Felix Geser und Gregor K. Wenning, Innsbruck
Multisystematrophie (MSA)
Klinik und Differenzialdiagnose
Die Multisystematrophie (MSA) ist eine degenerative Erkrankung des zentralen und autonomen Nervensystems. Die MSA ist als „Alpha-Synukleinopathie“ durch eine Alpha-Synuklein-Aggregation in der Oligodendroglia und in Neuronen charakterisiert. Die klinischen Hauptsymptome sind autonomes Versagen, Parkinson-Syndrom, zerebelläre Ataxie und Pyramidenbahnzeichen. Diese können in verschiedener Kombination auftreten.
Klinisch kann man zwei motorische Erscheinungsformen unterscheiden: Bei 80 % der Patienten ist die Parkinson-Symptomatik vorherrschend (MSA-P-Subtyp), während bei 20 % der Patienten eine zerebelläre Ataxie dominiert (MSA-C-Subtyp) [1]. Neuropathologisch ist die Parkinson-Symptomatik von MSA-P-Patienten mit striatonigraler Degeneration (SND) assoziiert. Der bei den MSA-C-Patienten vorherrschenden Ataxie liegt dagegen eine olivopontozerebelläre Atrophie (OPCA) zugrunde.
Das Parkinson-Syndrom bei MSA-Patienten ist durch progrediente Akinese und Rigidität gekennzeichnet. Irregulärer, zum Teil ruckartiger posturaler Tremor und – weniger häufig – Ruhetremor können noch dazukommen. Die Patienten zeigen oft eine orofaziale Dystonie (Abb. 1) assoziiert mit einer prominenten Dysarthrie, die sowohl extrapyramidale, zerebelläre und dystone Komponenten aufweisen kann. Die posturale Stabilität ist von Anfang an beeinträchtigt, jedoch sind wiederholte Stürze bei Krankheitsausbruch im Gegensatz zur progressiven supranukleären Paralyse (PSP) eher ungewöhnlich. Aufgrund einer Reihe von überlappenden Symptomen wie Tremor, asymmetrischer Akinese und Rigidität kann die Differenzialdiagnose von MSA-P und Parkinson-Krankheit in den frühen Stadien besonders schwierig sein. Darüber hinaus kann eine Levodopa-induzierte Besserung des Parkinson-Syndroms bei 30 % der MSA-P-Patienten beobachtet werden. In den meisten Fällen ist jedoch die Besserung nur vorübergehend, das heißt, auf lange Sicht sprechen 90 % der MSA-P-Patienten auf Levodopa nicht an. Levodopa-induzierte Dyskinesien der orofazialen Muskulatur und der Halsmuskulatur kommen bei 50 % der MSA-P-Patienten – manchmal ohne Besserung der Motorik – vor [2]. In den meisten Fällen entsteht innerhalb von fünf Jahren nach Krankheitsbeginn das Vollbild der MSA-P, so dass die klinische Diagnose im Rahmen von Verlaufskontrollen möglich wird [3].

Abb. 1. Eine MSA-P-Patientin mit Hypomimie, asymmetrischer orofazialer Dystonie (v. a. links) und zervikaler Dystonie des Platysmas [80]
Bei der MSA-C treten sowohl Gangataxie, Gliedmaßenataxie und Dysarthrie als auch zerebelläre Okulomotorik-Störungen auf. Patienten mit MSA-C entwickeln zusätzliche nichtzerebelläre Symptome. Bevor es jedoch dazu kommt, sind sie von Patienten mit idiopathischer, spät beginnender zerebellärer Ataxie nicht zu unterscheiden. Letztere Erkrankung ist bei vielen Patienten klinisch auf eine zerebelläre Symptomatik und pathologisch auf die Degeneration von Zerebellum und Oliven beschränkt.
Neben dem mangelnden Ansprechen auf Levodopa und dem zusätzlichen Vorhandensein von Pyramidenbahn- und zerebellären Zeichen (Ataxie) oder autonomer Dysfunktion als Kardinalsymptome, erheben oder verstärken eine Reihe von bestimmten anderen Symptomen den Verdacht auf eine MSA. Kürzlich wurden diese Warnsymptome der MSA, die so genannten „Red Flags“, für den Einsatz in der täglichen klinischen Praxis und prospektiven klinisch-pathologischen Studien operational definiert (Tab. 1) [4]. Die Sensitivität dieser Warnzeichen ist jedoch nicht immer hoch, so dass sie in den gängigen Diagnosekriterien nur bedingt berücksichtigt wurden. Jedoch ist die Häufigkeit der „Red Flags“ und deren diagnostische Wertigkeit in früher MSA nie prospektiv untersucht worden. Deshalb führt die Europäische MSA-Studiengruppe (EMSA-SG) eine „Natural History“-Studie durch, die unter anderem ein serielles „Red Flag“-Screening zum Inhalt hat. Vorläufige Querschnittsanalysen zeigen, dass axiale Deformität (wie beispielsweise ein disproportionierter Antecollis, Abb. 2), abnorme Atmung, atypische Dysarthrie und „Jerky Tremor“ die häufigsten dieser Warnzeichen bei der frühen MSA-P sind [5]. Diese sind daher besonders geeignet, die gegenwärtig existierenden klinischen Diagnosekriterien zu optimieren.
Tab. 1. „Red Flags“: Warnzeichen der MSA
|
Motorische „Red Flags“ |
|
|
Orofaziale Dystonien/Dyskinesien |
Atypische spontane oder Levodopa-induzierte Dystonien/Dyskinesien vorwiegend im Bereich der orofazialen Muskulatur, die gelegentlich an Risus sardonicus bei zephalem Tetanus erinnern |
|
Pisa-Syndrom |
Subakute axiale Dystonie mit schwerer tonischer Seitwärts-Neigung des Rumpfes, Nacken bzw. Kopfes |
|
Disproportionierter Antecollis |
Das Kinn ist auf der Brust, der Nacken kann nur mit Schwierigkeit, passiv und gewaltsam, zu der normalen Position gestreckt werden. Im Gegensatz zu dieser schweren chronischen Nackenbeugung ist die Beugung andersweitig nur geringfügig ausgeprägt |
|
„Jerky Tremor“ |
Irregulärer myoklonischer (jerky) posturaler oder Aktionstremor der Hände und/oder Finger |
|
Dysarthrie |
Atypische, zitternde, irreguläre, ausgeprägt hypophone Dysarthrie oft mit hoher Stimme, die sich im Vergleich zur Parkinson-Erkrankung früher und schwerwiegender entwickelt sowie meist mit Dysphagie assoziiert ist |
|
Nicht-motorische Red Flags |
|
|
Abnorme Atmung |
Nokturnaler bzw. diurnaler inspiratorischer Stridor (rauhe, gepresste, hohe inspiratorische Geräusche); unwillkürliche, tiefe inspiratorische Seufzer/Atemzüge; Schlaf-Apnoe (Arrest in der Atmung für > 10 s); exzessives Schnarchen (Zunahme vom prämorbiden Niveau bzw. neu auftretend) |
|
REM-Schlafstörungen |
Intermittierender Verlust der Muskel-Atonie und Auftreten von kunstvoller motorischer Aktivität (Schlagen mit Armen im Schlaf oft mit Reden/Schreien) assoziiert mit Träumen |
|
Kalte Hände/Füße |
Kälte und Farbänderungen (purpur/blau) der Extremitäten – die nicht auf Medikamente zurückzuführen sind – mit Erblassen bei Druck und schlechtem zirkulatorischem Return |
|
Raynaud-Phänomen |
Schmerzhafte „weiße Finger“, die von Ergot-haltigen Präparaten hervorgerufen werden dürften |
|
Emotionale Inkontinenz |
Unangemessenes Weinen (Weinen ohne Traurigkeit) oder unangemessenes Lachen (Lachen ohne Glücklichsein) |

Abb. 2. Disproportionierter Antecollis bei einer MSA-P-Patientin [81]
Die urogenitale Dysfunktion stellt eine wesentliche Manifestation der Dysautonomie bei MSA-Patienten dar. Bei Männern besteht nahezu ausnahmslos frühe Impotenz (erektile Dysfunktion). Harninkontinenz (71 %) oder Harnretention (30 %) bestehen oft schon im frühen Verlauf der Erkrankung [1]. Störungen der Miktion sind die Folge von Veränderungen in der komplexen peripheren und zentralen Innervation der Blase [6]. Sie treten bei MSA-Patienten häufiger, früher und stärker ausgeprägt als bei der Parkinson-Krankheit auf. Harnretention kann aber auch durch eine benigne Prostatahypertrophie oder bei Frauen durch perineale Schlaffheit infolge einer komplizierten Geburt oder Gebärmuttersenkung verursacht oder verstärkt werden.
Im Gegensatz dazu kommt Obstipation bei der Parkinson-Krankheit und bei MSA gleichermaßen vor.
Eine symptomatische orthostatische Hypotonie ist in 68 % der Fälle vorhanden. Sie verursacht jedoch nur bei 15 % der MSA-Patienten wiederholte Synkopen [1]. Levodopa oder Dopamin-Agonisten können die orthostatische Hypotonie provozieren oder verschlechtern.
Heute werden weitgehend Konsensuskriterien für die klinische Diagnose der MSA verwendet [7]. Diese MSA-Kriterien spezifizieren drei diagnostische Kategorien mit steigender Sicherheit: möglich, wahrscheinlich oder sicher. Die Diagnosen der möglichen und wahrscheinlichen MSA beruhen auf dem Vorhandensein klinischer Symptome, die in Tabelle 2 aufgelistet sind. Zusätzlich müssen noch die Ausschlusskriterien berücksichtigt werden (Tab. 3). Eine sichere Diagnose kann nur neuropathologisch gestellt werden. Sie erfordert den Nachweis typischer Läsionsmuster und von oligodendroglialen Alpha-Synuklein-haltigen zytoplasmatischen Einschlusskörpern.
Tab. 2. Richtlinien für die klinische Diagnose MSA [mod. nach 7]
|
A. Nomenklatur, klinische Domänen, Kriterien und Symptome |
||
|
Domäne |
Kriterium |
Symptom |
|
Autonome Dysfunktion und Blasenentleerungsstörungen |
Orthostatischer Blutdruckabfall (≥ 30 mmHg systolisch oder ≥ 15 mmHg diastolisch |
Orthostatische Hypotonie (≥20 mmHg systolisch oder ≥10 mmHg diastolisch) |
|
Parkinson-Syndrom |
Bradykinese plus |
Bradykinese (progrediente Verminderung der Geschwindigkeit und Amplitude repetitiver Willkürbewegungen) |
|
Zerebelläre Dysfunktion |
Gangataxie plus |
Gangataxie (breitbasig, irreguläre Schrittlänge) |
|
Pyramidenbahnläsion |
Es wird kein diagnostisches Kriterium definiert |
Babinski-Zeichen mit Hyperreflexie |
|
B. Diagnostische Kategorien der MSA |
|
|
Mögliche MSA-P |
Kriterium für ein Parkinson-Syndrom plus zwei weitere Symptome aus separaten Domänen. Mangelndes Ansprechen auf Levodopa zählt bereits als ein Symptom, daher ist nur noch ein zusätzliches Symptom erforderlich. |
|
Mögliche MSA-C |
Kriterium für zerebelläre Dysfunktion plus zwei weitere Symptome aus separaten Domänen |
|
Wahrscheinliche MSA-P |
Kriterium für autonome Dysfunktion/Blasenentleerungsstörungen plus Levodopa-refraktäres Parkinson-Syndrom |
|
Wahrscheinliche MSA-C |
Kriterium für autonome Dysfunktion/Blasenentleerungsstörungen plus zerebelläre Dysfunktion |
|
Gesicherte MSA |
Histopathologisch hohe Dichte an Alpha-Synuklein-positiven glialen zytoplasmatischen Einschlüssen (glial cytoplasmic inclusions, GCIs) sowie Degeneration der nigrostriatalen (SND) und olivopontozerebellären (OPCA) Projektionen |
Tab. 3. Ausschlusskriterien für die Diagnose von MSA [nach 7]
|
Anamnestisch |
|
Beginn vor dem 30. Lebensjahr, positive Familienanamnese, sekundäre Ursachen, spontane Halluzinose |
|
Neurologische Untersuchung |
|
DSM-IV-Kriterien für Demenz, Verlangsamung der vertikalen Sakkaden oder vertikale supranukleäre Blickparese, fokale kortikale Störungen wie Aphasie, „Alien-Limb“-Phänomen und parietale Dysfunktion |
|
Labor |
|
Metabolische, molekulargenetische und bildgebende Hinweise für eine andere Ursache der Krankheitssymptome, die in Tab. 2 angeführt sind |
Die klinische Diagnose MSA basiert hauptsächlich auf der Anamnese und der sorgfältigen neurologischen Untersuchung. Apparative zusatzdiagnostische Maßnahmen wie beispielsweise autonome Funktionstests, Sphinkter-EMG oder bildgebende Verfahren können laut der Konsenskonferenz über die Diagnose der MSA dazu verwendet werden, die Diagnose zu unterstützen oder andere Erkrankungen auszuschließen [7].
Bei eindeutigen klinischen Symptomen eines Morbus Parkinson und Ansprechen auf Dopamimetika wird keine Bildgebung empfohlen. Bestehen Zweifel an der Diagnose eines Morbus Parkinson, sollte ein 1,5-Tesla-Schädel-MRT mit Beurteilung durch einen in der Basalganglien-Diagnostik erfahrenen Neuroradiologen angestrebt werden. Bei der MSA findet sich gehäuft auf T2-gewichteten Aufnahmen eine Hypointensität des Putamens, vergesellschaftet mit einem hyperintensen Randsaum lateral des Putamens (Abb. 3) [8]. Weiterhin sind bei MSA-Patienten mit zerebellärer Symptomatik häufig neben einer infratentoriellen Atrophie pontozerebelläre Hyperintensitäten (so genannte Kreuzzeichen) zu beobachten [9].
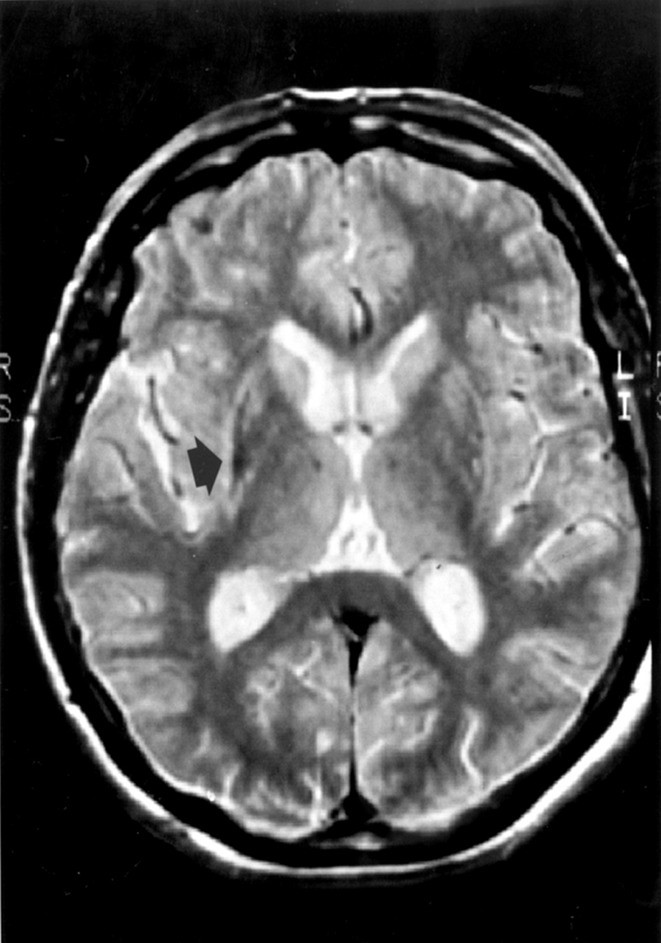
Abb. 3. Putaminale Atrophie, hyperintenser Randsaum (Pfeil) und putaminale Hypointensität im Vergleich zum Globus pallidus in einer T2-gewichteten Aufnahme (1,5 T) bei einem MSA-Patienten [9]
Diffusion-weighted Imaging (DWI) ist in der Lage, MSA-P-Patienten vom Morbus Parkinson und von gesunden Kontrollpersonen aufgrund von – bei MSA-P-Patienten – erhöhten putaminalen rADC(regional apparent diffusion coefficients)-Werten zu unterscheiden [10]. Schulz et al. [11] fanden eine signifikante Reduktion im durchschnittlichen striatalen und Hirnstamm-Volumen bei Patienten mit MSA-P, MSA-C und PSP, während Patienten mit MSA-C und MSA-P auch eine Reduktion des zerebellären Volumens aufwiesen. Unlängst bestätigte Voxel-basierte Morphometrie frühere Region-of-Interest (ROI)-basierte Volumetrie-Studien [11], indem Basalganglien- und infratentorialer Volumenverlust bei MSA-P-Patienten gezeigt werden konnte [12]. MR-basierte Volumetrie kann jedoch nicht für die routinemäßige diagnostische Abklärung von einzelnen Patienten herangezogen werden. Auch MR-Spektroskopie kommt in erster Linie im Rahmen wissenschaftlicher Protokolle zum Einsatz.
Präsynaptische Radioliganden zur Darstellung der dopaminergen Projektion sind zur Differenzierung degenerativer Parkinson-Syndrome nicht geeignet, werden aber in der Routinediagnostik zur Abgrenzung sekundärer Parkinson-Syndrome (Neuroleptika-Parkinsonoid, Lower-Body-Parkinson-Syndrom) sowie zur Differenzialdiagnose unklarer Tremores empfohlen. Postsynaptische Radioliganden zur Darstellung der Dopamin-D2-Rezeptoren wie Iodobenzamid (IBZM, SPECT) oder Racloprid (PET) tragen dagegen zur Differenzierung des Morbus Parkinson (erhaltener Rezeptorstatus) von atypischen Parkinson-Syndromen (verminderter Rezeptorstatus) bei und sollten entsprechend der Expertise des lokalen Zentrums bei Vorhandensein kameraspezifischer Normaldaten für den jeweiligen Liganden in der Routinediagnostik eingesetzt werden. Eine Differenzierung der verschiedenen atypischen Parkinson-Syndrome ist jedoch nicht möglich.
Die szintigraphische Darstellung der postganglionären kardialen Noradrenalin-Aufnahme mit dem Radioliganden Metaiodobenzylguanidin (MIBG) wird in den nächsten Jahren in der Differenzialdiagnose des Morbus Parkinson (reduzierte kardiale Aufnahme) von der MSA bzw. PSP (normale kardiale Aufnahme) an Bedeutung gewinnen.
Natürlicher Krankheitsverlauf
Die in jüngster Zeit durchgeführte epidemiologische Untersuchungen ergaben eine Prävalenzrate von 4,4/100000 [13] und eine Inzidenz von 3/100000/Jahr [14]. Die Krankheit betrifft sowohl Männer als auch Frauen, beginnt gewöhnlich im 6. Lebensjahrzehnt und schreitet unaufhaltsam bis zum Tod nach durchschnittlich neun Jahren fort [1]. Es gibt in manchen Fällen eine beträchtliche Variation in der Krankheitsprogression mit Überlebenszeiten von mehr als 15 Jahren. Wichtig ist, dass beide motorischen Erscheinungsformen von MSA mit ähnlichen Überlebenszeiten assoziiert sind [15], obgleich die Krankheitsprogression bei MSA-P rascher verläuft als bei MSA-C [16]. Die meisten MSA-Patienten sterben letztlich an Bronchopneumonie.
Therapieprinzipien
Autonomes Versagen
Leider steht keine kausale Therapie der autonomen Dysfunktion zur Verfügung. Deswegen orientiert sich das therapeutische Vorgehen an den klinischen Symptomen und ihrem Einfluss auf die Lebensqualität. Aufgrund des progredienten Krankheitsverlaufs ist es unbedingt notwendig, in regelmäßigen Abständen die Behandlung neu zu evaluieren und die Maßnahmen den klinischen Erfordernissen anzupassen.
Das Konzept der symptomatischen Therapie der orthostatischen Hypotonie beruht auf der Erhöhung des intravasalen Volumens und der Reduktion des venösen Pooling unter Orthostase. Selektion und Kombination der folgenden Optionen der Therapie hängen vom Grad der Symptome und der Anwendbarkeit dieser therapeutischen Möglichkeiten bei den einzelnen Patienten ab – und nicht vom Ausmaß des Blutdruckabfalls während der Kipptischuntersuchung.
Nichtpharmakologische Optionen beinhalten genügende Flüssigkeitsaufnahme, salzreiche Diät, häufige aber kleine Mahlzeiten tagsüber (um die postprandiale Hypotonie durch Verteilung der gesamten Kohlenhydrataufnahme zu vermindern) und Stützmieder. Das nächtliche Hochstellen des Bettkopfendes reduziert nicht nur den zerebralen Perfusionsdruck, sondern erhöht auch das intravasale Volumen innerhalb einer Woche bis zu einem Liter. Dies ist besonders hilfreich, um die Hypotonie am frühen Morgen zu verbessern. Diese Behandlung ist insbesondere erfolgreich in Kombination mit Fludrocortison, das die Natriumretention fördert.
Die nächsten Arzneimittel, die verwendet werden können, sind die Sympathomimetika. Ephedrin, mit direkten und indirekten Effekten, ist oft bei zentralen autonomen Störungen – wie die MSA – wertvoll. Bei höheren Dosen können Nebenwirkungen auftreten (Tremor, Appetitverlust, Harnretention bei Männern).
Unter der großen Zahl von vasoaktiven Substanzen, die bei MSA evaluiert wurden, entspricht nur eine den Kriterien der Evidenz-basierten Medizin, nämlich der direkt wirkende Alpha-Agonist Midodrin (Gutron®) [17–19]. Die Nebenwirkungen sind gewöhnlich leicht und führen nur selten zum Abbruch der Behandlung aufgrund von Harnretention oder Pruritus, der hauptsächlich an der Kopfhaut auftritt.
Ein anderes vielversprechendes Arzneimittel scheint der Norepinephrin-Vorläufer Droxidopa (L-Threo-Dihydroxy-Phenylserin, L-Threo-DOPS) zu sein, der in dieser Indikation in Japan schon seit Jahren verwendet wird und dessen Wirksamkeit in einer offenen Dosisfindungs-Studie gezeigt wurde [20].
Falls die genannten Arzneimittel den gewünschten Effekt nicht erreichen, ist ein gezieltes problemorientiertes Vorgehen erforderlich.
Das Somatostatin-Analogon Octreotid (Sandostatin®) hilft oft gegen die postprandiale Hypotonie [21]. Es wird vermutet, dass Octreotid die Freisetzung der vasodilatatorischen gastrointestinalen Peptide hemmt [22]. Die nächtliche Hypertonie wird dabei nicht gesteigert [21].
Das Vasopressin-Analogon Desmopressin (z.B. Minirin®), welches auf die renalen tubulären Vasopressin-2-Rezeptoren wirkt, reduziert die nächtliche Polyurie und verbessert die posturale Hypotonie am Morgen [23].
Das Peptid Erythropoetin (z.B. Erypo®) kann mit seiner Fähigkeit, die Bildung von Erythrozyten anzuregen, bei manchen Patienten eine Besserung der zerebralen Sauerstoffversorgung bewirken [24, 25].
Eine breite Palette von Arzneimitteln ist in der Behandlung der posturalen Hypotonie [26] angewendet worden. Der Nutzen und die Nebenwirkungen vieler dieser Arzneimittel wurden bei MSA-Patienten nicht ausreichend mit den entsprechenden Endpunkten bestimmt. Probleme, denen man in der Therapie begegnet, sind die starken Nebenwirkungen. Nur exemplarisch seien hier Herzinsuffizienz (Pindolol) und exzessive Hypertonie im Liegen (bei Kombination von Monoaminoxidase-Hemmer mit Tyramin) genannt.
Bei neurogener Blasenentleerungsstörung mit Restharn ist die intermittierende Selbst- oder Fremdkatheterisierung zwei- bis dreimal pro Tag eine weit verbreitete und wirksame Methode, um sekundäre Komplikationen zu vermeiden. Falls mechanische Obstruktionen in der Urethra oder motorische Symptome eine unkomplizierte Katheterisierung verhindern, kann es notwendig werden, den Patienten mit einem permanenten suprapubischen Katheter zu versorgen.
Pharmakologische Optionen mit anticholinergen, procholinergen oder alpha-adrenergen Substanzen bewirken im Allgemeinen keine adäquate Reduzierung des Post-Miktions-Residualvolumens bei MSA-Patienten. Anticholinerge Substanzen, wie Oxybutynin, können jedoch im frühen Verlauf der Krankheit die Symptome von Detrusor-Hyperreflexie oder Sphinkter-Detrusor-Dyssynergie bessern [6]. Zudem wurde gezeigt, dass alpha-adrenerge Rezeptor-Antagonisten (Prazosin – z.B. Minipress® – und Moxisylyt) die Blasenentleerung bei MSA-Patienten mit einer Reduktion des Residualvolumens verbessern [27]. Chirurgische Eingriffe sollten bei diesen Patienten vermieden werden, da es postoperativ fast immer zu einer Verschlechterung der Blasenkontrolle kommt [6].
Die Notwendigkeit einer spezifischen Behandlung der sexuellen Dysfunktion muss bei jedem MSA-Patienten individuell beurteilt werden. Vorläufige Evidenz bei Parkinson-Patienten [28] lässt darauf schließen, dass Sildenafil (Viagra®) auch in der Behandlung des Erektionsversagens bei MSA-Patienten erfolgreich sein dürfte. Tatsächlich konnte anhand einer randomisierten, doppelblinden, Plazebo-kontrollierten Cross-over-Studie bei zwölf Patienten mit der Parkinson-Erkrankung und zwölf MSA-Patienten gezeigt werden, dass Sildenafil in der Behandlung der erektilen Dysfunktion bei beiden Erkrankungen wirksam ist; jedoch kann es bei MSA-Patienten eine orthostatische Hypotonie demaskieren oder verstärken [29]. Erektiles Versagen bei MSA kann auch mit oralem Yohimbin (z. B. Yohimbin Spiegel®), durch intrakavernosale Injektion von Papaverin oder mit einem Penisimplantat verbessert werden [6].
Obstipation kann durch eine Erhöhung der intraluminalen Flüssigkeit mit Hilfe einer Macrogol-Wasser-Lösung behandelt werden [30].
Inspiratorischer Stridor entwickelt sich bei ungefähr 30 % der Patienten. Kontinuierliche Überdruck-Beatmung (CPAP) ist bei manchen Patienten nützlich [31]. In nur 4 % der Fälle wird eine Tracheostomie benötigt.
Motorische Störungen
Parkinson-Syndrom: Das Parkinson-Syndrom ist die vorherrschende motorische Störung bei MSA. Levodopa-Ersatz gilt als Goldstandard der Antiparkinson-Therapie beim Morbus Parkinson, nicht jedoch bei MSA. Eine doppelblinde kontrollierte Levodopa-Therapiestudie wurde zwar nie durchgeführt, offene Studien lassen jedoch darauf schließen, dass im Gegensatz zu Parkinson-Patienten bei den meisten MSA-Patienten – abgesehen von einem transienten Ansprechen bei bis zu 30 % der Patienten [1, 32] – keine Besserung durch die Behandlung mit Levodopa erzielt wird. Gelegentlich ist ein günstiger Effekt nur evident, wenn scheinbar nicht ansprechende Patienten nach Absetzen von Levodopa akinetischer werden [33].
Levodopa-assoziierte Dyskinesien können sogar bei Fehlen einer Antiparkinson-Wirkung vorkommen und sind vor allem orofazial und zervikal mit deutlich dystoner Komponente ausgeprägt [2]. Eine bestehende orthostatische Hypotonie wird oft durch die Levodopa-Behandlung verschlechtert oder sie wird erstmals manifest. Toxische Verwirrtheitszustände scheinen ungewöhnlich zu sein [1].
Das im Allgemeinen unzureichende oder fehlende Ansprechen auf Levodopa beruht sowohl auf dem Verlust von striatalen Dopamin-Rezeptoren als auch auf einer Pathologie stromabwärts von striatopallidalen Projektionen [34].
Die bislang publizierten Ergebnisse mit Dopamin-Agonisten bei MSA-Patienten sind enttäuschend [35]. Schwere psychiatrische Nebenwirkungen mit Alpträumen, visuellen Halluzinationen und Verwirrtheitszuständen traten in einer doppelblinden Cross-over-Studie mit Lisurid (Dopergin®) bei sechs Patienten auf [36]. Wenning et al. [1] berichteten über ein Ansprechen auf orale Dopamin-Agonisten nur bei vier von 41 Patienten. Keiner der 30 Patienten, die Bromocriptin erhielten, verbesserte sich. Jedoch zeigten drei von zehn Patienten, die mit Pergolid behandelt wurden, eine mäßige Besserung. 22 % der Levodopa-Responder, die mit Dopamin-Agonisten zusätzlich behandelt wurden, haben gut oder ausgezeichnet auf zumindest einen der oral aktiven Dopamin-Agonisten angesprochen. MSA-Patienten berichten häufig über das Auftreten oder die Verschlechterung der posturalen Hypotonie nach Beginn einer Therapie mit Dopamin-Agonisten, so dass die Dosissteigerung limitiert sein kann.
Antiparkinson-Effekte wurden bei vier von 26 MSA-Patienten beobachtet, die offen mit Amantadin (z. B. PK-Merz®) behandelt wurden [1], jedoch ergab sich keine signifikante Besserung bei einer offenen Studie von neun Patienten mit atypischem Parkinson-Syndrom, einschließlich fünf Patienten mit MSA [37].
Zerebelläre Ataxie: Es gibt keine effektive Therapie der MSA-assoziierten Ataxie, die bei MSA-C-Patienten klinisch im Vordergrund steht. Gelegentliche Erfolge wurden mit Cholinergika, Amantadin, 5-Hydroxytryptophan, Isoniazid, Baclofen und Propranolol berichtet. Für die Mehrheit von Patienten erwiesen sich diese Arzneimittel jedoch als unwirksam.
Eine interessante Beobachtung ist die scheinbar temporäre Exazerbation der Ataxie durch Zigarettenrauchen [38, 39]. Es ist bekannt, dass Nicotin die Freisetzung von Acetylcholin in vielen Bereichen des Gehirns erhöht, und wahrscheinlich führt es auch zur Freisetzung von Noradrenalin, Dopamin, Serotonin und anderen Neurotransmittern. Nicotinerge Systeme könnten deshalb bei zerebellären Störungen eine Rolle spielen. Studien mit Nicotin-Antagonisten könnten sich bei zerebellären Degenerationen lohnen.
Praktische Therapie
Aufgrund der geringen Anzahl an randomisierten kontrollierten Studien (↑↑ resp. ↓↓) beruht das praktische Management der MSA größtenteils auf empirischer Evidenz (↔) oder auf einzelnen randomisierten Studien (↑). Die Therapie-Empfehlungen sind in Tabelle 4 zusammengefasst.
Tab. 4. Praktisches Management der MSA [82]
|
A. Pharmakotherapie |
|
I. Akinesie-Rigidität |
|
Levodopa (bis zu 1000 mg/Tag, falls toleriert) (↔) |
|
Dopaminagonisten als Mittel der zweiten Wahl (Dosierung wie für Morbus-Parkinson-Patienten) (↔) |
|
Amantadin als Mittel der dritten Wahl (100 mg bis dreimal täglich) (↔) |
|
II. Fokale Dystonie |
|
Botulinumtoxin A (↔) |
|
III. Orthostatische Hypotonie |
|
Nächtliches Hochstellen des Bettkopfendes (↔) |
|
Elastische Stützstrümpfe (↔) |
|
Salzreiche Ernährung (↔) |
|
Fludrocortison (0,1–0,3 mg/Tag bzw. Nacht) (↔) |
|
Ephedrin (15–45 mg dreimal täglich) (↔) |
|
L-Threo-DOPS (300 mg zweimal täglich) (↔) |
|
Midodrin (2,5–10 mg dreimal täglich) (↑↑) |
|
IV. Postprandiale Hypotonie |
|
Octreotid (25–50 µg s. c. 30 min vor der Mahlzeit) (↔) |
|
V. Nächtliche Polyurie |
|
Desmopressin (Spray: 10–40 µg/Nacht od. Tablettenform: 100–400 µg/Nacht) (↔) |
|
VI. Blasenstörung |
|
Oxybutynin bei Detrusor-Hyperreflexie (2,5–5 mg zwei- bis dreimal täglich) (↔) |
|
Intermittierende Selbstkatheterisierung bei Retention (Restvolumen > 100 ml) (↔) |
|
B. Andere Therapien |
|
Physiotherapie (↔) |
|
Logopädie (↔) |
|
Ergotherapie (↔) |
|
Perkutane endoskopische Gastrostomie (PEG) (selten, im späten Stadium benötigt) (↔) |
|
Rollstuhl (↔) |
|
Kontinuierliche Überdruckbeatmung (CPAP) (↑) |
Autonome Störungen
Orthostatische Hypotonie ist ein Hauptmerkmal der MSA, das häufig die aus der progressiven motorischen Störung entstandene Behinderung steigert. Eine Anzahl von nicht pharmakologischen Strategien wie elastische Stützstrümpfe bzw. Strumpfhosen (↔), salzreiche Ernährung (↔), häufige kleine Mahlzeiten (↔), nächtliches Hochstellen des Bettkopfendes (↔) und langsames Aufstehen aus der Sitzposition (↔) können die orthostatischen Symptome verbessern und sollten unbedingt vor der Entscheidung für eine Arzneimitteltherapie versucht werden.
Wenn diese Maßnahmen erfolglos bleiben, dann kann nachts das Mineralocorticoid Fludrocortison (z.B. Astonin® H) verabreicht werden (0,1–0,3 mg) (↔). Falls die orthostatischen Blutdruckabfälle nicht abbrechen, sollten Sympathomimetika wie Ephedrin (15–45 mg dreimal täglich) (↔) oder Midodrin (100 mg dreimal täglich) (↑↑) zusätzlich zum Fludrocortison verabreicht werden. Alternativ kann Droxidopa (300 mg zweimal täglich) (↔), ein Vorläufer von Noradrenalin, die orthostatische Hypotonie verbessern. Droxidopa ist jedoch in vielen Ländern nicht verfügbar.
Bei den meisten MSA-Patienten stellen die Blasensymptome ein viel größeres Problem dar.
Desmopressin (Spray: 10–40 µg/Nacht oder Tablettenform: 100–400 µg/Nacht) (↔) reduziert die nächtliche Polyurie. Die Miktionshäufigkeit und Dranginkontinenz bessern sich oft mit Oxybutynin (z.B. Dridase®) (2,5–5 mg zwei- bis dreimal täglich) (↔). Dieses peripher wirksame anticholinerge Arzneimittel kann jedoch eine Harnretention hervorrufen.
Restharnbildung von >100 ml ist die Indikation für intermittierende Selbst- oder Fremdkatheterisierung (↔). In fortgeschrittenen Stadien der MSA kann ein transurethraler oder suprapubischer Katheter (↔) notwendig werden.
Erektiles Versagen kann durch orales Yohimbin (2,5–5 mg dreimal täglich) (↔), Sildenafil (50–100 mg/Tag) (↔), durch intrakavernosale Injektion von Papaverin (↔) oder durch ein Penisimplantat (↔) verbessert werden.
Inspiratorischer Stridor entwickelt sich bei 30 % der Patienten. CPAP kann sich günstig auswirken und sollte bei Patienten mit ausgeprägtem Stridor versucht werden (↑). Eine Tracheostomie wird selten benötigt (↔).
Motorische Störungen
Zunächst sollte als Antiparkinson-Therapie der ersten Wahl Levodopa (↔) bis zu 1000 mg/Tag (sofern toleriert) verabreicht werden.
Bei Nichtansprechen auf eine hoch dosierte Levodopa-Behandlung sind die Erfolgsaussichten mit Dopamin-Agonisten (↔) schlecht. Gelegentlich verbessert sich jedoch mit Dopamin-Agonisten der Zustand von MSA-Patienten, die auf Levodopa nicht angesprochen haben. Ein Versuch unter Verwendung von Titrations-Schemata, die für Parkinson-Patienten etabliert wurden (einschließlich Domperidon-Schutz mit 10 mg dreimal täglich. [↑]), ist deshalb wünschenswert.
Wenn Dopamin-Agonisten unwirksam sind, sollte Amantadin (100 mg dreimal täglich) (↔) verabreicht werden. Damit können – wenn auch nur in seltenen Fällen – günstige Antiparkinson-Wirkungen bei Patienten erzielt werden, die auf eine dopaminerge Therapie nicht ansprechen.
Es gibt keine effektive medikamentöse Therapie für die zerebelläre Ataxie bei der MSA.
Blepharospasmus und auch Gliedmaßendystonie, nicht jedoch Antecollis, können auf lokale Injektionen von Botulinumtoxin A (↔) gut ansprechen (Tab. 4).
Aufgrund der unzureichenden Wirksamkeit medikamentöser Therapie sollten nicht-medikamentöse Strategien ausgeschöpft werden. Physiotherapie (↔) hilft, die Mobilität zu erhalten und Kontrakturen zu verhindern. Logopädie (↔) kann Sprache und Schlucken verbessern und Kommunikationshilfen bereitstellen. Bei Dysphagie kann Ernährung durch eine nasogastrische Sonde (↔) oder perkutane endoskopische Gastrostomie (PEG) (↔) notwendig werden. Ergotherapie (↔) hilft die Beeinträchtigung zu limitieren, die durch die Behinderung des Patienten entsteht, und sollte auch Hausbesuche inkludieren. Viele MSA-Patienten werden etwa vier bis sechs Jahre nach Erkrankungsbeginn aufgrund zunehmender posturaler Instabilität mit Sturzgefahr rollstuhlpflichtig. Eine psychologische Betreuung (↔) für Patienten und Partner ist angesichts des unaufhaltsamen Krankheitsverlaufs von großer Bedeutung.
Progressive supranukleäre Paralyse (PSP)
Klinik und Differenzialdiagnose
Die progressive supranukleäre Paralyse (PSP), auch bekannt als Steele-Richardson-Olszewski (SRO)-Krankheit, ist eine sporadische neurodegenerative Störung, die neuropathologisch durch eine Tauopathie mit prominenter subkortikaler neurofibrillärer Degeneration gekennzeichnet ist. Ätiopathogenetisch wird zunehmend die Bedeutung subkortikaler Aggregate aus phosphoryliertem „4-repeat“-Tau hervorgehoben. Interessanterweise deuten genetische Studien darauf hin, dass ein spezifischer Haplotyp des Tau-Gens bei PSP und CBD (s. u.) überrepräsentiert ist. Dies legt einen gemeinsamen genetischen Hintergrund dieser Tauopathien nahe [40].
Klinisch stehen folgende Achsensymptome im Vordergrund: Levodopa-refraktäres Parkinson-Syndrom, supranukleäre vertikale Ophthalmoplegie (häufig nach unten mehr als nach oben), rezidivierende Stürze und subkortikale Demenz. Andere Symptome sind langsame vertikale und später auch horizontale Sakkaden (Frühsymptom der Blickparese), ein starrer Gesichtsausdruck (Abb. 4) mit deutlich reduziertem Lidschlag sowie frontaler Muskelüberaktivität, Nackenrigor, aufrechte Haltung, pseudobulbäre Parese mit Dysphagie und Dysarthrie, Palilalie oder Palilogie und pseudobulbäres Weinen oder Lachen. Die Kriterien des National Institute for Neurological Disorders and Stroke und der Society for PSP (NINDS-SPSP) zur klinischen Diagnose der PSP wurden in den letzten fünf Jahren als internationaler Standard etabliert [41] (Tab. 5). Obwohl die supranukleäre vertikale Blickparese nach den NINDS-Kriterien als ein obligates Kennzeichen für die Diagnose einer wahrscheinlichen PSP gilt, ist dieses klinische Symptom unspezifisch. Es kommt bei einer Reihe anderer ZNS-Erkrankungen – einschließlich vaskulärer Enzephalopathie, Demenz mit Lewy-Körpern (DLK), kortikobasaler Degeneration (CBD), Whipple-Krankheit und Huntington-Krankheit – vor. In manchen Fällen kann es während der ersten zwei bis drei Erkrankungsjahre schwierig sein, PSP-Patienten zu diagnostizieren, vor allem wenn keine posturale Instabilität und Ophthalmoplegie vorliegen und wenn Akinese und Rigor noch immer auf Levodopa ansprechen. Jedoch ist eine derartige Präsentation ungewöhnlich; in den meisten Fällen kann die PSP aufgrund klinischer Befunde gut vom Morbus Parkinson unterschieden werden (Tab. 6).

Abb. 4. Typischer „starrender“ und gefurchter Gesichtsausdruck eines PSP-Patienten [83]
Tab. 5. NINDS-SPSP klinische Kriterien zur Diagnose der progressiven supranukleären Paralyse [nach 41, 82]
|
Obligate Einschlusskriterien |
|
|
Mögliche PSP |
Langsam fortschreitende Erkrankung |
|
Erkrankungsbeginn über 40 Jahre |
|
|
Entweder vertikale supranukleäre Blickparese (nach oben oder unten) |
|
|
Ausschluss anderer Erkrankungen mit PSP-ähnlichem Verlauf (s. Ausschlusskriterien) |
|
|
Wahrscheinliche PSP |
Langsam fortschreitende Erkrankung |
|
Erkrankungsbeginn über 40 Jahre |
|
|
Vertikale supranukleäre Blickparese (nach oben oder unten) und ausgeprägte Gangunsicherheit mit Stürzen im ersten Erkrankungsjahr |
|
|
Ausschluss anderer Erkrankungen mit PSP-ähnlichem Verlauf (s. Ausschlusskriterien) |
|
|
Gesicherte PSP* |
Klinisch mögliche oder wahrscheinliche PSP mit typischen histopathologischen Veränderungen |
|
Nebenkriterien |
|
|
Symmetrische, proximal betonte Akinesie oder Rigor |
|
|
Retrocollis |
|
|
Fehlendes oder mangelhaftes Ansprechen auf Levodopa |
|
|
Frühzeitige Dysarthrie/Dysphagie |
|
|
Frühzeitiger kognitiver Abbau (subkortikale Demenz) mit mindestens zwei der folgenden Symptome: |
|
|
Obligate Ausschlusskriterien |
|
|
Zustand nach Enzephalitis |
|
|
„Alien Limb“-Phänomen, kortikale Sensibilitätsstörungen, fokale, frontale oder temporoparietale Atrophie |
|
|
Levodopa-unabhängige (primäre) Halluzinationen oder Wahnideen |
|
|
Kortikale Demenz vom Alzheimer-Typ (ausgeprägtes amnestisches Syndrom mit Aphasie oder Agnosie) |
|
|
Ataxie, ausgeprägte Dysautonomie zu Erkrankungsbeginn (orthostatische Hypotonie und Blasenentleerungsstörungen) |
|
|
Ausgeprägte asymmetrische Parkinson-Symptome |
|
|
Strukturanomalien im zerebralen CT oder MRT |
|
|
Morbus Whipple |
|
|
Frontalhirnzeichen |
|
* Gesicherte PSP ist eine klinisch-pathologische Diagnose
Tab. 6. Vergleich von PSP und Parkinson-Krankheit [82]
|
Symptome |
PSP |
Parkinson-Krankheit |
|
Akinese, Rigor |
Symmetrisch |
Asymmetrisch |
|
Ruhetremor |
Sehr selten |
Häufig |
|
Rigidität |
Stärker axial (Hals, Rumpf) ausgeprägt als an den Extremitäten |
Stärker ausgeprägt an den Extremitäten als an Hals und Rumpf |
|
Ansprechen auf Levodopa |
Wenig oder überhaupt nicht; selten mäßig, aber transient |
Ausgezeichnet |
|
Levodopa-induzierte Dyskinesie |
Keine |
Häufig |
|
Dystonie |
Hals > Extremitäten |
Extremitäten > Hals (Levodopa-induziert) |
|
Posturale Instabilität |
Früh |
Spät |
|
Stürze |
Früh |
Spät |
|
Axiale (Stamm-) Position beim Gehen |
Aufrecht |
Gebeugt |
|
Gesichtsausdruck |
Überrascht, die Augen weit geöffnet, Hypomimie, überaktiver Mb. frontalis (auf der Stirn gefaltete Haut, Augenbrauen erhöht) |
Hypomimie |
|
Lidschlagfrequenz |
Stark vermindert |
Vermindert |
|
Frontale Verhaltensstörung |
Früh, häufig |
Spät, nicht häufig |
Diagnostische Probleme ergeben sich bei Fehlen der klassischen neuroophthalmologischen Zeichen. In diesem Falle können elektronystagmographisch häufig eine mangelnde Fixationssuppression des vestibulookulären Reflexes sowie hypometrische Sakkaden normaler Latenz nachgewiesen werden. PSP-Patienten zeigen eine attenuierte oder fehlende Startle-Reaktion auf akustische Reize. Computer- und Kernspintomographie sind vor allem zum Ausschluss symptomatischer Blickparesen, zum Beispiel im Rahmen einer subkortikalen arteriosklerotischen Enzephalopathie, sinnvoll. Darüber hinaus lassen sich bildgebend PSP-typische Befunde wie eine Erweiterung des dritten Ventrikels und der Cisterna magna sowie eine diffuse Hirnstammatrophie, die im Bereich der Vierhügelplatte betont sein kann, bzw. eine Mittelhirnatrophie erheben [42]. Die Messung des anteroposterioren Mittelhirn-Durchmessers mit axialer T2-gewichteter Kernspintomographie ist ein zuverlässiges Mittel, um Patienten mit PSP von jenen mit der Parkinson-Erkrankung zu unterscheiden (Abb. 5). So zeigten PSP- (und auch MSA-)Patienten signifikant niedrigere Mittelhirn-Durchmesser als die Patienten mit Morbus Parkinson und die Kontrollgruppe, während keine Unterschiede im Mittelhirn-Durchmesser zwischen den Patienten mit Morbus Parkinson und der Kontrollgruppe gefunden wurden [43]. In einer Studie, bei der 123I-Iodobenzamid(IBZM)-SPECT (zur Darstellung der striatalen Dopamin-D2-Rezeptoren) und hoch auflösendes MRI bei PSP-Patienten eingesetzt wurden, korrelierte die Reduktion der IBZM-Bindung signifikant mit der Atrophie des Mittelhirns [44]. Gesteigerte putaminale rADC-Werte unterscheiden MSA-P- nicht von PSP-Patienten, da sich bei beiden im Vergleich zur Parkinson-Erkrankung signifikant erhöhte putaminale rADC-Werte mit DWI-Imaging nachweisen ließen [45]. Protonenmagnetresonanzspektroskopische Beobachtungen belegen im Vergleich zu gesunden Kontrollen bzw. zur MSA eine (mäßige, jedoch statistisch signifikante) Abnahme der N-Acetyl-Aspartat(NAA)-Amplitude im Bereich des Nucleus lentiformis bei der PSP [46]. Des Weiteren waren die Quotienten NAA/Cho (Cholin enthaltende Verbindungen) und NAA/Cr (Creatin und Phosphocreatin enthaltende Verbindungen) im Vergleich zu gesunden Kontrollen bei der PSP signifikant erniedrigt, wobei sich kein signifikanter Unterschied zwischen Patienten mit der Parkinson-Erkrankung und der Kontrollgruppe zeigte [47]. Eine reduzierte neuronale Aktivität lässt sich auch mit PET im Striatum und im frontalen Kortex beobachten [48].
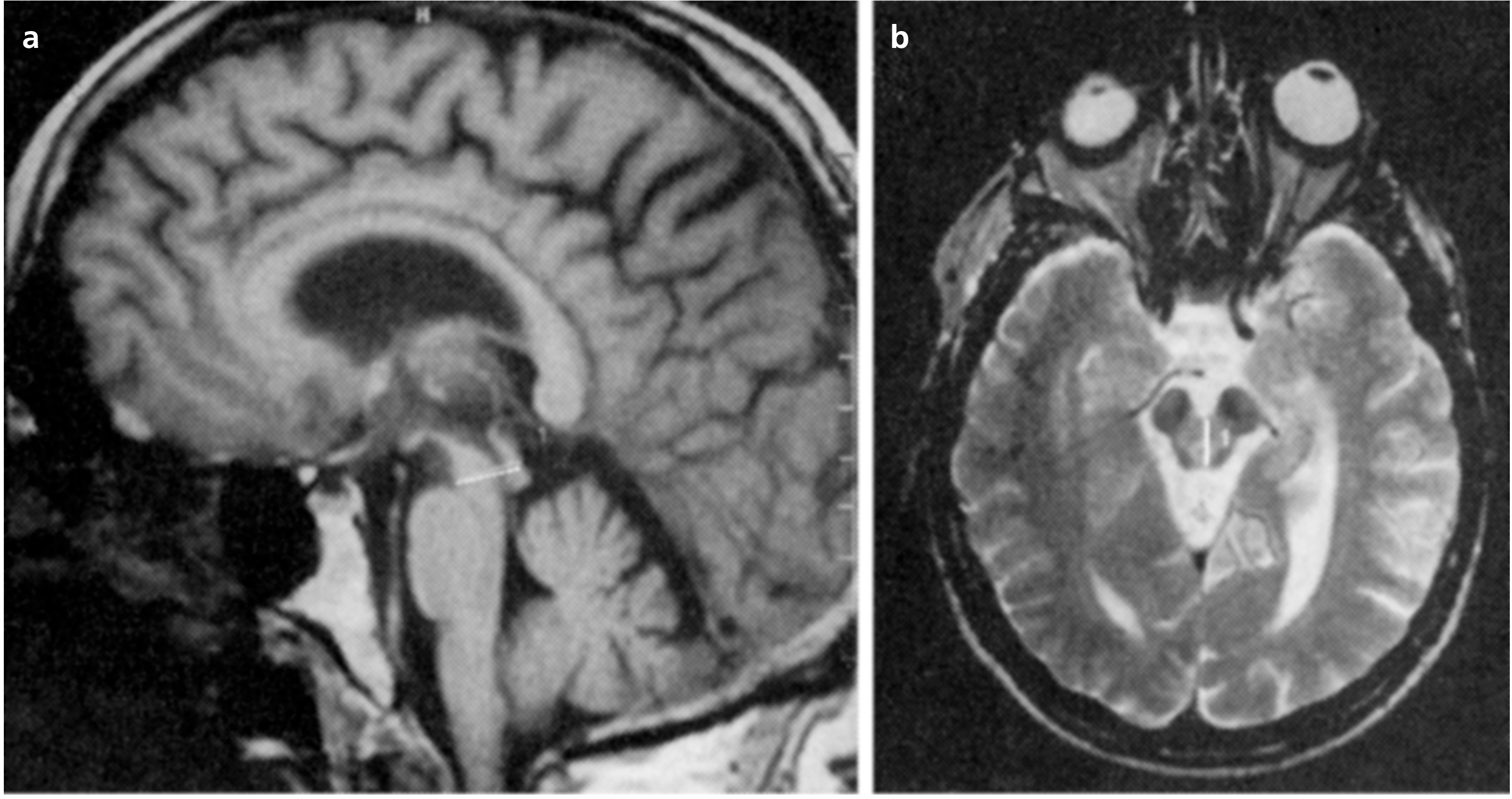
Abb. 5. Messung des sagittalen anterior-posterioren Mittelhirndiameters mit a) sagittalen T1- und b) axialen T2-gewichteten MRI-Aufnahmen bei der PSP [43]
Natürlicher Krankheitsverlauf
Die in jüngster Zeit durchgeführten epidemiologische Studien haben gezeigt, dass PSP eine häufige Ursache von atypischen Parkinson-Syndromen ist (Inzidenz: 5,3/100000 in der >50 Jahre alten Bevölkerung [14], altersadjustierte Prävalenz: 6,4/100000 [13]). Das Durchschnittsalter bei Krankheitsbeginn liegt um das 60. Lebensjahr (63 Jahre nach Litvan et al. [49]). Die durchschnittliche Überlebensdauer beträgt 5,6 Jahre [49]. Die Krankheit verläuft stetig progredient. In späten Stadien sind PSP-Patienten auf den Rollstuhl angewiesen oder bettlägerig. Viele Kranke klagen bereits frühzeitig über Dysphagie und zeigen eine erhöhte Aspirationsneigung.
Therapieprinzipien
Parkinson-Syndrom
Randomisierte doppelblind kontrollierte Studien zur dopaminergen Therapie (Levodopa und Dopamin-Agonisten) bei PSP-Patienten liegen nicht vor. Eine Ausnahme stellt eine Kurzzeit-Studie mit Pergolid (Parkotil®) dar, deren Aussagekraft allerdings durch die kleine Gruppengröße beschränkt ist [50]. So beruht die Evidenz lediglich auf anekdotischen Fallberichten, offenen Anwendungen und retrospektiven Kohorten-Studien unter Anwendung nicht validierter Diagnose-Kriterien. Die Ergebnisse deuten darauf hin, dass die dopaminerge Therapie bei PSP-Patienten gewöhnlich unwirksam ist.
Die mangelnde Wirksamkeit von Levodopa ist nicht nur auf den striatalen Dopamin-Rezeptorverlust zurückzuführen, sondern auch auf Läsionen in nicht-dopaminergen Neurotransmitter-Systemen (z.B. cholinerge Hirnstammkerngebiete). Insgesamt zeigen etwa 20 bis 40 % der PSP-Patienten eine transiente und leichte Besserung durch Levodopa [51–54]. Levodopa-induzierte motorische und psychiatrische Komplikationen scheinen selten zu sein. In einer Übersicht von 82 konsekutiven Patienten, die offen mit Levodopa behandelt wurden (durchschnittliche maximale tägliche Dosis 1,015 mg), zeigten nur drei Patienten leichte Dyskinesien und nur einer eine akute psychotische Reaktion nach Zugabe von Amitriptylin (z.B. Saroten®) zur laufenden Levodopa-Therapie [52].
Dopamin-Agonisten erwiesen sich ebenso als unwirksam. Williams et al. [55] konnten bei den meisten der neun PSP-Patienten, die in einer doppelblind Plazebo-kontrollierten Studie mit Bromocriptin (z.B. Pravidel®) behandelt wurden, keine signifikante Besserung demonstrieren. Jedoch wurde in einer retrospektiven Serie von post mortem gesicherten PSP-Fällen eine transiente Besserung mit Bromocriptin beobachtet [53].
Eine doppelblind kontrollierte Studie zeigte eine 20%ige Besserung der motorischen Behinderung bei drei mit Pergolid behandelten PSP-Patienten [50]. Im Gegensatz dazu waren Lisurid und Pramipexol in kleineren Studien wirkungslos [56, 57].
Trotz der enttäuschenden Erfahrungen mit Levodopa und Dopamin-Agonisten können diese Substanzen bei PSP-assoziierten Parkinson-Symptomen versucht werden, da transiente Effekte nicht ausgeschlossen werden können.
Eine leichte (selten dramatische) Besserung des Parkinson-Syndroms kann mit trizyklischen Antidepressiva bei wenigen PSP-Patienten mitunter erzielt werden [58–60]. In einer doppelblinden Cross-over-Studie mit vier PSP-Patienten wurde gezeigt, dass sowohl Amitriptylin als auch Desipramin (Petylyl®) die Parkinson-Symptomatik lindert [58]. Zusätzlich hatte sich eine Blickparese bei einem Patienten sowie eine Apraxie bei Augenlidöffnung bei zwei Patienten gebessert. Die Nebenwirkungen waren leicht, wie beispielsweise reversible Harnretention und Mundtrockenheit. In einer offenen Studie mit zwei PSP-Patienten wurde über die Antiparkinson-Wirksamkeit und bessere Verträglichkeit von niedrig dosiertem Amitriptylin berichtet [59]. Nortriptylin (Nortrilen®) eignet sich prinzipiell auch. Randomisierte kontrollierte Therapie-Studien sind notwendig, um die Rolle der Antidepressiva in der Behandlung der PSP zu klären.
Idazoxan ist ein potenter selektiver präsynaptischer Alpha-2-Antagonist, dessen Gesamtwirkung in der Erhöhung der Noradrenalin-Neurotransmission besteht. In einer doppelblinden Cross-over-Studie mit neun PSP-Patienten zeigte sich mit Idazoxan eine deutliche Besserung der Mobilität, des Gleichgewichts, des Gangs und der Fingergeschicklichkeit [61]. Im Gegensatz dazu zeigte Efaroxan, ein im Vergleich zu Idazoxan stärkerer Stimulator der Noradrenalin-Neurotransmission, keine Besserung bei 14 PSP-Patienten in einer doppelblinden Cross-over-Studie [62].
Der Serotonin-Antagonist Methysergid, der in einer offenen Therapie-Studie mit zwölf PSP-Patienten allein und in der Kombination mit Antiparkinson-Medikation eingesetzt wurde, führte bei acht Patienten zu Verbesserungen der Lebensqualität. Der Nutzen war bei vier Patienten mit schwerer Dysphagie besonders ausgeprägt. Auch zeigten die Patienten Besserungen des Sprechens, der Parkinson-Symptomatik sowie der Blickparese [63]. Diese Beobachtung konnte jedoch durch andere Studien nicht bestätigt werden [64, 65]. Serotonin-Antagonisten wie Methysergid können schwere Nebenwirkungen, wie retroperitoneale oder pleuropulmonale Fibrose, verursachen.
Aniracetam, ein Stoffwechselaktivator mit Acetylcholin-ähnlichen Eigenschaften, verbesserte die motorischen und kognitiven Funktionen bei zwei PSP-Patienten. Diese Beobachtungen sind jedoch bislang nicht bestätigt worden [66].
Andere Arzneimittel mit minimaler oder fehlender Wirksamkeit bei PSP sind Amantadin, Baclofen, Bupropion, Fluoxetin, Selegilin und Valproinsäure [37, 52, 67].
Es wurde berichtet, dass elektrokonvulsive Therapie die motorische Störung bei einigen PSP-Patienten bessert, jedoch wurde damit der Spitalaufenthalt verlängert. Bei diesen Patienten kam es zu einer behandlungsinduzierten Verwirrtheit. Dies limitiert die Nützlichkeit dieser Technik [68, 69].
Die Transplantation von adultem Nebennierenmark in den Nucleus caudatus wurde bei nur wenigen PSP-Patienten durchgeführt. Jedoch zeigte dieses Verfahren keinen Erfolg und war mit erheblicher perioperativer Morbidität und Letalität verknüpft [70, 71].
Okulomotorische und verwandte Störungen
Zolpidem (z. B. Stilnox®), ein kurzzeitwirksames Hypnotikum und selektiver Agonist des Benzodiazepin-Rezeptorsubtyps BZ1, besserte die sakkadischen Augenbewegungen und Parkinson-Symptome in einer doppelblinden Cross-over-Studie von zehn Patienten mit wahrscheinlicher PSP. Jedoch war der klinische Nutzen durch die dosisabhängige Somnolenz begrenzt, die besonders bei einer Dosis > 5 mg/Tag auftrat [72].
Buch- oder Zeitungslektüre kann durch das Hören von Tonträgern ersetzt werden.
Künstliche Tränen sind bei drohender Keratitis infolge der verminderten Lidschlagfrequenz hilfreich.
PSP-assoziierter Blepharospasmus oder eine Levator-Inhibition lassen sich mit Botulinumtoxin-Injektionen beherrschen [73–76]. Visuelle Prismen sind selten eine Hilfe.
Kognitive Störungen
Cholinerge Präparate sind nicht zu empfehlen. Durch diese Arzneimittel können sich der mentale Status und auch die Gangstörung verschlechtern [77–79]. Bis jetzt wurden nur drei Cholinergika zur Behandlung von PSP-Patienten verwendet. Foster et al. [77] untersuchten die Wirkung des M1/M2-muscarinergen Agonisten RS-86 auf motorische und kognitive Funktionen bei zehn PSP-Patienten in einer 9-wöchigen doppelblind randomisierten kontrollierten Studie. Es konnte keine Wirkung auf kognitive oder motorische Funktionen festgestellt werden. Außerdem zeigten Physostigmin und Scopolamin in einer doppelblind Plazebo-kontrollierten Studie bei neun PSP-Patienten keine therapeutische Wirksamkeit [78].
Fabbrini et al. [79] berichteten, dass der zentral wirksame Cholinesterase-Hemmer Donepezil (Aricept®) in einer offenen Studie mit sechs PSP-Patienten keine Wirkung zeigte.
Andere Symptome
Da die Ganginstabilität nur minimal oder überhaupt nicht auf Arzneimitteltherapie anspricht, sollten Gehhilfen in Betracht gezogen werden.
Aufgrund der Gefahr der Aspirationspneumonie sollten Schluckstörungen von Sprachtherapeuten regelmäßig untersucht und eine perkutane endoskopische Gastrostomie (PEG) frühzeitig angelegt werden. Bei Dysphagie werden Strohhalm, Verdickungsmittel oder pürierte Kost empfohlen. Obwohl Sialorrhö mit Anticholinergika behandelt werden kann, sollten diese Arzneimittel mit Vorsicht verwendet werden, da sie die Symptomatik des Patienten verschlechtern können.
Affektinkontinenz kann sich unter Amitriptylin bessern [58]. Wir beobachten deutliche Besserungen vor allem der Schluckstörungen und der Affektlabilität, auch der Dysarthrie (also des pseudobulbären Anteils), mit Amitriptylin.
Praktische Therapie
Aufgrund der geringen Anzahl der randomisierten kontrollierten Versuche (↑↑ resp. ↓↓) beruht das praktische Management der PSP-Therapie größtenteils auf empirischer Evidenz (↔) oder auf einzelnen randomisierten Studien (↑).
Obwohl selten eine Besserung erreicht wird, sollte zunächst mit Levodopa (bis zu 1000 mg/Tag, wenn toleriert) (↔) begonnen werden. Arzneimittel der zweiten Wahl sind Amitriptylin (oral bis zu 25 mg dreimal täglich, gelegentlich sind auch höhere Dosen bis zu 150 mg/Tag hilfreich) (↔) und Zolpidem (5–10 mg/Tag) (↑), wobei beide die motorische, vor allem die pseudobulbäre Behinderung transient für Wochen oder Monate verbessern können. Zum gegenwärtigen Zeitpunkt gibt es bei PSP-Patienten keine gesicherte Evidenz zur Wirksamkeit cholinerger, serotonerger oder noradrenerger Arzneimitteltherapie.
In Anbetracht der begrenzten pharmakologischen Optionen werden Physiotherapie (↔), Ergotherapie (↔) und Logopädie (↔) empfohlen. Bei Dysphagie kann Ernährung durch perkutane endoskopische Gastrostomie (PEG) (↔) notwendig werden. Nasensonden (↔) werden nur empfohlen, wenn es sich um eine kurzdauernde Intervention mit Überbrückungscharakter handelt. Periorbitaler und prätarsaler Blepharospasmus sowie Gliedmaßen-Dystonie sprechen in der Regel auf lokale Injektionen von Botulinumtoxin A (↔) gut an.
Danksagung
Ein Teil der hier berichteten Ergebnisse entstammt der wissenschaftlichen Tätigkeit der Innsbrucker MSA-Studiengruppe (IMSA-SG), welche unter der Leitung von Prof. Poewe die Koordinatorfunktion in der Europäischen MSA-Studiengruppe (EMSA-SG) wahrnimmt. IMSA-SG sowie EMSA-SG werden durch folgende kompetitive Drittmittel unterstützt: FWF-Projekte P11748-MED, P14633-PHA, P16128-B01; ÖNB-Projekt 8298; Forschungsauftrag des BMWV 70038/2-Pr4/98; EU-Projekt QLK6-CT-2000-00661 „European MSA-Study Group“ (www.emsa-sg.org).
Literatur
Das Literaturverzeichnis finden Sie im Internet unter www.ppt-online.de > Inhalt > 2005, Heft 2.
Univ.-Prof. Dr. Gregor Wenning (korrespondierender Autor), Dr. Felix Geser, Universitätsklinik für Neurologie, Anichstraße 35, 6020 Innsbruck, Österreich, E-Mail: gregor.wenning@uibk.ac.at
Clinical presentation and management of multiple system atrophy and progressive supranuclear palsy
During the last years the importance of atypical parkinsonian disorders and their differentiation to Parkinson’s disease (PD) have been increasingly appreciated. The clinical diagnosis is difficult especially in early disease stages. Besides cardinal diagnostic features, warning signs may help in the detection of early cases. Moreover, there is increasing interest in structural and functional neuroimaging. There is an almost ubiquitous presence of abnormal putaminal diffusion coefficients as measured by diffusion-weighted imaging in multiple system atrophy (MSA) and progressive supranuclear paralysis (PSP). The clinical differentiation of MSA and PSP from PD has also therapeutic and prognostic consequences. In the next years further phase II/III studies will be conducted and these require optimal clinical and surrogate markers. Two European research initiatives – European MSA-Study Group (EMSA-SG) and Neuroprotection and Natural History in Parkinson Plus Syndromes (NNIPPS) – have already been established with the aim to identify and evaluate surrogate markers of diagnosis and progression in MSA and PSP as well as to initiate neuroprotective intervention trials.
Keywords: Multiple system atrophy, progressive supranuclear palsy, clinical presentation, management
Psychopharmakotherapie 2005; 12(02)