Hans-Christoph Diener und Richard Dodel, Essen*
In der Pathophysiologie der Alzheimer-Erkrankung spielen Beta-Amyloid-Plaques und Neurofibrillen (Tau-Protein) eine zentrale pathogenetische Rolle. Aus diesem Grund wurden vor einiger Zeit Impfstoffe gegen Beta-Amyloide (Aβ) entwickelt, nach deren Gabe es tatsächlich zu einem Rückgang der Aβ-Ablagerungen im Gehirn kam. Da im Rahmen der aktiven Immunisierung einige der so behandelten Patienten eine Meningoenzephalitis entwickelten, wurde dieser Therapieansatz weitgehend verworfen [9a]. Parallel zur Entwicklung einer aktiven Immunisierung wurden monoklonale Antikörper gegen unterschiedliche Epitope von Aβ entwickelt und untersucht. Ein weiterer innovativer Ansatz sind Tau-gerichtete Antisense-Oligonukleotide [18] (siehe Ausblick).
Im Folgenden werden die neueren Ergebnisse der Studien zum Einsatz der monoklonalen Antikörper gegen Aβ dargestellt und diskutiert, welche Implikationen sich für die Behandlung von Patienten mit beginnender Alzheimer-Erkrankung ergeben.
Amyloid-Hypothese der Alzheimer-Erkrankung
Das Gehirn von Patienten mit Alzheimer-Erkrankung enthält eine extrazelluläre Anhäufung von Aβ-Plaques, die aus Aβ-Peptiden bestehen und eine intraneuronale Anhäufung von neurofibrillären Tangles (NFT), die hauptsächlich aus aggregierten Formen von hyperphosphoryliertem Tau-Protein bestehen (Abb. 1). Durch die Spaltung des Amyloid-Vorläuferproteins (APP) mittels Beta- und Gamma-Sekretasen entsteht das Aβ-Peptid. Das Peptid kann in unterschiedlichen Längen vorliegen und am N- bzw. C-terminalen Ende verändert sein. Die für die Alzheimer-Demenz relevantesten Peptide sind 42 bzw. 40 Aminosäuren lang und zeigen ein unterschiedliches Aggregationsverhalten. Aβ-Monomere aggregieren in verschiedenen wenig stabilen Zuständen, darunter Oligomere, Protofibrillen und Amyloid-Fibrillen. Amyloid-Fibrillen sind größer und unlöslich und können sich weiter zu Amyloid-Plaques zusammenlagern, wohingegen Amyloid-Oligomere löslich sind und sich im Gehirn verteilen können.
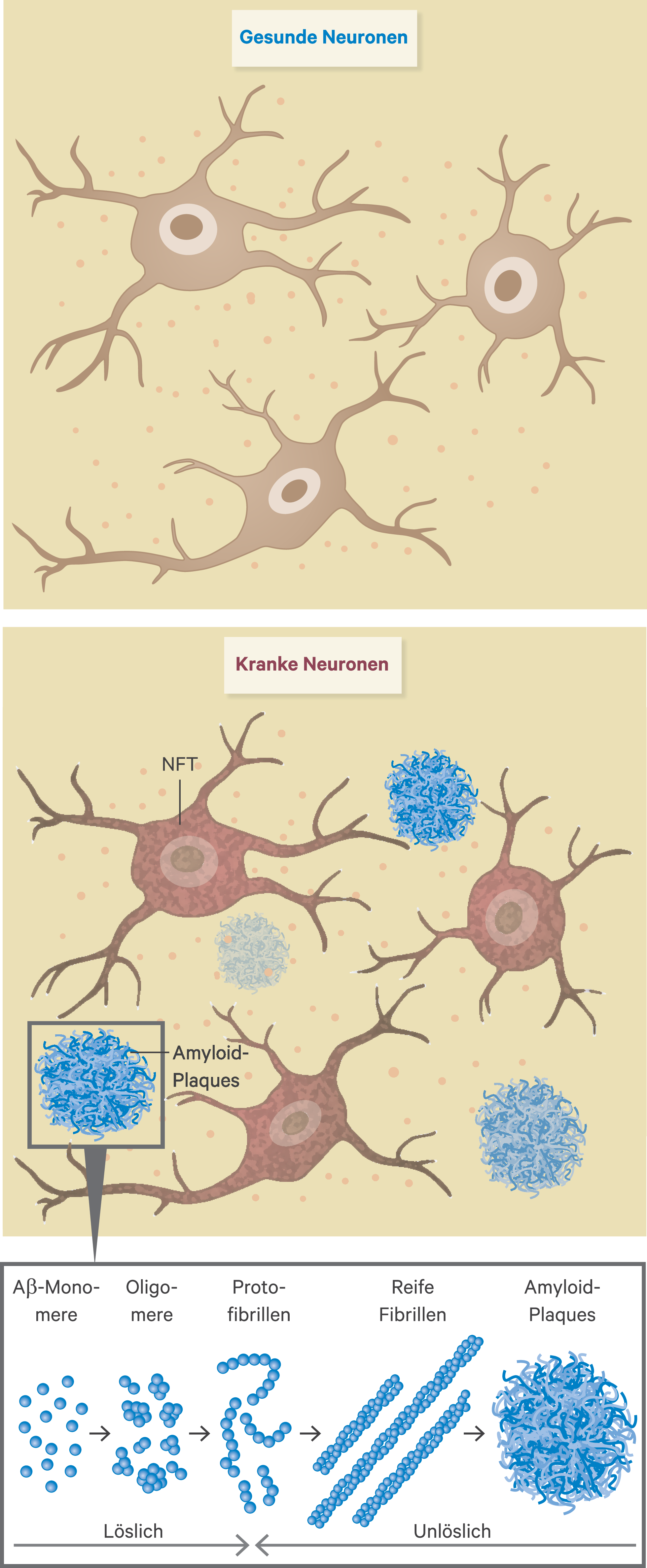
Abb. 1. Amyloid-Hypothese der Alzheimer-Erkrankung. Bei Patienten mit Alzheimer-Erkrankung liegt eine Anhäufung von Amyloid-Plaques und intrazellulären neurofibrillären Tangles (NFT) vor. Die kleinste Einheit der Amyloid-Plaques sind Aβ-Monomere, die zu weiteren löslichen Formen, darunter Oligomere und Protofibrillieren aggregieren, bis hin zu den größeren und unlöslichen Amyloid-Fibrillen und -Plaques. (modifiziert nach [Alzheimer Forschung Initiative e. V.] und [23a])
Während sich Aβ1–42-Peptide offenbar am frühesten im Gehirn anreichern und ihre freie Konzentration im Liquor lange vor dem Auftreten klinischer Symptome abnimmt, kann diese ursprüngliche Spezies im Laufe der Zeit in eine komplexe Reihe von verkürzten, isomerisierten und/oder phosphorylierten Peptiden modifiziert werden.
Das p3E-Peptid
Eine gut untersuchte Peptidvariante, die stark amyloidogen ist, ist das „p3E“-Aβ-Peptid; es wird an Position Asp(Aspartat)-1 und Ala(Alanin)-2 trunkiert sowie an Glu(Glutamat)-3 zyklisiert. In hAPP(humanes APP)-transgenen Mäusen konnte gezeigt werden, dass sich diese Variante erst relativ spät und in geringen Mengen anreichert [7]. Der gezielte Abbau mit einem spezifischen Antikörper fördert jedoch eine Art „Bystander-Clearance“ von auch früher abgelagerten Aβ-Spezies durch Mikroglia, was p3E trotz seiner geringen Häufigkeit zu einem attraktiven Ziel für die Aβ-Immuntherapie (siehe Donanemab, Remternetug) macht. Am C-terminalen Ende von Aβ ist die Variante Aβ43 sehr anfällig für eine Aggregation, und es ist unklar, wie lange lösliche Oligomere dieses Peptids als solche im Gehirn vorhanden sein können, bevor sie sich als unlösliche Amyloid-Plaques ablagern. Ein „Seeding“-Mechanismus der verschiedenen Aβ-Varianten wird als Voraussetzung für die Aggregation und Plaquebildung angenommen.
Die Amyloid-Kaskaden-Hypothese
Ergebnisse aus z. B. genetischen Studien (Mutationen im APP-Gen, Präsenilin-Genen) weisen stark darauf hin, dass die Aβ-Aggregation bestimmend für den Krankheitsprozess ist. Diese frühen Beobachtungen führten zur „Amyloid-Kaskaden-Hypothese“, die besagt, dass die Anhäufung von Amyloid-Plaques aus einem Ungleichgewicht zwischen der Produktion, Aggregation und Clearance von Aβ resultiert und den Krankheitsprozess auslöst [24]. Die Anhäufung von Aβ-Aggregaten begünstigt dann die Bildung von intrazellulären NFT, wodurch die Krankheit fortschreitet. Diese Erkenntnisse haben zu therapeutischen Strategien geführt, die Inhibitoren oder Modulatoren von Beta- und Gamma-Sekretasen bzw. Aktivatoren der Alpha-Sekretase umfassen, um die Aβ-Produktion zu hemmen oder die Aβ-Clearance zu fördern. Letzteres ist das Ziel der aktiven und passiven Immunisierung gegen Aβ.
Aβ-Clearance und Immunisierung
Der Ansatz der aktiven Immunisierung wurde mit wenigen Ausnahmen weitestgehend verlassen. Stattdessen wurden passive Immunisierungsansätze intensiv verfolgt. Verschiedene Epitope und unterschiedliche Aggregationen des Aβ wurden für die weitere Entwicklung der monoklonalen Antikörper differenziert. Die derzeit gängigen Antiköper und deren Zielstrukturen sind in Tabelle 1 zusammengestellt. Alle derzeit wirksamen Antikörper sind gegen das N-terminale Ende von Aβ gerichtet.
Tab. 1. Eigenschaften der Antikörper gegen Aβ
|
Substanz |
Zulassungsstatus |
Antikörper (Basis) |
Epitope |
Zielstruktur |
|
Aducanumab |
Z: USA Z: in EU abgelehnt |
Humanisierter IgG1 |
N-terminal (Aβ3–7) |
Oligomere oder fibrilläre Aggregate |
|
Donanemab |
EZ: USA EZ: EU |
Humanisierter IgG1 (mE8-IgG2a) |
N-terminal trunkiertes Aβ an Position 3 Glutamat, das durch Glutaminylzyklasen |
Plaques (AβP3–42) |
|
Lecanemab |
Z: USA EZ: EU |
Humanisierter IgG1 (mAb158) |
N-terminal |
Aβ-Oligomere und Protofibrillen |
|
Remternetug |
KP3 (B:2022/E:2026) |
Humanisierter IgG1 |
N-terminal trunkiertes Aβ an Position 3 Glutamat, das durch Glutaminylzyklasen |
Plaques (AβP3–42) |
Aβ: Beta-Amyloid; B: Beginn; E: Ende; EZ: eingereicht zur Zulassung; KP: klinische Studienphase; Z: Zulassung
Es gibt eine Reihe von Hypothesen zur Clearance von Aβ im Gehirn, die weitgehend davon abhängen, ob die Antikörper tatsächlich in das zentrale Nervensystem eindringen und dort ihre Wirkung entfalten, oder ob die in der Peripherie vorhandenen Antikörper ausreichen, um eine positive Wirkung zu erzielen („Peripheral-sink“-Hypothese, die erstmalig von Steven Paul und David Holtzman im Rahmen der Ergebnisse des Antikörpers m266 [„Solanezumab“] formuliert wurde). Bei der „Peripheral-sink“-Hypothese wird das Aβ durch den Antikörper in der Peripherie sequestriert, was die Konzentration von freiem Aβ im Blut senkt und zu einem Netto-Efflux von Aβ aus dem zentralen Nervensystem führt. Dies konnte insbesondere im Tiermodell nach Gabe des Antikörpers m266 gezeigt werden. Bei der zentralen Hypothese geht man davon aus, dass die Antikörper über die Blut-Hirn-Schranke hinweg in das Gehirn eindringen (etwa 0,1 % bis 0,2 % der IgG im Plasma sind im Liquor nachweisbar) und dort an die Plaques binden oder eine mikrogliaabhängige Phagozytose von Aβ bzw. eine Fc-abhängige phagozytische Clearance von Aβ-Plaques durch Mikroglia induzieren können. Neben diesen Ergebnissen wurde die Wirksamkeit von Anti-Aβ-Antikörpern ohne Effektor-Funktion nachgewiesen [2a, 3a]. Dieses Phänomen deutet darauf hin, dass neben der FcRγ-vermittelten mikroglialen Clearance mehr als ein Mechanismus der lokalen Clearance im Spiel sein könnte, wie etwa die direkte Auflösung von Plaques oder die Veränderung des Gleichgewichts zwischen Plaques und löslichen Aβ-Spezies. Derzeit geht man davon aus, dass nach Immunisierung mehrere der dargestellten Mechanismen eine Rolle in der Clearance von Aβ-Spezies spielen können.
Wirkungsmechanismus der monoklonalen Antikörper
Nach dem Scheitern der Studien zur aktiven Immunisierung gegen Aβ wurden Konzepte zu einer passiven Immuntherapie mit humanisierten monoklonalen Antikörpern entwickelt, um die Aβ-Clearance zu fördern. Die passive Immuntherapie gewährleistet relativ konstante Antikörpertiter, geht jedoch häufig mit einem vasogenen Ödem, einer zerebralen Amyloidangiopathie mit Mikroblutungen und anderen unerwünschten Reaktionen wie einer kortikalen Siderose einher. Die Mechanismen der passiven Immunisierung umfassen die Opsonisierung des Antigens durch den Antikörper, die eine Phagozytose durch Makrophagen und eine Komplementaktivierung verursacht. Die Antikörper hemmen die katalysierte Modifikation der Sekundärstruktur von Aβ-Monomeren und reduzieren so die Bildung von Oligomeren oder Fibrillen [26].
Klinische Studien zu Antikörpern mit Wirksamkeitsnachweis
Im Folgenden werden die Studien mit Wirksamkeitsnachweis eines monoklonalen Antikörpers gegen Aβ dargestellt (zusammengefasst in Tab. 2 und 3), gefolgt von den Studien ohne Nachweis der Wirksamkeit.
Tab. 2. Übersicht der Phase-III-Studien für monoklonale Antikörper mit Wirksamkeitsnachweis
|
Wirkstoff |
Studienregisternummer |
Teilnehmerzahl |
Interventionen |
Primärer Endpunkt |
Ergebnis primärer Endpunkt |
|
Aducanumab |
NCT02484547 |
n = 1638 |
Niedrigdosiertes Hochdosiertes Placebo 1 : 1 : 1-Randomisierung |
Veränderung des CDR-SB-Scores (Post-hoc-Analyse nach Abbruch aufgrund von Futility) |
Differenz niedrigdosiertes Aducanumab vs. Placebo: –0,26 (95%-KI –0,57 bis 0,04; p = 0,090) Differenz hochdosiertes Aducanumab vs. Placebo: –0,39 (95%-KI –0,69 bis –0,09; p = 0,012) |
|
NCT02477800 |
n = 1647 |
Placebo Niedrigdosiertes Aducanumab (i. v.) Hochdosiertes Aducanumab (i. v.) 1 : 1 : 1-Randomisierung |
Veränderung des CDR-SB-Scores (Post-hoc-Analyse nach Abbruch aufgrund von Futility) |
Differenz niedrigdosiertes Aducanumab vs. Placebo: –0,18 (95%-KI –0,47 bis 0,11; p = 0,225) Differenz hochdosiertes Aducanumab versus Placebo: 0,03 (95%-KI –0,26 bis 0,33; p = 0,833) |
|
|
Lecanemab |
NCT03887455 |
n = 1795 |
Lecanemab (i. v.) Placebo 1 : 1-Randomisierung |
Veränderung des CDR-SB-Scores nach 18 Monaten |
Differenz Lecanemab vs. Placebo: |
|
Donanemab |
NCT04437511 |
n = 1736 |
Donanemab (i. v.) Placebo 1 : 1-Randomisieurng |
Veränderung auf der iADRS nach 76 Wochen |
Differenz Donanemab vs. Placebo bei niedriger/mittlerer Tau-Pathologie: Differenz Donanemab vs. Placebo in der kombinierten Studienpopulation: |
|
Remternetug |
NCT05463731 |
n = 600 |
Remternetug (s. c.) Remternetug (i. v.) Placebo |
Prozentsatz der Patienten, bei denen die Amyloid-Plaques nach 52 Wochen abgebaut sind |
Wird für 2025/2026 erwartet |
CDR-SB: Clinical Dementia Rating Sum of Boxes; iADRS: integrierte Alzheimer Disease Rating Scale; i. v.: intravenös; KI: Konfidenzintervall; s. c.: subkutan
Tab. 3. Übersicht für die Verlangsamung der Krankheitsprogression in den klinischen Phase-III-Studien der Antikörper mit Wirksamkeitsnachweis. Gezeigt ist die Verbesserung des CDR-SB-Scores gegenüber dem Ausgangswert (ΔCDR-SB) von Verum (high dose und low dose) versus Placebo.
|
Antikörper |
Studie |
n (P/L/H) |
ΔCDR-SB-L |
ΔCDR-SB-H |
|
Aducanumab |
EMERGE [5] |
548/543/547 |
–0,26 |
–0,39 |
|
Aducanumab |
ENGAGE [5] |
545/547/555 |
–0,18 |
0,03 |
|
Lecanemab |
CLARITY-AD [30] |
875/–/859 |
– |
–0,45 |
|
Donanemab |
TRAILBLAZER [17] |
876/–/860 |
– |
–0,67 |
H: high dose; L: low dose; n: Anzahl der Patienten in den untersuchten Interventionsgruppen; P: Placebo
Aducanumab
EMERGE und ENGAGE waren zwei randomisierte, doppelblinde, Placebo-kontrollierte, globale Phase-III-Studien, die die Wirksamkeit von Aducanumab bei Patienten mit früher Manifestation der Alzheimer-Erkrankung untersuchten [5]. 1638 Patienten nahmen an der EMERGE-Studie und 1647 Patienten an der ENGAGE-Studie teil. Das Alter der Patienten betrug 50–85 Jahre. Einschlusskriterium war der Nachweis von Aβ im PET (Positronen-Emissions-Tomographie). Klinisches Einschlusskriterium war eine leichte kognitive Beeinträchtigung, definiert als ein Wert im Mini-Mental-Status-Test (MMST, Infokasten) ≥ 24 Punkte. Die Studie wurde an 348 Zentren in 20 Ländern durchgeführt. Die Patienten wurden im Verhältnis 1 : 1 : 1 randomisiert und mit einer niedrigen Dosis Aducanumab (3 oder 6 mg/kg), einer hohen Dosis (10 mg/kg) oder Placebo behandelt. Die Therapie erfolgte intravenös einmal im Monat über 76 Wochen.
Der primäre Endpunkt der Studien war die Veränderung auf der Clinical Dementia Rating Sum of Boxes (CDR-SB, Infokasten). Diese Skala misst die alltagsrelevanten Funktionen und die Kognition. Weitere Endpunkte waren Verträglichkeits- und Sicherheitsparameter wie ARIA (amyloid-related imaging abormalities/amyloidbezogene Bildgebungsanomalien) in der Kernspintomographie. Sekundäre Endpunkte umfassten die Kognition, Alltagsfunktion und Verhalten sowie Biomarker-Endpunkte wie die Amyloid- und Tau-Proteinlast im PET.
Die beiden Studien wurden aufgrund von „Futility“ – also einer nicht mehr vorhandenen Aussicht auf Erfolg – vom Sponsor abgebrochen, nachdem etwa die Hälfte der geplanten Patienen eingeschlossen wurde. In einer Post-hoc-Analyse wurde der primäre Endpunkt in der EMERGE-Studie erreicht. Die Verbesserung im CDR-SB betrug –0,39 Punkte für hochdosiertes Aducanumab vs. Placebo (95%-Konfidenzintervall [KI] –0,69 bis –0,09; p = 0,012). Die absolute Differenz betrug –22 Prozentpunkte. Der primäre Endpunkt war in der ENGAGE-Studie nicht signifikant. Der Unterschied betrug 0,03 Punkte im CDR-SB (95%-KI –0,26 bis 0,33; p = 0,833). Die Ergebnisse der Biomarkerstudien mit Messung von Amyloid- und Tau-Protein im PET zeigten eine dosisabhängige Reduktion mit Aducanumab. Die häufigsten unerwünschten Arzneimittelwirkungen waren amyloidbezogene zerebrale Ödeme im MRT (ARIA-E mit 29 % in EMERGE und 31 % in ENGAGE). Schwerwiegende ARIA-Symptome mit klinischer Manifestation waren selten, mit 1,4 % für die hohe Dosis und 0,6 % für die niedrige Dosis von Aducanumab sowie 0,2 % für die Placebo-Gruppe.
Erstaunlicherweise wurde Aducanumab von der FDA zur Therapie der beginnenden Alzheimer-Erkrankung zugelassen. Die Zulassung erfolgte aufgrund von Post-hoc-Analysen der beiden Phase-III-Studien und mit der Begründung, dass es positive Ergebnisse für die Surrogatparameter, eine Reduktion der Aβ-Plaques im PET, gab [9, 22]. Im Gegensatz zur FDA gab es ein negatives Urteil der europäischen Zulassungsbehörde. Vermutlich aufgrund der wenig überzeugenden Ergebnisse und der hohen Kosten wurde Aducanumab in den USA wenig eingesetzt. Die Firma Biogen hat im Mai 2022 mitgeteilt, dass sie die Vermarktung von Aducanumab einstellen wird.
Lecanemab
Lecanemab ist ein humanisierter monoklonaler Antikörper, der mit hoher Affinität an lösliche Aβ-Protofibrillen bindet, die nachweislich für Neurone toxischer sind als Monomere oder unlösliche Fibrillen. Eine Phase-IIb-Dosisfindungsstudie mit 854 Teilnehmern mit früher Alzheimer-Erkrankung zeigte allerdings keinen signifikanten Unterschied zwischen Lecanemab und Placebo in Änderungen eines zusammengesetzten Demenz-Scores nach 12 Monaten [28]. Weitere Analysen nach 18 Monaten zeigten aber eine dosis- und zeitabhängige Clearance von Amyloid mit Lecanemab im PET-CT, und das Arzneimittel war mit einer geringeren klinischen Verschlechterung bei einigen Demenzmesswerten verbunden als Placebo.
Lecanemab wurde in einer 18-monatigen, multizentrischen, doppelblinden Phase-III-Studie mit Personen im Alter von 50 bis 90 Jahren mit früher Alzheimer-Erkrankung (leichte kognitive Beeinträchtigung oder leichte Demenz aufgrund der Alzheimer-Erkrankung) mit Nachweis von Amyloid im PET oder durch Nachweis von Biomarkern im Liquor untersucht [30]. Die Studienteilnehmer wurden nach dem Zufallsprinzip im Verhältnis 1 : 1 einer intravenösen Lecanemab-Behandlung (10 mg pro Kilogramm Körpergewicht alle zwei Wochen) oder Placebo zugeteilt. Der primäre Endpunkt war die Veränderung der Punktzahl des CDR-SB gegenüber dem Ausgangswert nach 18 Monaten. Sekundäre Endpunkte waren die Veränderung der Amyloid-Belastung im PET, die Punktzahl auf der 14-teiligen kognitiven Unterskala der Alzheimer’s Disease Assessment Scale (ADAS-cog14), der Alzheimer’s Disease Composite Score (ADCOMS) und die Punktzahl auf der Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS-MCI-ADL) (Infokasten).
Insgesamt wurden 1795 Teilnehmer in die Studie aufgenommen, von denen 898 Lecanemab und 897 Placebo erhielten. Der mittlere CDR-SB-Score zu Studienbeginn lag in beiden Gruppen bei etwa 3,2. Die Patienten waren im Mittel 71 Jahre, und der Beginn der ersten Symptome lag im Schnitt vier Jahre zurück. Der mittlere Punktwert beim MMST betrug 25,5. Die mittlere Veränderung gegenüber dem Ausgangswert im CDR-SB nach 18 Monaten betrug 1,21 unter Lecanemab und 1,66 unter Placebo (Differenz –0,45; 95%-KI –0,67 bis –0,23; p < 0,001). In einer Unterstudie mit 698 Teilnehmern war die Verringerung der Amyloidbelastung des Gehirns im PET unter Lecanemab größer als unter Placebo (Differenz –59,1 Zentiloide; 95%-KI –62,6 bis –55,6). Weitere Unterschiede zwischen den beiden Gruppen für Veränderungen gegenüber dem Ausgangswert zugunsten von Lecanemab waren:
- für den ADAS-cog14-Score –1,44 (95%-KI –2,27 bis –0,61; p < 0,001),
- für den ADCOMS –0,05 (95%-KI –0,074 bis –0,027; p < 0,001)
- und für den ADCS-MCI-ADL-Score 2,0 (95%-KI 1,2 bis 2,8; p < 0,001).
Lecanemab führte bei 26,4 % der Teilnehmer zu infusionsbedingten Reaktionen und bei 12,6 % zu amyloidbedingten Bildgebungsanomalien im MRT mit Ödemen (ARIA-E) oder Mikrohämorrhagien (ARIA-H).
Die Studie mit Lecanemab hat erwartungsgemäß großes Aufsehen erregt. Es war die erste große klinisch positive Studie, in der ein Antikörper, der sich gegen β-Amyloid im Gehirn richtet, zu einer Reduktion der Amyloidkonzentration im PET und zu einem geringeren Fortschreiten der kognitiven Einschränkungen bei beginnender Alzheimer-Krankheit führte. Für die Wirksamkeit spricht, dass sich positive Effekte nicht nur für den primären Endpunkt, sondern für fast alle sekundären Endpunkte fanden. Numerisch wurde die Erkrankungsprogression um 27 % im Vergleich zu Placebo verlangsamt. Die Wirksamkeit wurde allerdings nur bei Patienten mit leichter kognitiver Beeinträchtigung und beginnender Alzheimer-Erkrankung gezeigt. Außerdem ist eine Behandlungsdauer von 18 Monaten relativ kurz.
Wie bei allen Antikörpern gegen Amyloid zeigten sich zentrale Nebenwirkungen in der Kernspintomographie des Gehirns, wie das Auftreten von Ödemen und Mikrohämorrhagien. Die meisten dieser zerebralen Veränderungen waren allerdings asymptomatisch. Hier ist ebenfalls nicht bekannt, ob bei einer längeren Behandlungsdauer das Risiko für zerebrale Blutungen erhöht ist. Weiterhin ist nicht bekannt, ob die Anwendung von Lecanemab bei antikoagulierten Patienten mit einem erhöhten zerebralen Blutungsrisiko einhergeht. Für eine endgültige Einschätzung des therapeutischen Nutzens sind weitere Langzeitstudien notwendig. Die wichtigste Einschränkung der Therapie ist allerdings, dass die Krankheit nicht kausal behandelt wird, sondern lediglich die Krankheitsprogression verlangsamt wird. Dies ist allerdings ein therapeutischer Durchbruch, da alle bisherigen Studien mit Antikörpern gegen Aβ negativ waren [2].
Lecanemab hat im Januar 2023 eine beschleunigte FDA-Zulassung erhalten, die auf der erheblichen Verringerung der Amyloid-Plaques basierte, gefolgt von einer erweiterten Zulassung im Juni 2023, die auf der Verlangsamung der klinischen Progression beruhte. Die Frage, ob das Ausmaß der Verlangsamung der klinischen Progression klinisch bedeutsam ist („minimal clinical important difference“; d. h. einen spürbaren Nutzen für Patienten/Caregiver bringt) [1, 4, 23], um die Risiken der Nebenwirkungen und die Kosten der Behandlung aufzuwiegen, ist derzeit ein viel diskutiertes Thema um die Behandlung der Alzheimer-Erkrankung mit krankheitsmodifizierenden Substanzen [14, 16, 31]. Aktuell wird in der Literatur für den CDR-SB – der den primären Outcome in der Lecanemab-Studie darstellte – eine Veränderung von 1–2,5 Punkten als bedeutender klinisch wichtiger Unterschied angegeben [17a].
Infokasten: Bewertungsskalen der kognitiven Beeinträchtigung
ADAS-cog14: Alzheimer’s Disease Assessment Scale; Spanne: 0–90; je höher der Wert, desto größer die Beeinträchtigung
ADCOMS: Alzheimer’s Disease Composite Score; Spanne: 0–1,97; je höher der Wert, desto größer die Beeinträchtigung
ADCS-MCI-ADL: Alzheimer’s Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment; Spanne: 0–53; je niedriger der Wert, desto größer die Beeinträchtigung
CDR-SB: Clinical Dementia Rating Sum of Boxes; Spanne: 0–18; je höher der Wert, desto größer die Beeinträchtigung
iADRS: integrierte Alzheimer Disease Rating Scale; Spanne: 0–144; je niedriger der Wert, desto größer die Beeinträchtigung
MMST: Mini-Mental-Status-Test; Spanne: 0–30; je niedriger der Wert, desto größer die Beeinträchtigung
Donanemab
Ziel der TRAILBLAZER-ALZ-2-Studie war die Bewertung der Wirksamkeit und der unerwünschten Wirkungen von Donanemab, einem Antikörper, der Amyloid-Plaques im Gehirn beseitigen soll, bei Patienten im Frühstadium der Alzheimer-Erkrankung. Es handelte sich um eine multizentrische, randomisierte, doppelblinde, Placebo-kontrollierte, 18-monatige Phase-III-Studie an 277 Forschungszentren/Krankenhäusern und in 8 Ländern. An der Studie nahmen 1736 Teilnehmer mit früher symptomatischer Alzheimer-Erkrankung (leichte kognitive Beeinträchtigung/leichte Demenz) mit Amyloid- und niedriger/mittlerer oder hoher Tau-Pathologie auf der Grundlage eines PET von Juni 2020 bis November 2021 teil [25]. Die Teilnehmer wurden 1 : 1 randomisiert und erhielten Donanemab (n = 860) oder Placebo (n = 876) intravenös alle 4 Wochen über 72 Wochen. Die Teilnehmer in der Donanemab-Gruppe wurden verblindet auf Placebo umgestellt, wenn die Kriterien für die Beendigung der Therapie nach PET-Kriterien erfüllt waren. Der primäre Endpunkt war die Veränderung der integrierten Alzheimer Disease Rating Scale (iADRS, Infokasten) von Studienbeginn bis Woche 76. Es gab insgesamt 24 Endpunkte (primär, sekundär und explorativ), einschließlich des sekundären Ergebnisses der Veränderung des CDR-SB-Scores.
Unter den 1736 randomisierten Teilnehmern mit einem Durchschnittsalter von 73 Jahren waren 57 % Frauen. 68 % der Teilnehmer hatten eine niedrige/mittlere Tau-Belastung im PET und 32 % eine hohe Tau-Last. 76 % der Teilnehmer schlossen die Studie ab. Von den 24 Endpunkten waren 23 statistisch signifikant zugunsten von Donanemab.
In der Studienpopulation mit niedriger/mittlerer Tau-Pathologie betrug der Mittelwert (LSM) der Veränderung des iADRS-Scores nach 76 Wochen –6,02 in der Donanemab- und –9,27 in der Placebo-Gruppe (Differenz 3,25; 95%-KI 1,88–4,62; p < 0,001). In der gesamten Studienpopulation lagen die Werte bei –10,19 unter Donanemab und bei –13,11 unter Placebo (Differenz 2,92; 95%-KI 1,51–4,33; p < 0,001).
Nach 76 Wochen betrug die LSM-Veränderung im CDR-SB-Score in der Studienpopulation mit niedriger/mittlerer Tau-Pathologie bei 1,20 unter Donanemab und 1,88 unter Placebo (Differenz –0,67; 95%-KI –0,95 bis –0,40; p < 0,001). In der Gesamtpopulation betrug die Veränderung des CDR-SB-Scores 1,72 unter Donanemab und 2,42 unter Placebo (Differenz –0,70; 95%-KI –0,95 bis –0,45; p < 0,001).
Amyloid-bezogene Bildgebungsanomalien wie Ödeme oder Mikroblutungen traten bei 205 Teilnehmern (24 %; 52 symptomatisch) in der Donanemab-Gruppe und 18 (2 %; 0 symptomatisch) in der Placebo-Gruppe auf. Infusionsbedingte Reaktionen traten bei 74 Teilnehmern (9 %) unter Donanemab und 4 (1 %) unter Placebo auf. Es gab drei Todesfälle in der Donanemab-Gruppe und einen in der Placebo-Gruppe. Alle verstorbenen Patienten hatten eine Amyloidangiopathie im MRT.
Donanemab ist der dritte monoklonale Antikörper, der nicht nur die Amyloid-Belastung im PET reduziert, sondern auch bei Patienten mit beginnendem Morbus Alzheimer die Progression verlangsamt. Die unerwünschten Wirkungen, die im MRT nachweisbar sind, entsprechen denen der anderen monoklonalen Antikörper.
Im Juli 2024 hat die amerikanische Arzneimittelbehörde FDA Donanemab (KisunlaTM) zugelassen. Einmal monatlich wird die Gabe als Injektion empfohlen und zwar für Erwachsene mit früher symptomatischer Alzheimer-Krankheit, zu der sowohl Menschen mit leichter kognitiver Beeinträchtigung (MCI) als auch Menschen mit leichter Demenz im Stadium der AD mit nachgewiesener Amyloid-Pathologie gezählt werden (https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-adults-alzheimers-disease). In den Verschreibungsinformationen wird spezifisch auf die Nebenwirkungen ARIAs hingewiesen und ein besonderes Vorgehen empfohlen, vorzugsweise bei Patienten mit ApoE-ε4-Homozygotie. In den Dosierungsanweisungen der FDA wird ausgeführt, dass ein Absetzen des Medikaments in Erwägung gezogen werden kann, wenn die Amyloid-Plaques auf minimale Werte zurückgegangen sind, wie sie bei der Amyloid-PET-Bildgebung beobachtet wurden.
Die Firma Lilly hat die Unterlagen bei der europäischen Arzneimittelbehörde vorgelegt. Es bleibt abzuwarten, wie sich die EMA hier entscheidet.
Remternetug
Remternetug (LY3372993) ist ein monoklonaler IgG1-Antikörper, der gegen die Pyroglutamat-Modifikation des Aβ-Peptids gerichtet ist, die nur in aggregierten Amyloid-Plaques im Gehirn vorhanden ist. Remternetug der Firma Eli Lilly, USA, ist ein Nachfolgeprodukt des Antikörpers Donanemab, der ebenfalls gegen pyroglutamiertes Aβ gerichtet ist.
Derzeit stehen nur Zwischenergebnisse der Phase-I-Studie zur Verfügung, die im April 2023 auf dem AD/PD Kongress präsentiert wurden [13]. Die Studie umfasste 41 Teilnehmer mit leichter kognitiver Beeinträchtigung aufgrund von Alzheimer oder leichter bis mittelschwerer Demenz (Einschlusskriterien u. a.: Alter: 55–85 Jahre; MMST-Wert ≥ 16; Florbetapir-PET ≥ 37 Zentiloide; MRT: keine Anzeichen von ARIA-E, ≤ 4 Mikroblutungen, ≤ 1 oberflächliche Siderose oder Anzeichen von Makroblutungen), die sechs Monate lang monatlich 250 mg, 700 mg, 1400 mg oder 2800 mg Verum bzw. Placebo (in einer Stratifizierung von 5 : 1) erhielten. Eine Kohorte wurde von 700 mg auf 1400 mg titriert. Nach sechs Monaten war die Plaquemenge im Florbetapir-PET in allen Kohorten dosisabhängig reduziert (bis zu 100 Zentiloide). Bei einer Dosis von 250 mg fiel niemand unter den Schwellenwert für die Amyloid-Positivität, der mit 24 Zentiloiden definiert ist. In der 700-mg-Gruppe war dies bei vier von zehn Personen der Fall; bei einer Dosierung von 2800 mg sank der Nachweiswert innerhalb von drei Monaten unter die Schwelle der Amyloid-Positivität (definiert als Wert < 24 Zentiloide). Das ist ein schnellerer Abbau als bei Donanemab, das die Plaques in sechs Monaten um 60 Zentiloide senkte und 40 % der Patienten unter den Schwellenwert brachte.
Die Daten zur Sicherheit sind verblindet, aber insgesamt gab es zehn ARIA-E- und sieben ARIA-H-Fälle, ohne offensichtliche Dosis-Korrelation. Alle ARIA-E-Fälle traten bei APOE4-Trägern auf; einer der Teilnehmer war symptomatisch. Interessant ist, dass bei keinem der Teilnehmer – im Gegensatz zur Behandlung mit Donanemab – ADAs (behandlungsbedingte, gegen das Arzneimittel gerichtete Auto-Antikörper) nachgewiesen oder systemische Reaktionen (zwei Personen zeigten lokale Reaktionen an der Injektionsstelle) festgestellt wurden. Remternetug befindet sich jetzt in der klinischen Phase III.
In der Phase-III-Studie (TRAILRUNNER-ALZ1) werden 600 Patienten mit früher symptomatischer Alzheimer-Krankheit an 75 Standorten in den USA und zwei Standorten in Japan eingeschlossen. Die Teilnehmer, die sich für die Studie qualifizieren, haben u. a. einen MMST-Wert von 20–28 Punkten, weisen einen Scan mit phosphorylierten Tau auf und zeigten mindestens sechs Monate vor dem Screening eine allmähliche und fortschreitende Veränderung der kognitiven Funktionen.
Primärer Endpunkt der Studie ist der Prozentsatz der Patienten, bei denen die Amyloid-Plaques bis zum Ende des Behandlungszeitraums abgebaut sind. Sekundäre Endpunkte sind u. a. die Amyloid-Clearance, die Pharmakokinetik und die Bildung von Auto-Antikörpern gegen das Arzneimittel. Das Ende der Studie wird für 2025/2026 erwartet.
Klinische Studien zu Antikörpern ohne Wirksamkeitsnachweis
Gantenerumab
Gantenerumab ist ein vollständig humaner monoklonaler Antikörper, der aggregiertes Aβ bindet und Aβ-Plaques durch Fc-Rezeptor-vermittelte Phagozytose beseitigt. In einer randomisierten, doppelblinden, Placebo-kontrollierten Phase-III-Studie wurde Gantenerumab über zwei Jahre untersucht [20]. Die Patienten erhielten randomisiert Gantenerumab 105 mg oder 225 mg oder Placebo alle vier Wochen durch subkutane Injektion. Primärer Endpunkt war die Veränderung des CDR-SB-Scores vom Ausgangswert bis zu Woche 104. Außerdem wurden die Auswirkungen der Behandlung auf Liquor-Biomarker (alle Patienten) und Amyloid-PET (Teilstudie) untersucht. Von den 3089 Patienten, die untersucht wurden, wurden 797 randomisiert. Die Studie wurde wegen Futility – Zwecklosigkeit – vorzeitig abgebrochen und entblindet. Es gab keine Unterschiede zwischen den Gruppen im primären Endpunkt, der CDR-SB-Veränderung vom Ausgangswert:
- Placebo: 1,60 (95%-KI 1,28–1,91)
- Gantenerumab 105 mg: 1,69 (95%-KI 1,37–2,01)
- Gantenerumab 225 mg: 1,73 (95%-KI 1,42–2,04)
Es gab auch keine Unterschiede für die sekundären Endpunkte. Die Inzidenz von asymptomatischen amyloidbezogenen Bildgebungsanomalien im MRT stieg mit steigender Dosis an. ARIAs hingen auch vom APOE ε4-Genotyp ab.
Crenezumab
Crenezumab, ein humanisierter monoklonaler Immunglobulin-G4-Antikörper, der auf Beta-Amyloid-Oligomere abzielt, wurde bei Teilnehmern mit prodromaler bis leichter (früher) Alzheimer-Krankheit untersucht. Die beiden multizentrischen, randomisierten, doppelblinden, Placebo-kontrollierten Parallelgruppenstudien zur Wirksamkeit und Sicherheit von Crenezumab bei Teilnehmern mit früher Alzheimer-Krankheit, CREAD und CREAD2, wurden 2016 bzw. 2017 initiiert [19]. CREAD (194 Standorte in 30 Ländern) und CREAD2 (209 Standorte in 27 Ländern) waren globale multizentrische Studien. Insgesamt wurden 3736 bzw. 3664 Teilnehmer untersucht. An beiden Studien nahmen Personen im Alter von 50 bis 85 Jahren mit Alzheimer im Frühstadium teil. Nach dem Ausschluss von 2923 bzw. 2858 Teilnehmern wurden 813 Teilnehmer in CREAD und 806 in CREAD2 nach dem Zufallsprinzip im Verhältnis 1 : 1 entweder zu Placebo oder Crenezumab randomisiert. In der endgültigen Analyse befanden sich 409 Teilnehmer in der Placebo-Gruppe und 404 in der Crenezumab-Gruppe in CREAD und 399 bzw. 407 Teilnehmer in CREAD2. Die Studienteilnehmer erhielten Placebo oder 60 mg/kg Crenezumab intravenös alle vier Wochen für bis zu 100 Wochen. Der primäre Endpunkt war die Veränderung des CDR-SB-Scores vom Ausgangswert bis Woche 105. Der Unterschied zwischen den Gruppen bei der mittleren Veränderung des CDR-SB-Scores (Placebo minus Crenezumab) betrug in Woche 105 in der CREAD-Studie –0,17 (95%-KI –0,86 bis 0,53; p = 0,63) (88 Placebo; 86 Crenezumab). Im Vergleich zu früheren Studien wurden keine neuen Sicherheitssignale festgestellt, und amyloidbedingte Bildgebungsanomalien mit Ödemen waren selten, leicht und vorübergehend. Es wurden keine bedeutsamen Veränderungen bei den Biomarkern beobachtet. Beide Studien wurden nach einer im Voraus geplanten Zwischenanalyse abgebrochen, die ergab, dass CREAD den primären Endpunkt wahrscheinlich nicht erreichen würde.
Solanezumab
Solanezumab, das auf monomeres bzw. lösliches Amyloid abzielt, wurde in einer Phase-III-Studie an Personen mit präklinischer Alzheimer-Krankheit untersucht [27]. Es wurden Personen im Alter von 65 bis 85 Jahren mit einem globalen Clinical Dementia Rating Score von 0 (Spanne: 0–3, wobei 0 keine kognitive Beeinträchtigung und 3 eine schwere Demenz anzeigt), einer Punktzahl im MMST von 25 oder mehr und erhöhten Amyloidwerten im Gehirn im 18F-Florbetapir-PET aufgenommen. Die Teilnehmer wurden nach dem Zufallsprinzip in einem Verhältnis von 1 : 1 entweder zu Solanezumab in einer Dosis von bis zu 1600 mg intravenös alle vier Wochen oder zu Placebo randomisiert. Der primäre Endpunkt war die Veränderung des Preclinical Alzheimer Cognitive Composite (PACC)-Scores (berechnet als Summe von vier z-Scores, wobei höhere Scores eine bessere kognitive Leistung anzeigen) über einen Zeitraum von 240 Wochen. Insgesamt wurden 1169 Personen randomisiert: 578 wurden der Solanezumab-Gruppe und 591 der Placebo-Gruppe zugewiesen. Das Durchschnittsalter der Teilnehmer betrug 72 Jahre, etwa 60 % waren Frauen und 75 % hatten eine Demenz in der Familie. Nach 240 Wochen betrug die mittlere Veränderung des PACC-Scores –1,43 in der Solanezumab-Gruppe und –1,13 in der Placebo-Gruppe (Differenz –0,30; 95%-KI –0,82 bis 0,22; p = 0,26). Die Amyloidwerte im PET des Gehirns stiegen in der Solanezumab-Gruppe um durchschnittlich 11,6 Zentiloide und in der Placebo-Gruppe um 19,3 Zentiloide. ARIA mit Ödemen traten bei weniger als 1 % der Teilnehmer in jeder Gruppe auf. ARIA mit Mikroblutungen oder Hämosiderose traten bei 29,2 % der Teilnehmer in der Solanezumab-Gruppe und bei 32,8 % der Teilnehmer in der Placebo-Gruppe auf. Solanezumab, das auf monomeres Amyloid bei Personen mit erhöhten Amyloidspiegeln im Gehirn abzielt, verlangsamte den kognitiven Abbau im Vergleich zu Placebo über einen Zeitraum von 240 Wochen bei Personen mit präklinischer Alzheimer-Krankheit nicht.
Zusammenfassung
In der Erforschung und Behandlung der Alzheimer-Krankheit haben neue Biomarker, die Aβ und phosphoryliertes Tau messen, die Früherkennung und biomarkergestützte Diagnose ermöglicht. Dieser Nachweis ist im Liquor, im Serum und im PET möglich. Die erste Klasse von Therapeutika, die in der Entwicklung von Arzneimitteln zur Behandlung der Alzheimer-Erkrankung in die Klinik gelangt sind, sind gegen Aβ-gerichtete monoklonale Antikörper, die an verschiedene Stellen der Aβ-Aggregation binden.
Der erste monoklonale Antikörper gegen Aβ war Aducanumab [11], der 2021 in den USA zugelassen wurde. Aufgrund der divergierenden Studiendaten wurde das Arzneimittel aber kaum verschrieben.
Im Juli 2023 wurde Lecanemab, der zweite monoklonale Antikörper gegen Aβ, in den Vereinigten Staaten zugelassen. In der dafür ausgewerteten CLARITY-Studie zeigten Patienten mit Mild Cognitive Impairment oder beginnender Alzheimer-Erkrankung eine um 27 % langsamere Verschlechterung kognitiver Funktionen verglichen mit Placebo [30]. Die Studie zeigte auch eine eindrucksvolle Reduktion von Aβ im Amyloid-PET.
Der dritte monoklonale Antikörper, der untersucht wurde, ist Donanemab. Nach 76 Wochen zeigte der Endpunkt in der Verum-Gruppe bei 35 % eine Verlangsamung des Fortschreitens der Erkrankung verglichen mit 22 % in der Placebo-Gruppe. Interessant war, dass ein Therapieeffekt nur bei den Patienten nachweisbar war, die eine geringe oder mittelgradige Belastung mit Tau-Protein hatten. Bei Patienten mit hoher Tau-Protein-Belastung ließ sich keine Wirksamkeit nachweisen, was daran liegen könnte, dass die Krankheit zu diesem Zeitpunkt bereits zu weit fortgeschritten war. Bezogen auf die verwendeten funktionellen Skalen würde eine Behandlung mit Donanemab bei Patienten in der Gruppe mit niedriger oder mittelgradiger Tau-Belastung die Krankheitsprogression im Vergleich zu Placebo um 7,5 Monate verzögern. Eindrucksvoll war die Reduktion von Amyloid-Plaques im PET. Bei drei Viertel der Patienten war kein Aβ mehr nachweisbar. Der Unterschied in der Wirksamkeit zu anderen monoklonalen Antikörpern könnte daran liegen, dass dieser Antikörper an das N-terminale Pyroglutamat-Epitop von Amyloid-Plaques bindet. Auffällig war, dass sich im PET keine Veränderung im frontalen Kortex fand. Als paradoxer Effekt fand sich in der Verum-Gruppe eine ausgeprägte Verringerung des Gesamtgehirnvolumens und eine Ausweitung der Ventrikel im Vergleich zu Placebo. Die Studie hatte darüber hinaus ein innovatives Design: das PET wurde nach 24 und 52 Wochen wiederholt. Bei Patienten unter Donanemab, bei denen sich eine ausgeprägte Reduktion des Beta-Amyloids fand, wurde die aktive Behandlung beendet und die Patienten auf Placebo umgestellt. Wie in den Studien zu anderen monoklonalen Antikörpern zeigten sich häufig amyloidbedingte Veränderungen in der Kernspintomographie (ARIA). Diese manifestieren sich entweder als Hirnödem oder als Mikroblutungen. Besonders bei Patienten mit Amyloidangiopathie ist dieses Risiko deutlich erhöht. Aufgrund des erhöhten Blutungsrisikos wurden Patienten mit Amyloidangiopathie ausgeschlossen. Patienten, die antikoaguliert wurden, durften allerdings an der Behandlung teilnehmen.
Warum sind nicht alle monoklonalen Antikörper wirksam?
Trotz der überwältigenden Evidenz der aktiven und passiven gegen Aβ-gerichteten Immuntherapien, um die Amyloidablagerung zu verhindern/aufzulösen und die Aβ-Toxizität zu verringern, zeigten die verschiedenen Antikörper nur eine geringe Wirksamkeit, den kognitiven Verfall bei leichter bis mittelschwerer Demenz zu verhindern. Dafür gibt es mehrere mögliche Erklärungen:
- Die Diskrepanz zwischen der Antikörper-vermittelten Plaque-Abräumung und dem weiteren Fortschreiten der Demenz deutet darauf hin, dass die Entfernung der Plaques nicht ausreicht, um den neurodegenerativen Prozess aufzuhalten, sobald eine Person ein fortgeschritteneres Stadium der Erkrankung erreicht. Möglicherweise lösen die Amyloid-Plaques, die fibrilläres, unlösliches Aβ enthalten, den neurodegenerativen Prozess der Alzheimer-Krankheit aus, während die nicht-fibrillären, oligomeren Formen von Aβ für das Fortschreiten der Krankheit verantwortlich sind. In Übereinstimmung mit diesen Beobachtungen könnte daher die Entfernung oder Sequestrierung von löslichen Aβ-Spezies die Wirksamkeit von Immuntherapien bei der Alzheimer-Krankheit erhöhen. Diese Annahmen sind alle nur gültig unter der Annahme der Amyloid-Kaskaden-Hypothese, die in den letzten Jahren immer wieder angezweifelt wurde [8, 10].
- Andererseits könnte die Immunisierung von Personen in früheren Studien bei Patienten mit leichter bis mittelschwerer Alzheimer-Krankheit zu spät erfolgt sein, um das Fortschreiten der Krankheit aufzuhalten. AD-Biomarkerstudien konnten eindeutig zeigen, dass die Neuropathologie der Alzheimer-Krankheit, insbesondere die Aβ-Plaque-Entstehungsphase, ca. 15–20 Jahre vor dem klinischen Ausbruch der Demenz beginnt [3, 12].
Probleme bei der Implementierung und Umsetzung der Therapie mit monoklonalen Antikörpern
Sollten antikörperbasierte Therapien wie Lecanemab oder Donanemab in Europa zugelassen werden, ergeben sich erhebliche praktische Konsequenzen und Probleme. Im Moment gibt es keine Infrastruktur, um für größere Patientenzahlen Aβ und Tau-Proteine im PET nachzuweisen. Die Behandlungskosten selbst sind erheblich (in den Vereinigten Staaten > 28 000 US-Dollar pro Jahr), und es ist nicht abzusehen, ob der Gemeinsame Bundesausschuss (G-BA) einer Erstattung zustimmen wird [14]. Wegen ARIA müssen regelmäßig kernspintomographische Kontrollen durchgeführt werden. Die i. v. Applikation muss entweder alle 14 Tage bei Lecanemab oder alle vier Wochen bei Donanemab erfolgen. Dafür müssten Infusionszentren eingerichtet werden. Es ist weiterhin unklar, welche Kontraindikationen für den Einsatz der monoklonalen Antikörper bestehen. Sehr wahrscheinlich müssten nicht nur Patienten mit Amyloidangiopathie in der Kernspintomographie ausgeschlossen werden, sondern auch Patienten, die antikoaguliert sind. Ob dies auch für Patienten unter Thrombozytenfunktionshemmern gilt, ist bisher nicht untersucht worden. Weiterhin ist nicht klar, ob eine Beendigung der Therapie bei Clearance von Beta-Amyloid und Tau-Proteinen im PET tatsächlich das weitere Fortschreiten der Erkrankung verlangsamt, oder ob zu einem späteren Zeitpunkt weitere Therapiezyklen notwendig sind.
Die Zulassung der monoklonalen Antikörper Lecanemab und Donanemab wird wahrscheinlich nur für Patienten mit beginnender Alzheimer-Erkrankung mit Mild Cognitive Impairment oder leichten Symptomen erfolgen. Ob die Zulassung den Nachweis von Aβ und/oder Tau-Protein im PET erfordert, oder ob auch Biomarker im Liquor und Serum als Einschlusskriterium akzeptiert werden, ist bisher nicht bekannt. Das größte Problem dürfte allerdings sein, ob die hier beobachteten, relativ geringen Effekte der Verlangsamung des Fortschreitens der Erkrankung die erheblichen Kosten für Diagnostik, Therapie und Nachsorge rechtfertigen.
Ausblick
Intrazelluläres Tau-Protein spielt eine Schlüsselrolle in der Pathophysiologie der Alzheimer-Krankheit.
Es gibt immer mehr Hinweise darauf, dass aggregiertes, hyperphosphoryliertes Tau ein Hauptfaktor für die Neurodegeneration bei der Alzheimer-Erkrankung sein könnte.
Das Tau-Protein wird durch das MAPT(mikrotubuliassoziiertes Protein Tau)-Gen kodiert und hauptsächlich in Neuronen exprimiert. Unter pathologischen Bedingungen akkumuliert das hyperphosphorylierte Tau intrazellulär und aggregiert zu Oligomeren und Fibrillen, was zu intraneuronalen neurofibrillären Ablagerungen führt und mit der Verschlechterung der kognitiven Fähigkeiten bei der Alzheimer-Erkrankung assoziiert ist. Tau wird auch von Neuronen ausgeschieden und verbreitet sich über spezifische neuronale Netzwerke über einen trans-synaptischen Weg aus. Diese Ausbreitung der Tau-Pathologie geht mit einer weiteren synaptischen Dysfunktion und dem Verlust von Nervenzellen einher. Präklinische Studien haben gezeigt, dass eine Tau-Reduktion spezifische Aβ-vermittelte Defizite verhindert, was eine zentrale Rolle von Tau bei der Vermittlung der Aβ-Toxizität in der frühen Pathogenese der Alzheimer-Erkrankung spielt. Es gibt Hinweise darauf, dass eine Senkung von Tau diese Pathologie reduzieren könnte. Die Autoren einer in Nature publizierten Studie versuchten, die MAPT-Expression mit einem auf Tau ausgerichteten Antisense-Oligonukleotid (MAPTRx) bei Patienten im Frühstadium der Alzheimer-Erkrankung zu hemmen [18]. Es handelte sich um eine randomisierte, doppelblinde, Placebo-kontrollierte, randomisierte, Phase-Ib-Studie. Die Studie untersuchte die Sicherheit, Pharmakokinetik und die mögliche Wirkung von MAPTRx. Vier aufsteigende Dosiskohorten wurden nacheinander aufgenommen und im Verhältnis 3 : 1 auf eine intrathekale Bolusgabe von MAPTRx oder Placebo alle 4 oder 12 Wochen randomisiert. Die Studiendauer betrug 13 Wochen, gefolgt von einem 23-wöchigen Nachbehandlungszeitraum. Der primäre Endpunkt der Studie war die Sicherheit. Der sekundäre Endpunkt war die Pharmakokinetik von MAPTRx im Liquor. Das vorab festgelegte explorative Ergebnis war die Gesamt-Tau-Protein-Konzentration im Liquor.
46 Patienten nahmen an der Studie teil, von denen 34 auf MAPTRx und 12 auf Placebo randomisiert wurden. Unerwünschte Ereignisse wurden bei 94 % der mit MAPTRx behandelten Patienten und 75 % der mit Placebo behandelten Patienten beobachtet. Alle Ereignisse waren leicht oder mittelschwer und nicht mit der Behandlung assoziiert. Es wurden keine schwerwiegenden unerwünschten Ereignisse bei den mit MAPTRx behandelten Patienten beobachtet. In dieser Sicherheitsstudie wurde eine dosisabhängige Verringerung der Gesamt-Tau-Konzentration im Liquor von mehr als 50 %, 24 Wochen nach der letzten Dosis in den Gruppen mit 60 mg und 115 mg MAPTRx beobachtet. Es gab keine Sicherheitsbedenken. Es handelt sich hier um eine der innovativsten Studien für eine mögliche zukünftige Behandlung der frühen Alzheimer-Krankheit.
Es wird davon ausgegangen, dass Aβ zu etwa 50 % für die Krankheitsentstehung und Progression der Alzheimer-Erkrankung verantwortlich ist.
Die neuen Daten zu den monoklonalen Antikörpern gegen Aβ zeigen, dass einige dieser Substanzen den Krankheitsverlauf bei der frühen Erkrankung verlangsamen können [30]. PET-Studien zeigten, dass diese monoklonalen Antikörper zu einer dramatischen Reduktion der Aβ-Ablagerungen im Gehirn führen. Therapieansätze bei neurodegenerativen Erkrankungen wie dem Morbus Parkinson mit monoklonalen Antikörpern gegen Tau waren bisher leider nicht erfolgreich [15, 21]. Daher ist der neue Therapieansatz, ein Antisense-Oligonukleotid zu verwenden, neu und innovativ. Die kleine Studie zeigt als Proof-of-Concept, dass mit dem Antisense Oligonukleotid MAPTRx bei Patienten mit einer frühen Alzheimer-Erkrankung die Tau-Konzentration im Liquor dramatisch reduziert werden kann. Die Studie war allerdings viel zu klein und viel zu kurz, um ermessen zu können, ob die Therapie auch Auswirkungen auf kognitive Funktionen und die Verschlechterung von kognitiven Funktionen im Verlauf hat. Um dies zu untersuchen, müssen größere Langzeitstudien durchgeführt werden, die jetzt geplant sind. Zusätzlich sind weitere vielversprechende therapeutische Substanzen in der Entwicklung [6].
Die Pipelines für die medikamentösen Therapieoptionen für die Alzheimer-Krankheit im industriellen und akademischen Bereich sind weiterhin gut gefüllt und so sind weitere Neuerungen für die Behandlung der Alzheimer-Krankheit in den nächsten Jahren zu erwarten.
Interessenkonflikterklärung
HCD hat keine Interessenkonflikte beim Thema Demenz.
RD ist Miterfinder auf Patenten der Philipps-Universität Marburg zur passiven Immunisierung bei neurodegenerativen Erkrankungen. Er hat Vorträge mit Honorar für die Firmen Eisai, Lilly, Roche u. a. zum Thema neurodegnerative Erkankungen durchgeführt. Er ist Berater bei der Firma Lilly. Alle genannten Honorare der letzten Jahre werden für Forschungszwecke verwendet.
Literatur
1. Andrews JS, Desai U, Kirson NY, Zichlin ML, et al. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer’s disease clinical trials. Alzheimers Dement (N Y) 2019;5:354–63.
1a. Adolfsson O, Pihlgren M, Toni N, Varisco Y, et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci 2012;32:9677–892.
2. Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res Rev 2021;68:101339.
2a. Bacskai B, Kajdasz S, McLellan M, Games D, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci 2002;22:7873–8.
3. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012;367:795–804.
4. Borland E, Edgar C, Stomrud E, Cullen N, et al. Clinically relevant changes for cognitive outcomes in preclinical and prodromal cognitive stages: implications for clinical alzheimer trials. Neurology 2022;99:e1142–53.
5. Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis 2022;9:197–210.
6. Cummings J, Zhou Y, Lee G, Zhong K, et al. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement (N Y) 2023;9:e12385.
7. Demattos RB, Lu J, Tang Y, Racke MM, et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012;76:908–20.
8. Fedele E. Anti-amyloid therapies for Alzheimer’s Disease and the amyloid cascade hypothesis. Int J Mol Sci 2023;24:14499.
9. Fillit H, Green A. Aducanumab and the FDA – where are we now? Nat Rev Neurol 2021;17:129–30.
9a. Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553–62.
10. Granzotto A, Sensi SL. Once upon a time, the Amyloid Cascade Hypothesis. Ageing Res Rev 2023;93:102161.
11. Haeberlein S, Aisen P, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s Disease. J Prev Alz Dis 2022;9:197–210.
12. Jack CR, Jr., Bennett DA, Blennow K, Carrillo MC, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–62.
13. Jin L, Gueorguieva I, Cheng Yun-Ju, et al. Safety and amyloid plaque reduction effects of remternetug in patients with alzheimer’s disease: Interim analysis from a phase 1 study. PowerPoint-Präsentation bei der International Conference on Alzheimer’s and Parkinson’s Diseases (AD/PD) Gothenburg, Schweden 2023.
14. Jönsson L, Wimo A, Handels R, Johansson G, et al. The affordability of lecanemab, an amyloid-targeting therapy for Alzheimer’s disease: an EADC-EC viewpoint. Lancet Reg Health Eur 2023;29:100657.
15. Lang AE, Siderowf AD, Macklin EA, Poewe W, et al. Trial of Cinpanemab in Early Parkinson’s Disease. N Engl J Med 2022;387:408–20.
16. Liu KY, Villain N, Ayton S, Ackley SF, et al. Key questions for the evaluation of anti-amyloid immunotherapies for Alzheimer’s disease. Brain Commun 2023;5:fcad175.
17. Mintun MA, Lo AC, Duggan Evans C, Wessels AM, et al. Donanemab in early Alzheimer’s Disease. N Engl J Med 2021;384:1691–704.
17a. Muir RT, Hill MD, Black SE, Smith EE. Minimal clinically important difference in Alzheimer’s disease: Rapid review. Alzheimers Dement 2024. doi: 10.1002/alz.13770.
18. Mummery CJ, Börjesson-Hanson A, Blackburn DJ, Vijverberg EGB, et al. Tau-targeting antisense oligonucleotide MAPTRx in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat Med 2023;29:1437–47.
19. Ostrowitzki S, Bittner T, Sink KM, Mackey H, et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early alzheimer disease: two phase 3 randomized placebo-controlled trials. JAMA Neurol 2022;79:1113–21.
20. Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 2017;9:95.
21. Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, et al. Trial of prasinezumab in early-stage Parkinson’s Disease. N Engl J Med 2022;387:421–32.
22. Rabinovici GD. Controversy and Progress in Alzheimer’s Disease – FDA Approval of Aducanumab. N Engl J Med 2021;385:771–4.
23. Rentz DM, Wessels AM, Annapragada AV, Berger AK, et al. Building clinically relevant outcomes across the Alzheimer’s disease spectrum. Alzheimers Dement (N Y) 2021;7:e12181.
23a. Scheidt HA, Morgado I, Rothemund S, Huster D, et al. Solid-state NMR spectroscopic investigation of Aβ protofibrils: implication of a β-sheet remodeling upon maturation into terminal amyloid fibrils. Angew Chem Int Ed Engl 2011;50:2837–40.
24. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608.
25. Sims JR, Zimmer JA, Evans CD, Lu M, et al. Donanemab in early symptomatic alzheimer disease: The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 2023;330:512–27.
26. Song C, Shi J, Zhang P, Zhang Y, et al. Immunotherapy for Alzheimer’s disease: targeting β-amyloid and beyond. Transl Neurodegener 2022;11:18.
27. Sperling RA, Donohue MC, Raman R, Rafii MS, et al. Trial of solanezumab in preclinical Alzheimer’s Disease. N Engl J Med 2023;389:1096–107.
28. Swanson CJ, Zhang Y, Dhadda S, Wang J, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 2021;13:80.
29. Thakur A, Bogati S, Pandey S. Attempts to develop vaccines against Alzheimer’s disease: a systematic review of ongoing and completed vaccination trials in humans. Cureus 2023;15:e40138.
30. van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med 2022;388:9–21.
31. Wu W, Ji Y, Wang Z, Wu X, et al. The FDA-approved anti-amyloid-β monoclonal antibodies for the treatment of Alzheimer’s disease: a systematic review and meta-analysis of randomized controlled trials. Eur J Med Res 2023;28:544.
* mod. Nachdruck aus Arzneimitteltherapie 2024;42:94–104.
Prof. Dr. Hans-Christoph Diener, Leiter der Abteilung für Neuroepidemiologie, Institut für Medizinische Informatik, Biometrie und Epidemiologie (IMIBE), Medizinische Fakultät der Universität Duisburg-Essen, Hufelandstraße 55, 45147 Essen, E-Mail: hans.diener@uk-essen.de
Univ.-Prof. Dr. med. Richard Dodel, Lehrstuhl für Geriatrie, Virchowstraße 171, 45147 Essen, E-Mail: richard.dodel@uk-essen.de
Monoclonal antibodies in the therapy of Alzheimer’s disease
Alzheimer’s disease is the most common cause of dementia. In the pathophysiology of Alzheimer’s disease, β-amyloid plaques play a central pathogenetic role. For the treatment of early stages of Alzheimer’s disease, monoclonal antibodies against different epitopes of β-amyloid were developed and investigated in large, placebo-controlled clinical trials. Two of these antibodies, lecanemab and donanemab, showed a significant slowing of disease progression. However, side effects such as cerebral edema, microbleeds and iron deposits are problematic. In addition, complex primary diagnostics with PET-CT or biomarkers in the cerebrospinal fluid are necessary. The high costs of the therapy are not only due to the cost of the medication itself, but also by the costs for infusion centers and regular follow-up examinations including cerebral imaging. It is also currently unclear why some other monoclonal antibodies were not effective in the treatment of Alzheimer’s disease.
Key words: Alzheimer’s disease, beta-amyloid, monoclonal antibodies, primary diagnostics, practical application
Psychopharmakotherapie 2024; 31(04):128-138