Tjalf Ziemssen und Claudia Schmidt, Dresden
Obwohl mit dem idiopathischen Parkinson-Syndrom (IPS) in der Regel motorische Symptome assoziiert werden, setzt sich in Klinik und Wissenschaft in der letzten Zeit immer mehr die Ansicht durch, dass viele klinische Symptome beim idiopathischen Parkinson-Syndrom nichts oder nur wenig mit dem motorischen Funktionssystem zu tun haben. Diese „nicht-motorischen“ Symptome der Parkinson-Erkrankung schließen Verhaltens-, Schlaf- oder Wahrnehmungsstörungen ebenso ein wie Funktionsstörungen des autonomen Nervensystems. Dabei treten diese Störungen überraschend häufig auf, sie können Erstsymptome der Erkrankung sein und manchmal sogar das klinische Bild der Erkrankung dominieren. So berichtete schon James Parkinson in seinem 1817 erschienenen „Essay on the shaking palsy“ wiederholt von autonomen Regulationsstörungen und weist am Ende des Buches mit der Formulierung „mysterious sympathetic influence“ schon auf einen möglichen Zusammenhang mit dem autonomen Nervensystem hin [1]. Lewy beschrieb etwa 100 Jahre später Rigor, Tremor und „Sympathicusstörungen“ als typische Parkinson-Symptome, worunter er unter anderem „eine Incontinentia urinae et alvi, Speichel-, Tränen- und Nasenfluss, Ödeme und Cyanosen einer oder mehrerer Extremitäten, Stellwag und Graefe, ein- oder doppelseitiges Schwitzen“ verstand [2].
Das autonome Nervensystem
Das autonome Nervensystem steuert – in der Regel für uns nicht bewusst – die verschiedensten Funktionen unseres Organismus, um die innere Homöostase des menschlichen Körpers aufrechtzuerhalten [3]. Die Bedeutung des autonomen Nervensystems liegt unter anderem darin begründet, dass jedes Organ des menschlichen Körpers vom autonomen Nervensystem innerviert und somit reguliert wird. Verschiedene Bereiche der Gehirns gelten als Bestandteile eines komplexen zentralen autonomen Netzwerks, das ankommende Informationen aus der Peripherie (autonome Afferenzen) verarbeitet und eine entsprechende Reizantwort an die peripheren Zielorgane generiert (autonome Efferenz). Innerhalb dieses efferenten Systems werden traditionell zwei zumeist gegenläufige Komponenten unterschieden:
Das sympathische Nervensystem ist das so genannte „Notfallsystem“. Nach Aktivierung führt es unter anderem zur Pupillenerweiterung, Beschleunigung der Herzfrequenz, Zunahme der Herzkraft und des Gefäßwiderstands. Nachdem die sympathischen Nerven das Rückenmark im Brust- und Lendenwirbelbereich verlassen haben, müssen sie noch entweder in prä- oder paravertrebralen Ganglien auf das zweite sympathische Neuron umgeschaltet werden. Liegt eine Störung vor dieser ganglionären Umschaltung vor, spricht man von einer präganglionären, andernfalls von einer postganglionären Schädigung. An allen präganglionären Nervenendigungen sowie postganglionär an den Schweißdrüsen wird Acetylcholin als Transmitter freigesetzt, während postganglionär an den Effektororganen mit Ausnahme der Schweißdrüsen Noradrenalin ausgeschüttet wird.
Das parasympathische Nervensystem versteht man vereinfacht als Gegenspieler des sympathischen Systems, also etwa als „Ruhe- oder Erholungssystem“, das beispielsweise wesentlich an der Steuerung der Verdauung beteiligt ist. Nach Aktivierung führt es unter anderem zur Pupillenverkleinerung, Abnahme der Herzfrequenz und Aktivierung der Verdauung. Im oberen Teil versorgt es ebenfalls nach ganglionärer Umschaltung die Augen, Tränen- und Speicheldrüsen, Herz, Lunge sowie den Verdauungstrakt. Die im Bereich des Steißbeins austretenden Nervenfasern sind entscheidend an der Kontrolle des Harntrakts sowie des unteren Verdauungstrakts beteiligt. Der primäre Neurotransmitter der postganglionären parasympathischen Neuronen ist Acetylcholin.
Bereits Langley beschrieb als dritten eigenständigen Teil des autonomen Nervensystems das enterische Nervensystem, das ein von Sympathikus- bzw. Parasympathikus beeinflussbares komplexes neuronales Netzwerk zur Regulation von Motilität und Sekretion im Magen-Darm-Trakt bildet.
Zur Aufrechterhaltung der inneren Homöostase benötigt das autonome Nervensystem eine Vielzahl von autonomen Reflexbögen als Regelkreise, die vereinfacht jeweils aus einer afferenten, einer zentral verarbeitenden und einer efferenten Komponente bestehen: Das afferente Signal stammt meist von spezialisierten Sensoren wie beispielsweise dem Barorezeptor, der Veränderungen des Blutdrucks feststellen und in Nervenimpulse umwandeln kann. Über periphere Nerven oder Hirnnerven erreicht das Signal das zentrale Nervensystem. Dort wird die Afferenz unter mehrfacher neuronaler Umschaltung im Vergleich mit anderen Signalen und Steuerungssignalen von höheren Regulationszentren weiterverarbeitet, wobei für jeden Reflexbogen meistens mehrere spezifische zentralnervöse Verarbeitungszentren existieren. Von definierten zentralnervösen Zentren wird dann eine efferente Antwort zu den jeweiligen spezifischen Stellgliedern des Regelkreises wie beispielsweise der glatten Muskulatur der Blutgefäße generiert. Die Reaktion des Efferenzorgans trägt dazu bei, den von den Sensoren vorher festgestellten gestörten Zustand mit Hilfe eines spezifischen gegenregulatorischen Mechanimus zu beheben.
Eine Fehlregulation innerhalb eines Reflexbogens und somit eine Funktionsstörung des autonomen Nervensystems kann somit durch eine Störung oder einen Defekt im afferenten, zentralen oder efferenten Teil des autonomen Reflexbogens bedingt sein.
Idiopathisches Parkinson-Syndrom und autonome Funktionsstörungen
Die Angaben über die Häufigkeit von autonomen Funktionsstörungen beim idiopathischen Parkinson-Syndrom (IPS) schwanken je nach Kollektiv und Methodik zwischen 14 und 80 %, subjektive Beeinträchtigungen des Alltags treten bei mehr als der Hälfte der Patienten mit idiopathischem Parkinson-Syndrom auf [4, 5]. Die autonomen Funktionsstörungen nehmen in der Regel mit Fortschreiten der Erkrankung zu, sie gewinnen entsprechend Einfluss auf das subjektive Beschwerdebild, die Lebensqualität und auch die Behandelbarkeit der Erkrankung [5, 6]. Welche Ursachen kommen für die autonome Dysfunktion beim IPS-Patienten differenzialdiagnostisch in Betracht (Abb. 1)?
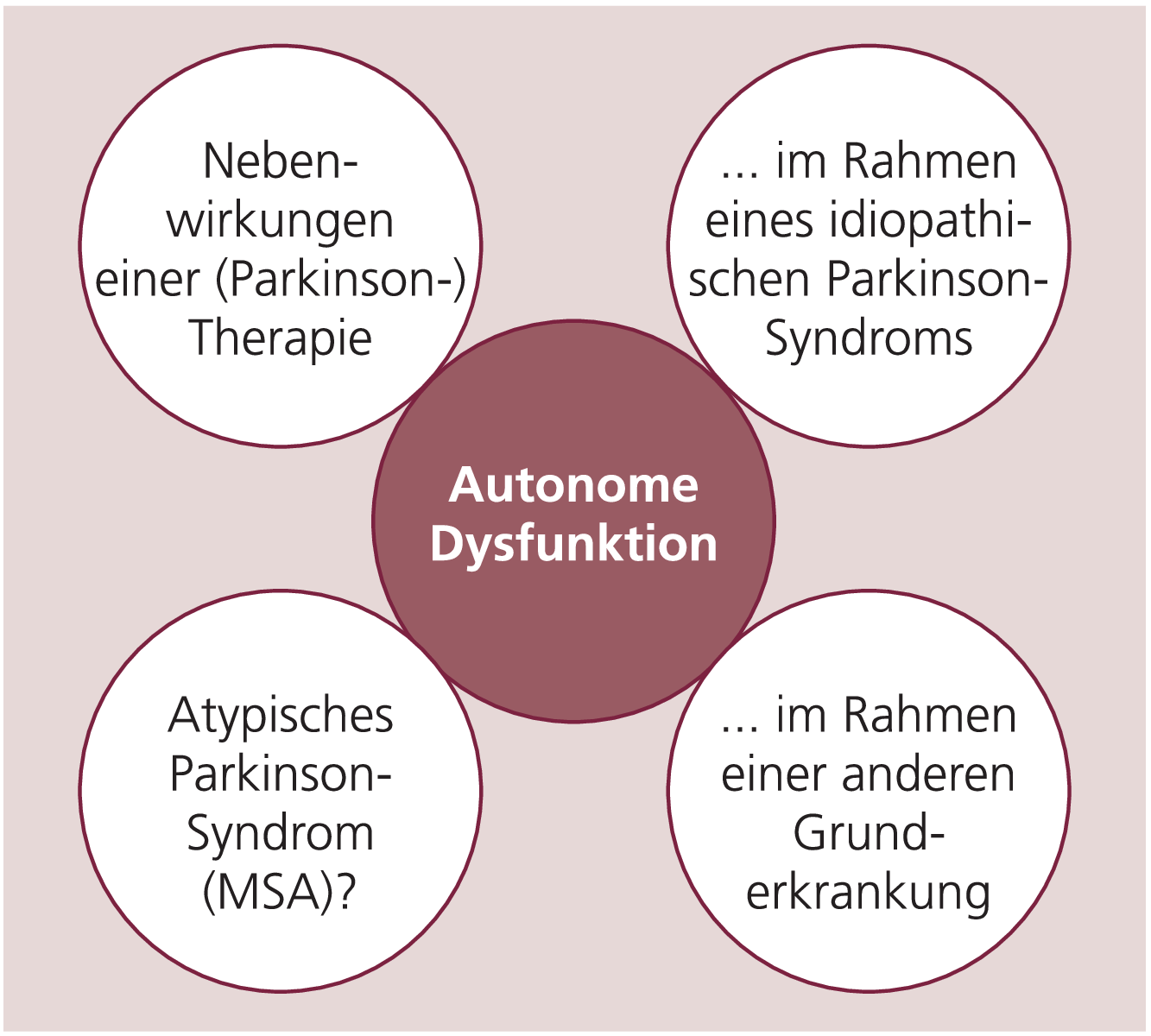
Abb. 1. Rolle der autonomen Dysfunktion beim idiopathischen Parkinson-Syndrom (Erläuterungen im Text)
Als primäre Ursache dieser Dysautonomie kann das fast ubiquitäre Auftreten von Neuronenverlusten und das Auftreten von Lewy-Körperchen in ganz unterschiedlichen Anteilen des peripheren und zentralen autonomen Nervensystems gelten. So beschreiben Braak et al. bei IPS-Patienten bereits zu präklinischen Zeitpunkten und vor dem Auftreten der charakteristischen histopathologischen Veränderungen in der Substantia nigra Läsionen im Bereich des dorsalen Vaguskerns und anderen autonomen Hirnstammzentren; dabei wurde das periphere autonome Nervensystem noch nicht mit in die Untersuchungen eingeschlossen [7].
Als wichtige Differenzialdiagnose muss insbesondere bei früh ausgeprägter autonomer Funktionsstörung die Multisystematrophie (MSA) bedacht werden, wobei die Differenzierung zwischen einem idiopathischen Parkinson-Syndrom mit ausgeprägter autonomer Dysfunktion und einer Multisystematrophie durchaus nicht einfach sein kann und besonderer innovativer diagnostischer Techniken bedarf (s.u.). Die Multisystematrophie ist eine sporadische neurodegenerative Erkrankung, die klinisch durch eine Kombination von extrapyramidalen, autonomen, zerebellären und Pyramidenbahnzeichen und histologisch durch Neuronenverlust, Gliose und gliale zytoplasmatische Einschlusskörperchen charakterisiert ist [8].
Darüber hinaus kann die autonome Funktionsstörung auch sekundär durch eine andere (Grund-) Erkrankung des IPS-Patienten wie beispielsweise Diabetes mellitus verursacht sein.
Am häufigsten führen jedoch Medikamente zu sekundären autonomen Funktionsstörungen, die durch zumeist unerwünschte Interaktionen mit den unterschiedlichen Komponenten des autonomen Nervensystems Fehlfunktionen hervorrufen können. Als wichtiges Beispiel seien die Psychopharmaka mit ihren vielfältigen unter anderem anticholinergen oder antiadrenergen Effekten genannt.
Autonome Funktionsdiagnostik
Es gibt eine Vielzahl von Untersuchungsverfahren zur Erfassung autonomer Funktionsstörungen, die zum Teil technisch und zeitlich aufwendig oder für den Patienten sehr belastend sein können. Im praktischen Alltag verwenden wir deshalb ein zweistufiges diagnostisches Verfahren, um zum einen möglichst früh autonome Funktionsstörungen diagnostizieren und somit therapieren zu können, aber auf der anderen Seite den Patienten nicht unnötig zu belasten [3].
Wir verwenden dabei einen von uns entwickelten, standardisierten Fragebogen (QAM-System), der die verschiedenen autonomen Funktionssysteme getrennt voneinander abfragt. Zusätzlich wird mit Hilfe einer speziell für diese Fragestellung entwickelten autonomen Testbatterie eine einfache autonome Funktionstestung durchgeführt, die vor allem auf der Untersuchung des kardiovaskulären autonomen Nervensystems (metronomische Atmung mit 6 Atemzyklen pro Minute, Valsalva-Manöver, pressorische Funktionstests, Orthostase-Test) basiert.
Bei auffälliger Screening-Untersuchung oder Anamnese wird speziell für das individuelle medizinische Problem eine Funktionsuntersuchung durchgeführt, die bedeutend komplexer und technisch aufwendiger als die Screening-Untersuchung sein kann, die aber notwendig ist, um die richtige Diagnose zu stellen und eine geeignete Therapie einzuleiten.
Pathophysiologie der Dysautonomie beim IPS
Mit Hilfe moderner Techniken wie zum Beispiel der Szintigraphie mit 123I-Metaiodobenzylguanidin (MIBG), einem pharmakologisch inaktiven Harnstoffderivat, das wie Noradrenalin aktiv von postganglionären adrenergen Neuronen verstoffwechselt wird, konnte bei über 250 IPS-Patienten eine deutlich reduzierte MIBG-Aufnahme ausschließlich in kardialen sympathischen Efferenzen nachgewiesen werden [9, 10]. Dieser Befund ließ sich konstant bei praktisch allen IPS-Patienten mit einem Hoehn-und-Yahr-Stadium von II oder höher nachweisen, unabhängig vom Vorliegen klinischer Zeichen einer autonomen Funktionsstörung, von der Dauer der Erkrankung und von der jeweiligen Medikation. Mit Hilfe anderer innovativer Methoden wie beispielsweise des 18F-Dopa-PET konnte gezeigt werden, dass der Verlust der postganglionären sympathischen Innervation spezifisch für den Herzmuskel ist, weil zum Beispiel Leber, Milz und Nasenschleimhaut keine veränderten 18F-Dopa-Konzentrationen aufweisen [11].
Damit zeigt sich beim idiopathischen Parkinson-Syndrom eine relevante Schädigung der postganglionären sympathischen Efferenzen als wesentliche Ursache der Dysautonomie. Die postganglionären Läsionen treten bei Patienten mit Multisystematrophie nicht auf, so dass die Darstellung einer kardialen sympathischen Denervierung auf das Vorliegen eines idiopathischen Parkinson-Syndrom hinweist. Die relativ häufig in Kardiologie und Diabetologie angewandte und deshalb breit verfügbare MIBG-Szintigraphie kann also somit als zurzeit validester Test zur Differenzierung zwischen idiopathischen Parkinson-Syndrom und Multisystematrophie angesehen werden. Parallel zur kardialen Denervierung finden sich bei idiopathischen Parkinson-Syndrom-Patienten auch histologisch Auffälligkeiten mit Nervenzellverlust und Lewy-Körperchen in peripheren Anteilen des sympathischen Nervensystems wie beispielsweise in den sympathischen Grenzstrangganglien oder ein Verlust an Tyrosinhydroxylase-Färbung im Epikardbereich [11]. Darüber hinaus konnte neuroendokrinologisch bei IPS-Patienten ein signifikant erniedrigter Noradrenalin-Spiegel im Serum nachgewiesen werden ebenso wie eine postganglionäre Denervierungshypersensitivität adrenerger Neuronen, die sich zum Beispiel in einer verstärkten Blutdruckantwort auf eine definierte applizierte Noradrenalin-Menge zeigt [11].
Daraus ergibt sich eine neue pathophysiologische Klassifikation von Störungen mit primärer Dysautonomie, wobei IPS-Patienten in charakteristischer Weise eine postganglionäre, sympathische noradrenerge Dysfunktion aufweisen (Tab. 1).
Tab. 1. Schematische Darstellung der klinischen Hauptsymptome bei primär chronischen autonomen Erkrankungen [nach 11]
|
Erkrankung |
Präganglionäre Dysautonomie |
Postganglionäre Dysautonomie |
Extrapyramidale Funktionsstörung |
Zerebelläre und/oder pyramidale Funktionsstörung |
|
Multisystematrophie – Parkinson-Typ MSA-P |
|
|
||
|
Multisystematrophie – Kleinhirn-Typ MSA-C |
|
|
||
|
Multisystematrophie – gemischter Typ MSA-M |
|
|
|
|
|
Idiopathisches Parkinson-Syndrom IPS |
|
|
||
|
Reines autonomes Versagen (PAF) |
|
Kardiovaskuläres System
Unter den kardiovaskulären Störungen ist vor allem die orthostatische Hypotension von klinischer Relevanz. So klagen etwa 50 % der IPS-Patienten über die prinzipiell unspezifischen Symptome einer orthostatischen Hypotonie wie Schwindel, Benommenheit, Kopfleere, Übelkeit, zumeist jeweils im Gehen oder Stehen [10]. Die orthostatische Hypotonie resultiert aus einer Störung der sympathisch-noradrenergen Innervation der kardiovaskulären Zielorgane [11]. Neben der primären Dysautonomie kommen als weitere Ursachen eine unzureichende Flüssigkeitsaufnahme sowie medikamentöse Nebenwirkungen gerade der Dopamin-Agonisten und von Selegilin in Frage [4]. Die Hypotonieneigung kann durch Nahrungsaufnahme verstärkt werden mit etwa 30 bis 50 Minuten danach verstärktem postprandialem Blutdruckabfall. Des Weiteren kann in der Erholungspause nach körperlicher Belastung bei IPS-Patienten eine charakteristische Postbelastungshypotonie auftreten.
Nach der Definition der American Autonomic Society wird die orthostatische Hypotension operational definiert durch einen anhaltenden Abfall des systolischen Blutdrucks um ≥ 20 mmHg bzw. des diastolischen Blutdrucks um ≥ 10 mmHg innerhalb von drei Minuten nach dem aktiven Hinstellen oder dem passiven Aufrichten auf dem Kipptisch [12]. Die orthostatische Hypotonie kann asymptomatisch sein, wenn der Patient dabei keine Symptome entwickelt, oder symptomatisch, wenn es zur Entwicklung von beispielsweise Schwindel, Schwäche, Übelkeit, Schmerzen oder Verschwommensehen kommt. Robertson hat deshalb einen für die klinische Diagnostik sehr praktischen Parameter vorgeschlagen, die sog. „Stehzeit“ oder „standing time“, das heißt die Zeit, die nach aktivem Aufstehen vergeht, bis ein Patient durch orthostatische Symptome gezwungen wird, sich wieder zu setzen. Patienten sind nach Robertson in den Aktivitäten ihres täglichen Lebens deutlich behindert, wenn die „standing time“ weniger als 30 Sekunden beträgt, während eine „standing time“ von mehr als einer Minute zumeist ein unabhängiges Leben erlaubt.
Ist die orthostatische Hypotonie symptomatisch und kann mit vermehrter Flüssigkeitszufuhr und Modifikation der Parkinson-Medikation keine ausreichende Symptomverbesserung erreicht werden, stehen verschiedene nicht-medikamentöse und medikamentöse Therapieverfahren zur Verfügung, die in Tabelle 2 dargestellt sind. Diese werden je nach Durchführbarkeit und Akzeptanz individuell in Abhängigkeit der Schwere der orthostatischen Hypotonie – eventuell in Kombination – eingesetzt. Klagen Patienten vor allem über eine postprandiale Hypotonie, empfehlen sich mehrere kleine Mahlzeiten mit einer gleichmäßigen Verteilung der Kohlehydrateinnahme.
Tab. 2. Nichtmedikamentöse und medikamentöse Therapie der orthostatischen Hypotonie
|
Nichtmedikamentöse Maßnahmen |
|
Vermeiden eines plötzlichen Wechsels der Körperposition, von langem Liegen sowie von Situationen, die zu einer Vasodilatation der Hautgefäße führen (z. B. heiße Bäder) |
|
Einnahme kochsalzreicher Diät (3–6 g NaCl) und gleichzeitig Aufnahme von 2 bis 3 l Flüssigkeit täglich |
|
Nahrung mit eher geringem Kohlenhydratanteil; keine einzelne große Mahlzeit, sondern eher in kleinen Portionen über den Tag verteilt |
|
Leichte körperliche Aktivität (eher isotonisch) wie zum Beispiel Schwimmen, Aerobic-Training, Fahrradfahren oder Gehen ohne anstrengende körperliche Übungen |
|
Verwendung von Gegenmanövern wie beispielsweise Hinhocken oder „Derbychair“-Verwendung |
|
Tragen elastischer Strümpfe oder eines elastischen Anzugs |
|
Nachtschlaf mit erhöhtem Oberkörper (bis zu 15 bis 30 cm) |
|
Medikamentöse Maßnahmen |
|
Erhöhung des Blutvolumens |
|
Fludrocortison zu Anfang 0,1–0,2 mg/Tag; bis maximal 1 mg/Tag. Cave: Herzinsuffizienz, Hypokaliämie, Ödeme |
|
Erythropoetin 4 000 I.E. s. c. zweimal/Woche. Cave: ausreichende Eisensubstitution, Hämatokrit-Anstieg, Hypertonie |
|
Desmopressin als Nasenspray besonders bei Nykturie. Cave: Hyponatriämie, Hypertonie |
|
Steigerung der peripheren Vasokonstriktion |
|
Midodrin dreimal 2,5–10 mg/Tag, bis maximal 40 mg/Tag; letzte Gabe um 17 Uhr. Cave: Hypertonie im Liegen, Pruritus |
|
Ephedrin dreimal 12,5–25 mg/Tag. Cave: Tachykardie, Tremor, Hypertonie im Liegen |
|
Yohimbin zwei- bis dreimal 8 mg/Tag p. o. Cave: Diarrhö, Nervosität, Angstzustände |
|
Coffein 250 mg (= 2 Tassen Kaffee) morgens. Cave: Tachyphylaxie |
|
Verschiedenes |
|
Methylphenydat zweimal 5–10 mg/Tag p. o.; letzte Gabe vor 18 Uhr. Cave: Agitation, Tremor, Schlaflosigkeit |
|
Metoprolol 12,5–100 mg/Tag p. o. bzw. Pindolol bei Bradykardie zwei- bis dreimal 2,5–5,0 mg/Tag p. o. Cave: Hypotonie, Bradykardie, Herzinsuffizienz |
|
Clonidin zweimal 0,1–0,3 mg/Tag p. o. oder ein Pflaster pro Woche. Cave: Mundtrockenheit, Bradykardie, Hypotonie |
|
Dihydroxyphenylserin (DOPS) zweimal 250–500 mg p. o.; vor allem bei Dopamin-beta-Hydroxylase-Defizienz |
Neben der orthostatischen Hypotonie kann außerdem quasi paradox im Liegen eine so genannte „supine hypertension“ bei IPS-Patienten mit ausgeprägter Dysautonomie sowie vor allem bei MSA-Patienten auftreten, die sich beispielsweise in einem inversen Verhalten des Blutdrucks über den Tag-Nacht-Verlauf hin äußern kann [11]. In Abbildung 2 findet sich das Beispiel eines IPS-Patienten mit ausgeprägtem Blutdruckanstieg während der Nachtphase, in der der Blutdruck normalerweise abnimmt. Dieses Faktum muss bei der Behandlung der orthostatischen Hypotonie dringend beachtet werden. So darf die blutdrucksteigernde Medikation nicht später als 17 Uhr eingenommen werden. Es kann sogar nötig werden, neben einer blutdrucksteigernden Therapie tagsüber im Gegensatz dazu nachts eine blutdrucksenkende Therapie (am besten mit Clonidin) einzuleiten, um die zum Teil ausgeprägten nächtlichen Blutdruckanstiege zu vermeiden.
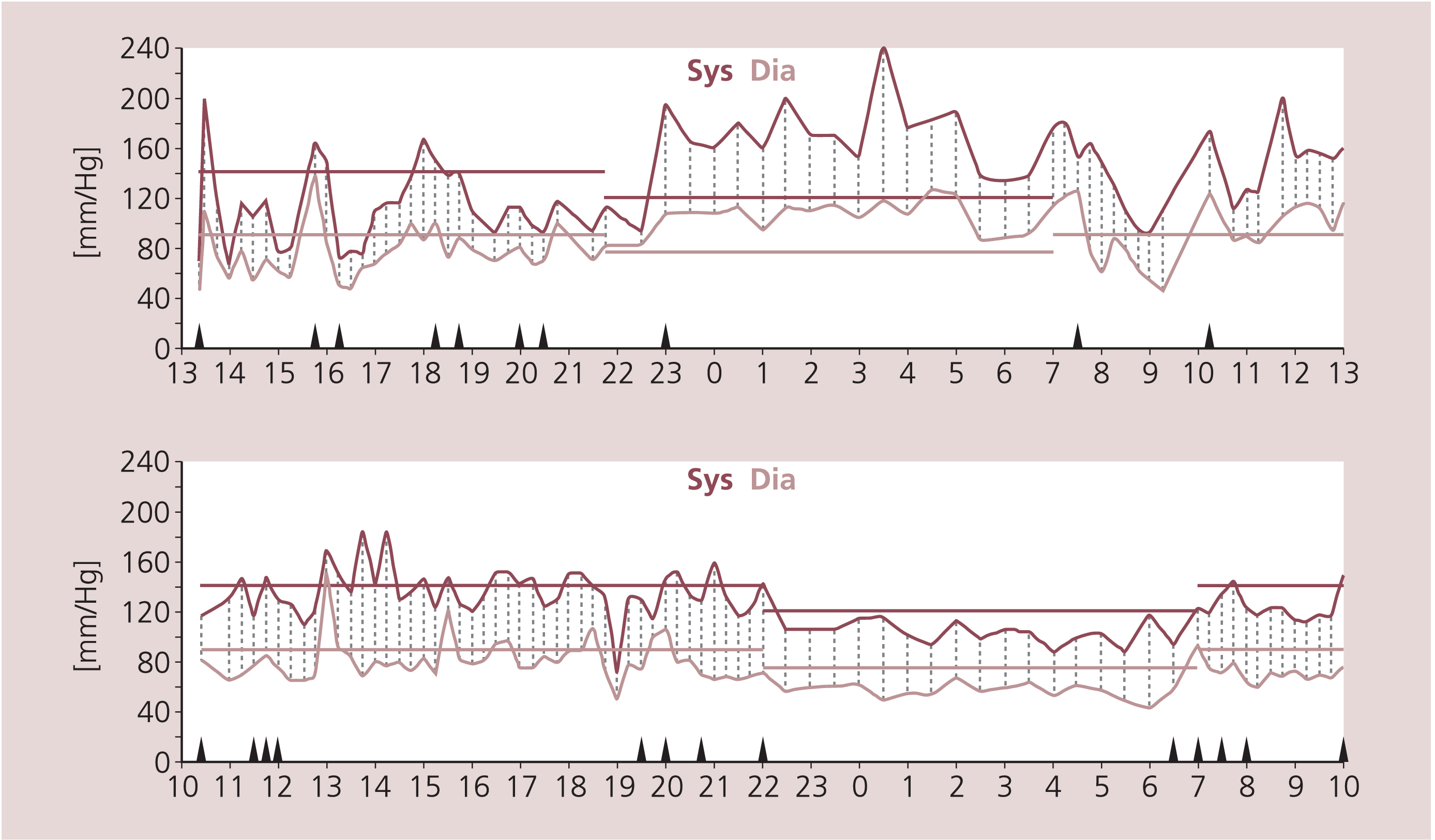
Abb. 2. Pathologisches 24-h-Blutdruckprofil beim IPS-Patienten mit nächtlichem Blutdruckanstieg und wiederholten hypotonen Phasen (oben) im Vergleich zu einem physiologischen Profil mit Nachtabsenkung (unten)
Gastrointestinales System
Gastrointestinale Funktionsstörungen spielen eine zentrale Rolle unter den charakteristischen autonomen Funktionsstörungen bei IPS-Patienten, wobei es zu Überschneidungen mit den Symptomen der Akinese kommt [13]. Die Funktionsstörungen können dabei den gesamten Gastrointestinaltrakt betreffen. Ursächlich dürften neurodegenerative Veränderungen in den gastrointestinalen Plexus mit Neuronenverlusten und Nachweis von Lewy-Körperchen im Bereich des oberen Ösophagus bis hin zum Rektum sein.
Oropharyngeale Dysfunktion
Die klinisch häufig auffällige Hypersalivation (70 bis 80 % aller IPS-Patienten) ist Folge einer Schluckstörung und nicht einer erhöhten Speichelproduktion [14]. Die Speichelproduktion selbst ist bei IPS-Patienten sogar eher vermindert im Vergleich zu entsprechenden Vergleichskollektiven. Weil die ursächlich zugrunde liegende Schluckstörung therapeutisch nur unbefriedigend angegangen werden kann, werden Anticholinergika zur Reduktion der Speichelproduktion eingesetzt, die entsprechende systemische Nebenwirkungen wie Konstipation, Harnretention, Gedächtnisprobleme und sogar Halluzinationen hervorrufen. In zwei Studien konnte ein positiver Effekt einer intraparotidalen Botulinumtoxin-Injektion gezeigt werden, auch hier müssen jedoch mögliche Komplikationen (z. B. trockener Mund, mögliche Injektionsfehler, Muskelschwäche) beachtet werden [13]. Nach unserer Erfahrung spielen Sprachtherapie, praktische Hilfen (Kaugummikauen zur Erhöhung der Schluckfrequenz) sowie eine Optimierung der motorischen Funktion durch geeignete Medikamente eine entscheidende Rolle.
Die Schluckstörung selbst (bis zu 95 % aller IPS-Patienten) nimmt ihren Ursprung bereits in einem unzureichenden Kauprozess. Aufgrund einer verminderten Peristaltik und einer inadäquaten Koordination der einzelnen muskulären Komponenten des Schluckakts kommt es zur so genannten oropharyngealen Dysphagie, die neben einer chronischen Laryngitis als schlimmste Komplikation zu einer Aspiration führen kann [15], die bei etwa 15 bis 56 % der Patienten beschrieben wurde [13]. Im Gegensatz zu diesen alarmierenden Zahlen kommt es nur selten zur Entwicklung einer Aspirationspneumonie, vor allem bei den zu Hause lebenden IPS-Patienten. Auch im Bereich der Speiseröhre selbst werden beim IPS-Patienten vielfältige Funktionsstörungen wie beispielsweise ein verlangsamter ösophagealer Transit, segmentale Spasmen oder eine Aperistaltik, eine ösophageale Dilatation oder ein insuffizienter Sphinkter beschrieben.
Therapeutisch lassen sich die Schluckstörungen nur schlecht beeinflussen. Eine dopaminerge oder anticholinerge Medikation zeigt nur inkonsistente Ergebnisse, in Einzelfällen kann eine Bougierung oder Injektion von Botulinumtoxin im Bereich der Sphinkter in Frage kommen. In Anbetracht der sekundären Komplikationen sollte rechtzeitig die Anlage eines perkutanen endoskopischen Gastrostomas (PEG) erwogen werden.
Gastrointestinale Dysfunktion
Störungen der Magenentleerung bis hin zur Gastroparese führen bei IPS-Patienten zu subjektiven Beschwerden wie Sodbrennen, Übelkeit, Völlegefühl und Appetitlosigkeit [13–15]. Neben der verminderten Nahrungsaufnahme hat die verzögerte Magenentleerung entscheidende pharmakokinetische Implikationen, weil ein verzögerter Levodopa-Transport in den Dünndarm, den Ort der Levodopa-Resorption, einen verzögert einsetzenden oder auch ganz ausbleibenden Medikamenteneffekt zur Folge hat. Diese Motilitätsstörung ist damit mitverantwortlich für die On-off-Symptomatik.
Während Metoclopramid wegen seiner zentralen Wirkung mit Verschlechterung der Parkinson-Symptomatik kontraindiziert ist, kommt als peripherer Dopamin-Antagonist Domperidon (Motilium®) in Dosierungen von 3-mal 10 bis 20 mg zum Einsatz [16]. Domperidon führt zu einer beschleunigten Magenentleerung und somit Bereitstellung von Levodopa im Dünndarm zur Resorption und Erhöhung des Levodopa-Plasmaspiegels. Darüber hinaus kann Domperidon als Zusatzmedikation einer Levodopa-Therapie die peripheren Nebenwirkungen wie beispielsweise die Übelkeit vermindern. Das ebenfalls wirksame Cisaprid wird aufgrund seiner möglichen Kardiotoxizität in den meisten Ländern nicht mehr eingesetzt [13].
Die Obstipation gilt seit der Erstbeschreibung als häufiges Problem bei IPS-Patienten (60–70 %), oft schon vor Beginn der Erkrankung [17]. Eine aktuelle Studie aus Hawaii berichtete gar von einem um den Faktor 2,7 bzw. 4 erhöhten Risiko, an idiopathischen Parkinson-Syndrom zu erkranken, wenn man weniger als einmal Stuhlgang pro Tag habe im Vergleich zu Personen mit einmaligem bzw. mehrmaligem Stuhlgang pro Tag [18]. Das pathophysiologische Korrelat der Obstipation scheint die verlängerte Transitzeit im Kolon zu sein, die im Verlauf der Erkrankung sogar noch zunehmen soll. Die Obstipation kann bis hin zu einem Megakolon, zur Pseudoobstruktion, zum Volvulus, Ileus oder einer Perforation führen. Therapeutisch sollte aufgrund der schlechten Studienlage eine eskalierende Therapie zum Einsatz kommen. Zunächst empfehlen sich eine ballaststoffreiche Kost, die Gabe von Plantago afra oder ovata, ein Absetzen der anticholinergen Medikation sowie ausreichend Bewegung, Flüssigkeitsaufnahme und Physiotherapie. Bei ausgeprägteren Fällen mit Passagezeiten von mehreren Tagen müssen zusätzliche medikamentöse Maßnahmen zum Einsatz kommen: Während sich die gute Wirksamkeit von Prokinetika am oberen Gastrointestinaltrakt bei der Obstipation nicht bestätigt hat, lassen sich zur Zeit die besten Erfolge mit Macrogol oder anderen Laxanzien erzielen [13].
Anorektale Dysfunktion
Auch der Defäkationsprozess ist aufgrund einer abdominopelvinen Dyssynergie im Rahmen des idiopathischen Parkinson-Syndrom pathologisch verändert. Es kann zum so genannten Anismus kommen, das heißt unwillkürlichen Kontraktionen des Analsphinkters im Sinne einer Dystonie [13]. Die Behandlung einer gestörten Defäkation ist schwierig, da herkömmliche Laxanzien die gestörte anorektale muskuläre Koordination nicht verbessern und eher zu einer Verschlechterung der gesamten Symptomatik führen können. Dopaminergika oder Apomorphin-Injektionen können im Einzelfall eine Verbesserung bewirken.
Urogenitales System
Urogenitale Funktionsstörungen sind ein häufiges Symptom bei bis zu 93 % der IPS-Patienten, wobei imperativer Harndrang, Pollakisurie und Harninkontinenz zu den häufigsten Symptomen gehören [19, 20]. Blasenentleerungsstörungen haben eine entscheidende soziale Bedeutung für IPS-Patienten. Zum einen stören sie den Nachtschlaf, zum anderen erschweren sie ein freies öffentliches Leben der Patienten. Die Blasenstörungen können einerseits unabhängig von der Primärerkrankung, andererseits als Folge der verabreichten Medikation oder der Primärerkrankung auftreten [4]. Die Therapie muss in Kooperation mit einem Urologen nach ausführlicher neurourologischer Diagnostik erfolgen. Ziel der Therapie ist eine kontrollierte Blasenentleerung, kein unwillkürlicher Harnverlust sowie eine komplette Blasenentleerung. Neben den oben beschriebenen Medikamenten kommt dabei insbesondere dem intermittierenden Selbstkatheterismus eine wichtige Rolle zu.
Bei der Beurteilung der Blasenfunktion kommt besonders dem Austreibungs- und dem Schließmuskel eine besondere Bedeutung zu.
Detrusorfunktion
Die meisten urodynamischen Studien beschreiben eine Überaktivität des Blasenaustreibungsmuskels, des M. detrusor vesicae, das heißt, es kommt bereits bei kleinen Füllungsdrücken zu unwillkürlichen Kontraktionen des Austreibungsmuskels mit Miktionseinleitung und eventuell sogar Urinabgang [21]. Das Auftreten einer Detrusorhyperreflexie wird mit der fehlenden Inhibition der Blasenentleerung durch die gestörten Basalganglien erklärt. Zur allgemeinen Verwirrung finden sich jedoch auch Studien, die eine Detrusorhyporeflexie zeigen [10]. Hier stellt sich die Frage, ob nicht vielleicht die Blase durch eine Obstruktion sekundär schlaff geworden ist oder ein möglicher Einfluss der Anticholinergika besteht. Insgesamt muss die Auswirkung eines obstruktiven Prozesses beispielsweise durch eine Prostatahypertrophie kritisch beurteilt werden und eine operative Maßnahme nach urodynamischer Evaluation gut überlegt sein. Für die Hemmung eines gesteigerten Detrusormuskels werden Anticholinergika wie beispielsweise Trospiumchlorid (z. B. Spasmex®) oder muskulotrop-anticholinerge Substanzen wie Oxybutynin (Dridase®), Flavoxat (Spasuret®) oder Imipramin (Tofranil®) eingesetzt. Zur Stimulation des Austreibungsmuskels können Cholinergika wie Carbachol oder Bethanechol (Myocholine-Glenwood®) oder Cholinesterasehemmer wie Physostigmin (Anticholium®) oder Neostigmin eingesetzt werden.
Sphinkterfunktion
Neben der jeweiligen Detrusoraktivität kann auf der anderen Seite der Harnröhrenverschluss durch den komplexen Sphinkterapparat normal, hyperaktiv oder hypoaktiv sein. Während ein hyperaktiver Verschlussmechanismus zu einer so genannten Detrusor-Sphinkter-Dyssynergie mit Blasenentleerungsstörungen bis hin zur so genannten Urge-Inkontinenz führt, bedeutet ein hypoaktiver Verschlussmechanismus die Inkontinenz. Zur Erweiterung des inneren Sphinkters können Substanzen wie Tamsulosin (z. B. Alna®), Alfuzosin (Urion®, UroXatral®), Phenoxybenzamin (Dibenzyran®) oder Doxazosin (z.B. Cardular®) eingesetzt werden.
Sexualfunktion
Störungen der Sexualfunktion im Sinne von Störungen der Libido, Erektion und Ejakulation sind häufig Probleme der Patienten mit Parkinson-Erkrankung [22]. Häufig ist die erektile Dysfunktion nicht nur bei der Multisystematrophie anzutreffen, sondern auch Frühsymptom des idiopathischen Parkinson-Syndroms. Uns allen sind Patienten im höheren Lebensalter bekannt, die wir durch Levodopa oder Dopamin-Agonisten-Therapie zu einer erhöhten Libido bei unveränderter Einschränkung der Erektionsfähigkeit führten, was zu besonderen psychischen Belastungen in der Familie dieser Patienten führt. Viele Patienten halten die Störung der Sexualfunktion für nicht besonders belastend, bei anderen wird Sildenafil (Viagra®) bei Ausschluss der wichtigen Kontraindikationen erfolgreich eingesetzt.
Sudomotorisches System
Bei einem Drittel bis zur Hälfte der IPS-Patienten liegt eine gesteigerte oder auch reduzierte Schweißsekretion vor [5, 23, 24]. Ein episodenhaftes starkes Schwitzen ohne Auslöser kommt bei bis zur Hälfte der Patienten vor, betrifft vorwiegend das Gesicht und den Oberkörper und tritt gehäuft in der Nacht auf [24]. Bei etwa zwei Drittel der Patienten treten diese Schweißanfälle interessanterweise in Assoziation mit einer schweren Akinese auf [25]. Bei den Parkinson-Patienten, die vielfach auch an einer gestörten Thermoregulation (z. B. Unwohlsein bei hohen Umgebungstemperaturen) leiden, scheint insbesondere das thermoregulatorische Schwitzen wahrscheinlich aufgrund einer hypothalamischen Störung gestört zu sein [4, 26].
Ob sich die sudomotorische Dysfunktion bei Parkinson-Patienten unter Parkinson-Medikation verbessert oder verschlechtert, wird in der Literatur unterschiedlich beurteilt [24]. Kommt es bei Patienten zu ausgeprägten, behandlungsbedürftigen Schweißsekretionsstörungen, können therapeutisch Anticholinergika wie beispielsweise Pirenzepin (z.B. Gastrozepin®) 2 x 50 mg oder nebenwirkungsärmer auch Salbeiextrakt 3 bis 4 x 100 mg erfolgreich eingesetzt werden. Anfallsartige Hyperhidrosen behandeln wir bevorzugt mit Betablockern wie zum Beispiel Propranolol (z.B. Dociton®). Bei seltenen fokalen Hyperhidrosen können topische Externa auf Aluminiumbasis oder eine Botulinumtoxin-Injektion eingesetzt werden.
Weitere autonome Funktionsstörungen verbergen sich unter Symptomen wie kalten Händen, Seborrhö, verminderter Tränensekretion, gestörter Pupillomotorik und -akkumulation, Atem- und Schlafstörungen [10].
Zusammenfassend soll noch einmal unterstrichen werden, dass das idiopathische Parkinson-Syndrom auch eine Erkrankung des autonomen Nervensystems darstellt. So hatte schon Lewy 1913 geschrieben: „Die Verschiedenheit des klinischen Bildes bei Sitz einer Erkrankung im Linsenkern muss wohl aus der Mitbeteiligung anderer Systeme, über die wir noch nicht genügend orientiert sind, resultieren“ [2]. Geben wir also diesen bisher vernachlässigten Systemen, über die wir gerade heute mehr und mehr wissen, die Beachtung, die sie aufgrund ihrer Bedeutung für Diagnose und Therapie unserer IPS-Patienten verdienen.
Literatur
1. Parkinson J. An essay in the shaking palsy. London: Sherwood, Neely, and Jones, 1817.
2. Lewy FH. Zur pathologischen Anatomie der Paralysis agitans. Dtsch Z Nervenheilk 1913;50:50–5.
3. Ziemssen T, Süß M, Reichmann H. Funktionsdiagnostik des autonomen Nervensystems – eine interdisziplinäre Herausforderung. Sächsisches Ärzteblatt 2001;8:363–79.
4. Jost WH. Autonome Regulationsstörungen beim Parkinson-Syndrom. Aachen: Shaker-Verlag, 1999.
5. Mesec A, Sega S, Kiauta T. The influence of type, duration, severity and levodopa treatment of Parkinson’s disease on cardiovascular autonomic responses. Clin Autonom Res 1993;3:339–44.
6. Meco H, Pratesi L, Bonifati V. Cardiovascular reflexes and autonomic dysfunction in Parkinson’s disease. J Neurol 1991;238:195–9.
7. Braak H, Del Tredici K, Rueb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurol Aging 2003;24:197–211.
8. Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol 2004;3:93–103.
9. Braune S, Reinhardt M, Schnitzer R, et al. Cardiac uptake of [123I]MIBG separates Parkinson’s disease from multiple system atrophy. Neurology 1999;53:1020-5.
10. Jost WH, Braune S. Autonome Regulationsstörungen beim idiopathischen Parkinson-Syndrom. Akt Neurol 2001;28(S3):S235–41.
11. Goldstein DS. Dysautonomia in Parkinson’s disease: neurocardiological abnormalities. Lancet Neurol 2003;2:669–75.
12. Low PA. Clinical autonomic disorders. Evaluation and management. Boston: Little Brown and Company, 1993.
13. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 2003;2:107–16.
14. Eady MJ, Tyrer JH. Alimentary disorder on parkinsonism. Australas Ann Med 1965;14:13–22.
15. Edwards L, Quigley EMM, Hofman R, Pfeiffer TF. Gastrointestinal symptoms in Parkinson disease: 18-month follow-up study. Mov Disord 1993;8:83–6.
16. Indo T, Ando K. Metoclopramide-induced parkinsonism. Arch Neurol 1982;39:494–6.
17. Martignoni E, Pacchetti C, Godi L, Micieli G, et al. Autonomic disorders in Parkinson’s syndrome. J Neural Transm 1995;45(Suppl):11–9.
18. Abbott RD, Petrovich H, White LH, et al. Frequency of bowel movements and future risk of Parkinson’s disease. Neurology 2001;57:456–62.
19. Aminoff MJ, Wilcox CS. Assessment of autonomic function in patients with a Pakinsonian syndrome. Br Med J 1971;4:80–4.
20. Hess CW, Enderli JB, Fröhlich-Egli F, Ludin HP. Neurogene Blasenfunktionsstörungen beim Morbus Parkinson. Nervenarzt 1987;58:55–60.
21. Fowler CJ. Urinary disorders in Parkinson’s disease and multiple system atrophy. Funct Neurol 2001;16:277–82.
22. Wermuth L, Stenager E. Sexual problems in young patients with Parkinson’s disease. Acta Neurol Scand 1995;91:453–5.
23. Mano Y, Nakamuro T, Takayanagi T, Mayer RF. Sweat function in Parkinson’s disease. J Neurol 1994;241:573–6.
24. Goetz CG, Lutge W, Tanner CM. Autonomic dysfunction in Parkinson’s disease. Neurology 1986;36:73–5.
25. Taly AB, Muthane UB. Involvement of peripheral nervous system in juvenile Parkinson’s disease. Acta Neurol Scand 1992;85:272–5.
26. Appenzeller O, Goss JE. Autonomic deficits in Parkinson’s syndrome. Arch Neurol 1971;24:50–7.
Dr. Tjalf Ziemssen, Dr. Claudia Schmidt, Autonomes und neuroendokrinologisches Funktionslabor (ANF), Neurologische Universitätsklinik Dresden, Fetscherstr. 74, 01307 Dresden, E-Mail: Ziemssen@web.de, Web: www.neuro.med.tu-dresden.de/anf
Dysautonomia in Parkinson’s disease
The non-motor features of Parkinson’s disease include as one important clinical component the derangement of autonomic function, which is quite predominant in the cardiovascular, gastrointestinal und urogenital autonomic subsystem. If there is severe autonomic failure at early stages of disease in patients with extrapyramidal disorders, multiple system atrophy has to be taken into account as an important differential diagnosis which can today be differentiated from Parkinson’s disease by new innovative techniques. Appropriate and symptom-oriented diagnosis and symptomatic treatment can greatly benefit the patient as part of an interdisciplinary approach to Parkinson’s disease.
Keywords: Parkinson’s disesase, autonomic dysfunction, multiple system atrophy, neurocardiology, dysautonomia
Psychopharmakotherapie 2005; 12(01)