (ADRs).
Patients and methods: 610 patients with an ICD-10 diagnosis of depression were treated with the generic citalopram for up to 12 weeks by 206 psychiatrists or neurologists. Outcome measures included the 17-item Hamilton Depression Scale (HAMD); the Clinical Global Impression Scale (CGI) and efficacy and safety/adverse drug reaction assessments.
Results: Overall, HAMD and CGI severity score decreased significantly. About 75 % of the patients showed therapeutic response (HAMD reduction >50 %) and 65 % showed remission (HAMD at end of study ≤ 7). 7.7 % of the patients had ADRs; only 0.5 % had serious ADRs. Gender did not influence the efficacy of the citalopram treatment, but younger persons, patients with a first episode of depression, patients without co-morbidities as well as patients with psychotropic co-medication had a significantly greater benefit of the citalopram treatment. The efficacy judgements of specialists and patients were highly correlated.
Conclusion: Even under real-life conditions, the generic citalopram showed a comparable efficacy and a comparable safety as the original product citalopram investigated in previous clinical and placebo-controlled studies.
Keywords: Citalopram, depression, post-marketing surveillance, safety, generic medicaments
Psychopharmakotherapie 2005; 12(03)