Werner Kropf und Dorothée Möller, Klingenmünster
Epidemiologie: Vorkommen – Dimension
Mit einem Vorkommen von etwa 1 zu 5000 und einer Inzidenz von 10 (Neuerkrankungen pro einer Million Bewohner) ist die Myasthenie eine eher seltene Krankheit, die allerdings während der letzten Jahre etwas häufiger aufzutreten scheint. Das liegt womöglich an einer verbesserten Diagnostik und einer allgemein höheren Lebenserwartung [11, 13, 19, 31].
Die Ausprägung der Symptomatik und Schwere der Muskelschwäche variiert von Fall zu Fall sehr stark, sodass von einem höchst individuell verlaufenden, heterogenen Geschehen mit marginaler Beeinträchtigung bis hin zum intubationsbedürftigen Notfall die gesamte Bandbreite vorkommen kann. Insgesamt ist davon auszugehen, dass sich die Muskellähmungen über die Zeitspanne hinweg, das heißt progredient, ausweiten: Nach anfänglichen Effekten auf die kleineren Augen- und Gesichtsmuskeln werden in der Folge die des Kopfhalteapparats sowie die proximalen Muskeln der Extremitäten beeinträchtigt. Sehr schwere Fälle zeigen eine Beteiligung der Atemmuskulatur. Noch vor etwa 80 Jahren wurden letale Verläufe von immerhin mehr als 30 % gesehen [16, 31].
Grundsätzlich können alle Altersgruppen betroffen sein, wobei sich zwei Erkrankungsgipfel, einmal um das 30. Lebensjahr (mit deutlichem Überwiegen des Frauenanteils) und dann um das 70. Lebensjahr (eher Männer) erkennen lassen.
Als gut gesichert gilt, dass es sich um eine Autoimmunerkrankung handelt, wobei sich nahezu regelhaft sogenannte Autoantikörper gegen Proteine der neuromuskulären Endplatte, insbesondere den Acetylcholin-Rezeptor nachweisen lassen [13].
Die zugrunde liegende Ursache ist bis heute unbekannt. Obwohl eine gewisse familiäre Häufung zur Ausbildung autoimmun vermittelter Erkrankungen zu bestehen scheint, kann kein wirklich gut gesicherter genetischer Anhaltspunkt für eine Erblichkeit nachgewiesen werden.
Die Myasthenia gravis kann auch – weit seltener – mit weiteren Autoimmunerkrankungen (z. B. Thyreoiditis, rheumatoide Arthritis, Lupus erythematodes) gemeinsam vorkommen, eine differenzialdiagnostische Abklärung ist obligat [31].
Es erscheint sehr lohnend, sich näher mit der Myasthenie zu befassen, da sie ganz im Sinne einer Modellerkrankung für weitere Autoimmunkrankheiten dienen kann. Nicht zuletzt auch deshalb, da ein dynamisches Zusammenspiel der Steuerungsmechanismen mit nervaler, immunologischer und hormoneller Beteiligung vorgefunden wird und zudem gute therapeutische Interventionen bereitstehen.
Merkmale der neuromuskulären Transmission
Als Teil des somatischen Nervensystems im Bereich der neuromuskulären Übertragung kommt dem Transmitter Acetylcholin (ACh) mit seinen nicotinergen Acetylcholin-Rezeptoren (AChR) der motorischen Endplatte die tragende Rolle zu (Abb. 1).
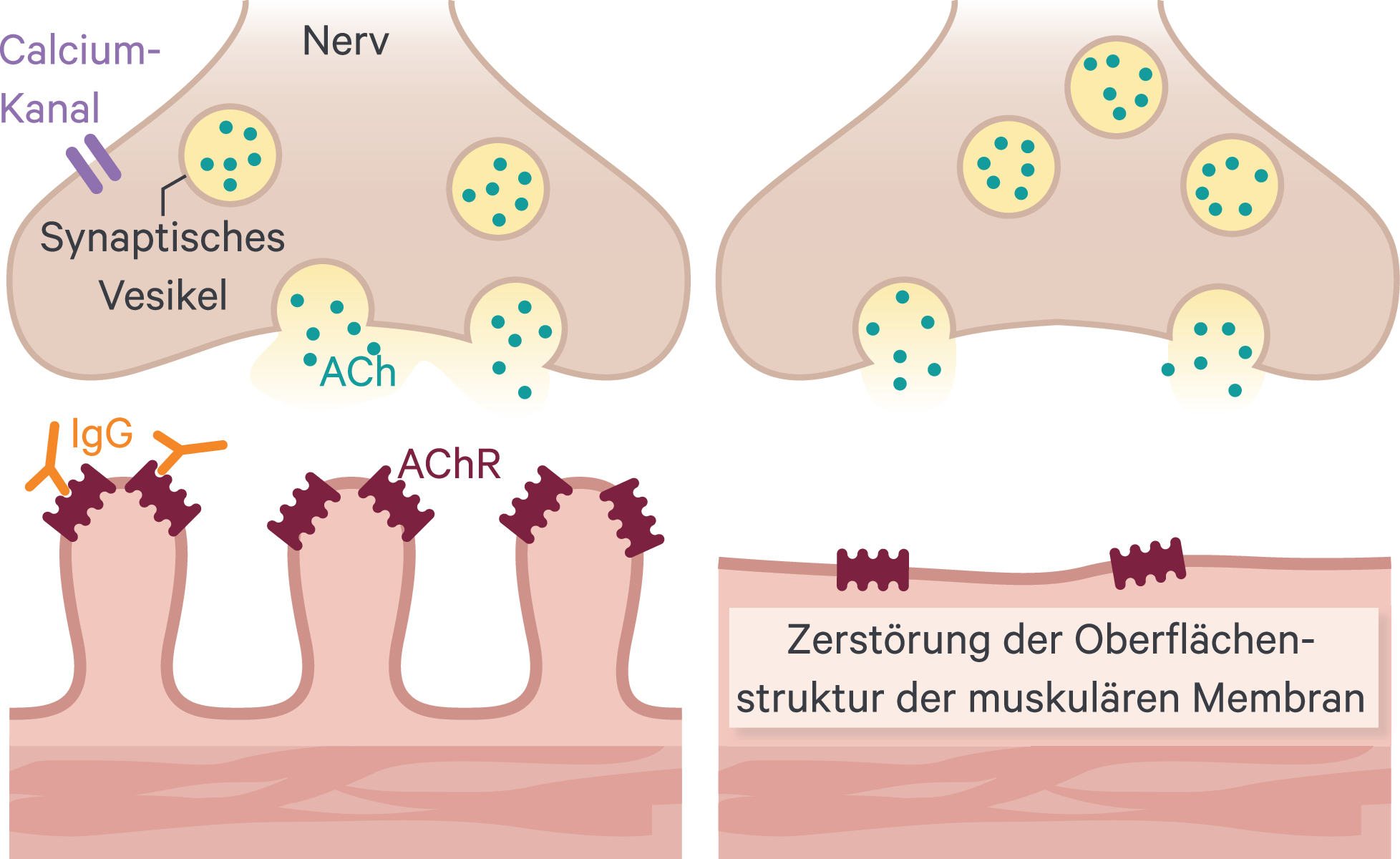
Abb. 1. Neuromuskuläre Synapse und ihr pathophysiologischer Zustand bei MG (adaptiert nach [1]) ACh: Acetylcholin; AChR: ACh-Rezeptor; IgG: Immunglobulin G
Das Signal des Motoneurons löst (nach Aktivierung des präsynaptisch gelegenen Calciumkanals) die Verschmelzung von Acetylcholin-speichernden Vesikeln mit der Membran aus; die sodann freigesetzten ACh-Moleküle diffundieren zu den Rezeptoren der nachgeschalteten Muskelzelle, um dort anzukoppeln. Die Stimulierung führt dort zur Öffnung von Natriumkanälen und in der Folge innerzellulärer Prozessvermittlung kommt es zur Muskelkontraktion [1].
Das ACh verlässt die Bindung und wird rasch durch eine Esterase gespalten, wobei das Cholin in die Nervenzelle rückresorbiert und nach Acetyl-CoA-Aktivierung recycelt wird, um erneut zur Verfügung zu stehen.
Im Falle von pathophysiologischen Veränderungen werden durch den Thymus (Auto-)Antikörper produziert, die gegen die eigenen AChR gerichtet sind und deren Zerstörung bewirken. Wenn mehr als 50 % der AChR inaktiviert sind, zeigen sich die Muskelermüdungserscheinungen bzw. Lähmungen, da trotz Neusynthese keine Kompensation mehr möglich ist. Neben diesen AChR-Antikörpern können auch solche gegen die abbauenden Folgeprozesse vorkommen (z. B. Anti-MuSK) sowie eine Komplement-Aktivierung erfolgen. In der Konsequenz findet sich eine Zerstörung der postsynaptischen Muskelzellmorphologie (Glättung der sonst üblichen wellenartigen Struktur, vgl. Abb. 1). Deren Degeneration geht mit einer Weitung des synaptischen Spalts sowie letztlich mit dem Funktionsverlust der entsprechenden Endplatte einher [12].
Exkurs: Lambert-Eaton-Myasthenie-Syndrom (LEMS)
Hierbei finden sich spezifische Auto-AK gegen den präsynaptisch gelegenen Calciumkanal (vergl. Abb. 1), dessen Funktionsfähigkeit erfolgskritisch für die Freisetzung von ACh aus den Vesikeln ist. Die neuromuskuläre Transmission wird somit bereits vorzeitig präsynaptisch beeinträchtigt. Klinisch zeigen die Betroffenen eine rasche Ermüdbarkeit der rumpfnahen Extremitäten, was sich beispielsweise im Alltag beim Treppensteigen sehr eindrücklich bemerkbar macht. ACh-AK können allerdings gleichwohl beim LEMS zusätzlich vorkommen [13, 23, 31].
Es ist bis dato nicht bekannt, wieso es im Thymus zur Fehlfunktion mit Bildung der Autoantikörper kommt. Denkbar erscheint, dass der Pathomechanismus infolge prägender Kontakte durch Viren bzw. Bakterien (molekulare Mimikry) oder auch durch sogenannte Myoidzellen des Thymus in Gang gesetzt wird [16] und später die korrigierende Selektion durch regulierende Immunzellen unterbleibt, was sich in der gestörten Toleranz zeigt. Bei Patienten mit Myasthenia gravis findet sich fast regelmäßig ein morphologisch veränderter Thymus (90 %) mit einer Dys- bzw. Hyperplasie, mitunter als Thymom imponierend (etwa 15 %). Die Entfernung dieses Tumors führt immerhin bei etwa einem Drittel der Betroffenen zur vollständigen Ausheilung. Die Thymektomie ist insbesondere für jüngere Erwachsene eine aussichtsreiche therapeutische Option [31].
Klinische Merkmale, Symptomatik und Verlauf
Die Mehrzahl der Betroffenen berichtet zunächst über okuläre Probleme mit Sehstörungen (Doppelbilder) und/oder einer Ptosis (hängendes Augenlid). Nach einigen Wochen bis Monaten können sogenannte bulbäre Symptome (Kau-, Schluck-, Sprechstörungen) hinzukommen. Nur selten bleibt die wahrgenommene Muskelschwäche auf eine funktionelle Partie beschränkt, sondern breitet sich im Krankheitsverlauf auf weitere Bezirke der Willkürmotorik aus: Nacken, Schulter, Oberarm, Oberschenkel, bis hin zur Atemmuskulatur. Dann wird von einer generalisierten Myasthenia gravis gesprochen, die sich bei der Mehrzahl der Betroffenen nach etwa zwei bis drei Jahren entwickeln kann. Das Suffix gravis ist hinweisgebend auf das Krankheitsbild in seiner schweren Ausprägung [11, 23, 31].
Ein wichtiges diagnostisches Charakteristikum zeigt sich in der raschen Ermüdbarkeit beanspruchter Muskeln, die allerdings nach einer ausreichenden Ruhephase wieder ihre Funktion zurückgewinnen können. Auch der Aspekt, noch am Morgen mit Muskeltätigkeiten wenig beeinträchtigt zu sein (z. B. Essen, d. h. Kauen und Schlucken sind gut möglich), was sich hingegen im Tagesverlauf deutlich verschlechtert, kann auf eine Myasthenia gravis hinweisen [23].
Da sich die Myasthenia gravis nicht homogen präsentiert, wurde ein Klassifizierungssystem entworfen, das die klinische Situation mit ihrer Schwere-Ausprägung der betroffenen Muskelgruppen abbildet. Hiernach werden fünf aufsteigende Grade festgelegt (Tab. 1) [31].
Tab. 1. Schweregrade der Myasthenia gravis – Klassifizierung nach Ossermann (nach [31])
|
Schweregrad |
|
1. Rein okuläre Form, auf äußere Augenmuskeln und Lidschluss beschränkt |
|
2. Leichtgradig generalisierte Form (ggf. additive okuläre Form) |
|
3. Mittelgradig generalisierte Form (ggf. additive okuläre Form) |
|
4. Schwere generalisierte Form |
|
5. Sehr schwere generalisierte, intubationsbedürftige Form |
|
Die Grade 2 bis 4 können nach Schwerpunkt der Symptomausprägung noch weiter unterteilt werden: |
|
a) Insbesondere die Muskulatur der Extremitäten und des Stammes sind betroffen |
|
b) Insbesondere die oropharyngeale Muskulatur und/oder Atemmuskulatur imponiert |
Andere Möglichkeiten der Klassifikation beziehen den Krankheitsbeginn, die Antikörper-Spezifitäten und die Thymus-Histologie ein. Vor allem der Laborbefund zur Serologie mit dem Nachweis spezieller Antikörper gewinnt immer mehr an Beachtung und kann hinweisgebend auf das Vorliegen beispielsweise eines Thymoms sein [11, 13].
Zur Diagnostik
Grundlage der Diagnostik bilden die gründliche Anamnese, die klinischen Befunde, insbesondere die körperliche Untersuchung: das wiederholte Durchführen lassen von Muskelbewegungen mit dem Ergebnis rascher Ermüdbarkeit (z. B. Augen-Lidschluss, Handöffnen), der Simpson-Test (längeres Nachobenblicken mit Ptosis-Provokation) oder auch der Einsatz von Kälte-Packs, die eine Verbesserung der Muskelleistung bewirken [11, 13, 16, 19, 31].
Pharmakologische Testung: Der sogenannte Tensilon®-Test mit Injektion des kurzwirkenden Acetylcholinesterase-Inhibitors Edrophoniumchlorid, der unmittelbar eine Besserung zeigt, ist heute mangels Verfügbarkeit faktisch bedeutungslos geworden. Einzig ein Präparat mit japanischer Zulassung (Antirex®) wäre als Import beziehbar. Ausweichend kann Pyridostigmin (p. o.) zur pharmakologischen Testung verwendet werden. Etwa eine Stunde nach Gabe sollte sich eine grundlegende Besserung zeigen. Auch Neostigmin (s. c.) erzeugt relativ rasch eine etwa halbstündig andauernde Besserung der präsentierten Muskellähmung.
Elektrophysiologische Untersuchung: Es zeigt sich nach repetitiver Nervenstimulation eine typische Amplitudenabnahme (Dekrement von > 10 %) des Nervs, der den Muskel versorgt. Dies ist zwischen erster und fünfter Reizung deutlich ausgeprägt und kann danach wieder steigen.
Besonders bei generalisierter Myasthenia gravis kann mit einem derartigen Messergebnis gerechnet werden (80 %), während es bei der rein okulären Form wesentlich seltener gefunden wird (etwa 20 %).
Labor: Ein Nachweis von Antikörpern im Serum, insbesondere gegen AChR, ist aufschlussreich, da sie bei etwa 80 bis 85 % der Patienten vorkommen. Dieses Ergebnis sollte durch weitere serologische Befunde gesichert werden. Ein positiver AChR-AK-Nachweis ist noch nicht beweisend, da er auch beim Lambert-Eaton-Syndrom, der amyotrophen Lateralsklerose und einigen weiteren Erkrankungen vorliegen kann. Es sollten auch das Alter und die Krankheitsdauer des Patienten beachtet werden, da im Verlauf zunehmend weitere Antikörper gegen Proteine der quergestreiften Muskulatur exprimiert werden. An Autoantikörpern sind bei Myasthenia gravis evident:
- AChR-AK (bei etwa 80 bis 85 % der Patienten)
- MuSK-AK (bei etwa 5 % nachweisbar; muskelspezifische Tyrosinkinase des AChR)
- Anti-Lrp4-AK (bei etwa 2 %, ein Transmembranprotein, Low-Density-Lipoproteine)
Weitere AK-Nachweise sind gegebenenfalls noch relevant und Gegenstand aktueller Evaluation:
- Anti-Titin-AK (Titin ist ein Strukturprotein des Muskelsarkomers)
- Anti-RyR-AK (Ryanodin-Rezeptor findet sich am Calciumkanal des Sarkomers)
- Anti-Agrin-AK (Agrin ist ein Proteoglycan-Protein, das die Aggregation des ACh mit seinem Rezeptor fördert)
Interesse erfährt das Vorliegen von Titin-AK (Eiweiß des Muskelsarkomers) bei jüngeren Patienten (< 40 Jahre), da sich hier eine hohe Korrelation zu einem Thymom belegen lässt. Ähnliches wird für den Anti-Ryanodin-Rezeptor-AK (RyR-AK) beschrieben.
Immerhin verbleiben heute noch etwa 5 % als sogenannte seronegative MG-Patienten, bei denen oben beschriebene Antikörper nicht nachweisbar sind bzw. als solche noch nicht bekannt sind [20a].
Bildgebung: Bildgebende Verfahren (CT, MRT) des Brustkorbs sind zur Feststellung von Veränderungen am Thymus stets angebracht, da ein eventuell vorliegendes Thymom erkennbar wird. Eine Indikation zur raschen operativen Entfernung (Thymektomie) ist zunehmend Konsensus, lediglich bei der leichten/okulären Myasthenie älterer Patienten wird Zurückhaltung empfohlen.
Therapeutische Möglichkeiten – Überblick
Die Therapie orientiert sich am Verlauf, am aktuellen Krankheitszustand, der Verträglichkeit und dem Ansprechen auf die Maßnahmen. Wesentliche Ziele werden dabei in der Remission von Muskellähmungen, der Verhinderung weiterer Progredienz sowie einer Vorbeugung von lebensbedrohlichen Krisen gesehen. In Bezug auf den zeitlichen Verlauf wird man bei nur leichten Beeinträchtigungen zunächst eher abwartend vorgehen, zumal Spontanremissionen mit etwa 10 % vorkommen. Im weiteren Krankheitsgeschehen – einhergehend mit Symptomverstärkung – ist allerdings eine Medikation oder weitere Interventionen angezeigt. An Therapieprinzipien können folgende Optionen grundsätzlich in Betracht gezogen werden [19, 15, 27, 31]:
- Symptomatische Basis-/Dauermedikation mit Hemmstoffen der Acetylcholinesterase
- Immunsuppressive Dauertherapie
- Thymektomie
- Plasmapherese
- Gabe von Immunglobulinen
- Reserve-Optionen (Gabe von Antikörpern bzw. Komplement-Inhibitoren)
Da die Myasthenia gravis überwiegend leicht und mit unsteten Funktionsstörungen kleinerer Muskeln beginnt, kann sich die Zeit bis zur sicheren Diagnose über Monate oder sogar Jahre hinziehen. Auch nach Diagnosestellung wird bei leichterer Präsentation häufig noch abgewartet. In der Regel wird medikamentös zunächst mit Acetylcholinesterase-Inhibitoren behandelt. Im weiteren Krankheitsverlauf wird dann ein Glucocorticoid gebraucht. Bei Bedarf wird ein Immunsuppresivum hinzugefügt, nicht zuletzt, um Glucocorticoide zu sparen, was die Verträglichkeit insgesamt verbessert. Die immunsuppressive Therapie zielt darauf ab, die pathogen agierenden Autoantikörper zu reduzieren, um die fortgesetzte Schädigung der neuromuskulären Endplatte weitgehend zu verhindern. Da bei nahezu 80 % der Betroffenen ein progredienter Krankheitsverlauf erwartet werden kann, der zu einer Verstärkung der Symptomatik mit Generalisierung führen kann, sind konsequente Interventionen im Rahmen der sogenannten Basistherapie wichtig [23].
Dennoch kann es infolge besonderer Umstände (z. B. Stress, Infektionen, Medikamente) zu krisenhaften Zuständen kommen, bei denen eine intensivmedizinische Behandlung mit Beatmung erforderlich wird. Für die Versorgung einer myasthenen Krise ist die Gabe von Immunglobulinen (0,4 g/kg Körpergewicht [KG] an fünf sukzessiven Tagen) ein gut durchführbares Verfahren. Die Plasmapherese, mit der ein Herauswaschen zirkulierender Autoantikörper temporär erfolgt, ist technisch deutlich anspruchsvoller und belastender für den Patienten (Shaldon-Katheter) [4].
Eine zunehmende Bedeutung erlangt die Thymektomie, die nach Detektion eines Thymoms, insbesondere bei jungen Erwachsenen innerhalb eines Jahres nach Krankheitsbeginn und Ausbleiben einer Remission – erwogen werden sollte [17, 32]. Das Entfernen der organischen Struktur, die für die Antikörperbildung verantwortlich ist, kann als kausaler Ansatz betrachtet werden und überzeugt mit beeindruckenden Ausheilungsraten (etwa 70 %). Lediglich die lange Zeitspanne bis zur Abheilung von bis zu einem Jahr muss durch Medikation (Glucocorticoide ± Azathioprin) gestützt werden [23] (Abb. 2).
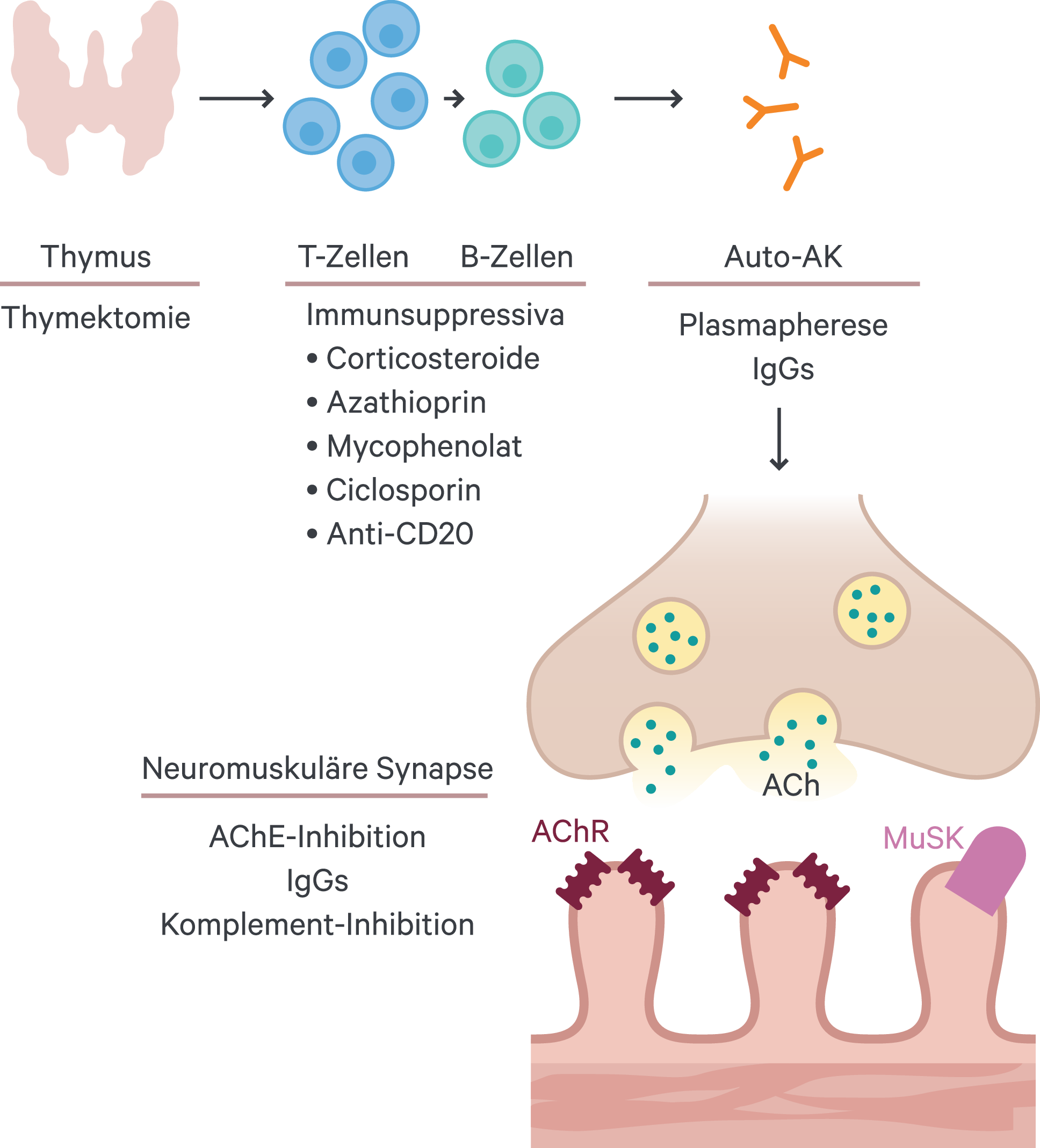
Abb. 2. Die physiologische Abfolge des pathologischen MG-Geschehens und die Interventionsmöglichkeiten. AChE: Acetylcholinesterase; AK: Antikörper; MuSK: muskelspezifische Tyrosinkinase des AChR
Acetylcholinesterase-Hemmstoffe (AChE-Inhibitoren)
Acetylcholin (ACh) stimuliert unter anderem die nicotinergen Rezeptoren der quergestreiften Muskulatur. Da ACh durch das Enzym Acetylcholinesterase inaktiviert wird, ist gerade die Hemmung dieses Enzyms ein probates Mittel, um die ACh-Verfügbarkeit zu erhöhen. Die heute gebräuchlichen Inhibitoren der Acetylcholinesterase blockieren das abbauende Enzym reversibel, womit ein vermehrtes Angebot des Neurotransmitters ACh erreicht wird.
Es sind die Klassiker in der Therapie der Myasthenia gravis: Eine erste Fallbeschreibung mit erfolgreichem Einsatz von Physostigmin geht auf das Jahr 1934 zurück (Walker, nach [11]). Aus der Erkenntnis, dass die Lähmung der Myasthenie-Patienten teilweise derjenigen mit Curare-Intoxikationen glich, kam es zu diesem erfolgreichen Therapieversuch. Ein Roche-Lehrfilm (mit Neostigmin®) aus dieser frühen Epoche veranschaulicht den Durchbruch mit drastischen Verbesserungen der zuvor noch gezeigten MG-bedingten Muskellähmungen (siehe [38]).
Die Gabe der AChE-Hemmstoffe erzielt bei gegebener Myasthenia gravis sehr rasch deutliche Symptomverbesserungen (Abb. 3). Für diese Art der Basis-/Akutbehandlung wird heute im Wesentlichen Pyridostigmin genutzt, das schnell anflutet (30–60 min), jedoch infolge kurzer Halbwertszeit alle vier bis fünf Stunden eingenommen werden muss. Bewährt hat sich ein Eintitrieren mit dreimal 10 mg/Tag, sodann 30 bis 90 mg alle vier bis fünf Stunden mit einer Erhaltungsdosis von etwa 360 mg/Tag. Von Vorteil ist die Verfügbarkeit einer halbierbaren Retardformulierung (à 180 mg), mit der die Einnahme-Intervalle verringert werden können oder die – am Abend genommen – auch die nächtliche Zeitspanne abdeckt (Tab. 2). Bei zu erwartender Belastung kann bedarfsadaptiert kurzfristig eine geringe Dosiserhöhung (10–20 %) erfolgen. Bei leichteren Formen der rein okulären Myasthenia gravis genügen bereits maximal 200 mg/Tag [23, 31]. Eine Tagesmaximaldosis von 600 mg soll allerdings wegen der Gefahr cholinerger Nebenwirkungen keineswegs überschritten werden.
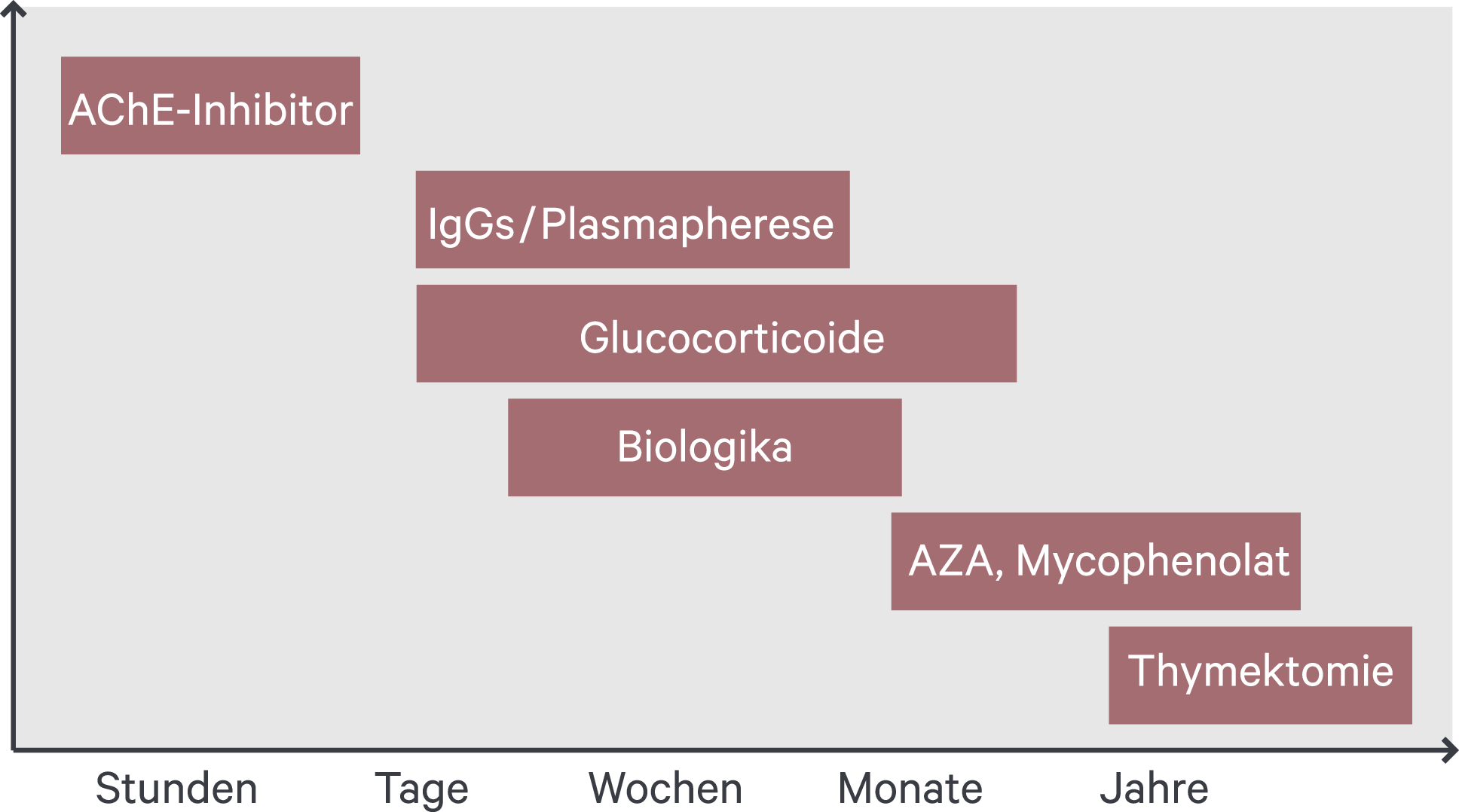
Abb. 3. Die Übersicht zeigt die potenziell nutzbaren Verfahren im Zeitverlauf ihres jeweiligen Wirkeintritts. Bei Glucocorticoiden kann es zu Beginn zu einer Verschlechterung kommen (adaptiert nach [24]). AZA: Azathioprin
Tab. 2. Acetylcholinesterase-Inhibitoren
|
AChE-Inhibitor |
Präparat (Handelsname) |
Dosis p. o. |
Dosis i. m. |
Pharmakokinetik |
|
Pyridostigminbromid |
Mestinon 1 mg Tbl. Mestinon 60 mg Ftbl. Kalymin 10 mg Ftbl. Kalymin 60 mg Ftbl. |
60 mg |
– |
Wirkeintritt ~ 30 min Wirkdauer ~ 3–6 h |
|
Mestinon 180 mg Ret. Kalymin 180 mg Ret. |
90 mg |
– |
Wirkeintritt ~ 1 h Wirkdauer ~ 6–10 h |
|
|
Mestinon 25 mg/5 ml Amp. Kalymin 5 mg/ml Amp. |
– |
2 mg |
Wirkeintritt ~ 10 min Wirkdauer ~ 2–3 h |
|
|
Distigminbromid |
Ubretid 5 mg Tbl. |
10 mg |
- |
Wirkeintritt ~ 30 min Wirkdauer ~ 10 h |
|
Neostigmin-Metilsulfat |
Neostigmin 0,5 mg Amp. Neostigmin Nasenspray NRF |
– |
1 mg |
Wirkeintritt ~ 10 min Wirkdauer ~ 1–2 h |
|
Ambenoniumchlorid |
Mytelase 10 mg Tbl. |
7,5 mg |
– |
Wirkeintritt ~ 30 min Wirkdauer ~ 8–10 h |
|
Edrophoniumbromid |
Antirex 10 mg Amp. |
– |
10 mg |
Wirkeintritt ~ 1 min Wirkdauer ~ 2–5 min |
Die Übersicht zeigt die potenziell nutzbaren Mittel. Die Pyridostigmin-Präparate werden in vielfältigen Varianten angeboten, wodurch sie die Therapie gut abdecken. In Ausnahmen kann auf Ambenonium (nur als Import!) zurückgegriffen werden. Neostigmin kann als Notfall-Option erwogen werden, ob nun als NRF-Rezeptur oder via MAD-Zerstäuber (mucosal atomization device, nasaler Zerstäuber). Distigmin wird als obsolet eingestuft [23, 31].
Die indirekt wirkenden Parasympathomimetika zeigen ihre Effekte nicht nur an der neuromuskulären Endplatte, sondern sind überdies im autonomen Nervensystem bei der Innervation der glatten Muskulatur wirksam, sodass es zu unerwünschten Effekten kommen kann. Magen-Darm-Störungen, Schwitzen, Hypersalivation, Blasendrang und Bradykardie sind potenzielle Begleiteffekte, auf die die Patienten aufmerksam zu machen sind. Im Therapieverlauf werden häufig Immunsuppressiva hinzugefügt, die den Substanzbedarf an Pyridostigmin etwas verringern. Trotz der beeindruckenden Symptomverbesserung bleibt die alleinige Gabe von Acetylcholinesterase-Inhibitoren lediglich für nur etwa 10 % der Patienten dauerhaft eine Option, sodass Kombinationen mit weiteren Arzneimitteln, den Immunsuppressiva, erforderlich sind.
Exkurs cholinerge Krise: Die Auswirkungen eines Überangebots an Acetylcholin infolge von Überdosierung des Cholinesterasehemmers mit den Anzeichen von Bradykardie, Miosis, Hypersalivation, Krämpfen und einer akuten allgemeinen Muskelschwäche sind nicht trivial und können sich zu einem medizinischen Notfall entwickeln (vgl. Kampfmittelintoxikation). Eine rasche Gabe von Atropin als Antidot und Intubation mit Beatmung müssen bereitstehen und sollten situativ erwogen werden [11, 13].
Das Immunsystem beeinflussende Therapeutika
Wie bereits oben ausgeführt, liegt der Myasthenie eine Autoimmunstörung zugrunde: Komponenten und Teile des Immunsystems zeigen dabei eine veränderte, pathologische Toleranz gegenüber körpereigenen Strukturen und bilden sogenannte Autoantikörper. Es ist somit konsequent, in das fehlgeleitete immunvermittelnde Geschehen einzugreifen. Mit Glucocorticoiden, Immunsuppressiva, Antikörpern oder auch der Entfernung pathogener Autoantikörper durch Plasmapherese sind gleich eine ganze Reihe interessanter Möglichkeiten gegeben, die im Folgenden vorgestellt werden (Tab. 3) [3, 16, 15, 23, 25].
Tab. 3. Übersicht der medikamentösen Optionen zur Therapie der Myasthenia gravis
|
Therapieoption |
Wirklatenz |
Bemerkungen |
|
Basis-/symptomatische Dauertherapie |
||
|
Acetylcholinesterase-Inhibitoren |
½ h |
Krankheitsverlauf wird nicht beeinflusst! |
|
Pyridostigmin |
Verteilt auf mehrere ED (4 × 60 mg); Überdosis kann cholinerge Krise hervorrufen |
|
|
Immunsuppressive Dauertherapie (Günstig für den Krankheitsverlauf) |
||
|
Azathioprin 100–200 mg/Tag |
6–9 Monate |
Azathioprin ist 1. Wahl, max. 3 mg/kg KG/Tag, Blutbild-, Leberwertkontrolle |
|
Mycophenolat 1000–2500 mg/Tag |
5–6 Monate |
Ausweichmittel bei Azathioprin-Unverträglichkeit; Blutbild-, Leberwertkontrolle |
|
Glucocorticoid-intermediär-Therapie |
||
|
Prednisolon 60 mg/Tag als Zieldosis |
3–4 Wochen |
Als Kombinationsoption/Cushing-Schwelle |
|
Reserve-Optionen |
||
|
Ciclosporin 200–400 mg/Tag |
2–3 Monate |
Calcineurin-Inhibitor; Nephrotox.; Blutbild, Nieren- und Leberwerte |
|
Tacrolimus 4–10 mg/Tag |
2–3 Monate |
Calcineurin-Inhibitor; Nephrotox.; Blutbild, Nieren- und Leberwerte |
|
Methotrexat (MTX) 7,5 mg/Woche |
2–3 Monate |
Blutbild, Nieren- und Leberwerte |
|
Rituximab |
2 Monate |
AK, der an das CD20-AG der Lymphozyten bindet, bei refraktärer Anti-MuSK-MG |
|
Eculizumab |
2 Monate |
Komplement-Hemmstoff, extrem teuer |
|
Cyclophosphamid |
2–3 Monate |
Erhebliche Toxizität schränkt seinen Nutzen sehr stark ein |
Basisversorgung
Glucocorticoide: Prednison bzw. Prednisolon sind hochbewährt, entfalten allerdings sehr vielfältige, komplexe Wirkungen [5]. Bekanntlich spielt Cortison während Stresszuständen des Körpers eine elementare Rolle, indem es zur Mobilisierung von Ressourcen mit Bereitstellung von Energie kommt. Andererseits greifen die Glucocorticoide in entzündliche Prozesse ein und erreichen dort über den Zellstoffwechsel eine beachtliche Hemmwirkung. Hieraus leitet sich der im Kontext der MG-Therapie gewünschte immunsuppressive Effekt ab. Im Detail wird die Zytokinproduktion unterdrückt sowie ein durch einen Transkriptionsfaktor aktivierter B-Lymphozyten-Vorgang gehemmt, der für die Immunantwort wichtig ist. Letztendlich ist die Hemmung der die Myasthenia gravis fördernden Autoantikörper die eigentlich gewünschte Intention.
Der therapeutische Gebrauch von Glucocorticoiden bei Myasthenia gravis ist lange schon etabliert [11]. Als Zieldosis hat sich 1 mg Prednisolon/kg KG bewährt, das zu Therapiebeginn mit 10 bis 20 mg einschleichend und mit 5 mg pro Folgetag aufdosiert werden muss. Allerdings kann es zunächst zu einer leichten, vorübergehenden Verschlechterung der MG-Symptomatik kommen. Die morgendliche Gabe (analog zur physiologischen Ausschüttung) ist Standard. Nach Stabilisierung und Symptomkontrolle (Cushingschwelle beachten) wird sodann wieder eine Reduktion der Dosis durch langsames Herabtitrieren (z. B. Gabe nur jeden zweiten Tag) erreicht. Später im Verlauf erfolgt die additive Gabe eines (reinen) Immunsuppressivums, um ein stabiles Regime für die notwendige Langzeitkontrolle der Myasthenia gravis zu erreichen, was beispielsweise mit Azathioprin geschieht. Azathioprin zeigt seine Wirkung allerdings mit deutlicher Latenz, kann aber die gewünschte Glucocorticoid-Einsparung erbringen. Auf die unerwünschten Arzneimittelwirkungen (UAW) einer langfristigen Glucocorticoid-Gabe (z. B. in Form von beispielsweise Osteoporose, Stoffwechselstörungen, Magen-Ulzera, Ödeme) muss geachtet werden [31, 23].
Azathioprin ist ein Purinanalogon, das als Prodrug im Organismus rasch in die aktive Form 6-Mercaptopurin metabolisiert wird. Das Nukleosid interagiert mit der RNA- und DNA-Synthese der Immunzellen, insbesondere wird die Zahl an Lymphozyten gesenkt (zytotoxische Wirkung). Es ist eine hochbewährte, gut verträgliche und kostengünstige Substanz, die noch weit verbreitet ist (Zulassungsstatus bei Myasthenia gravis ist gegeben und erste Wahl). Die Standarddosis beträgt 1,5 bis 2 mg/kg KG. Der Wirkeintritt erfolgt mit einer erheblichen Latenz nach etwa sechs bis neun Monaten.
Auf eine mögliche gleichzeitige Einnahme von Allopurinol ist zu achten, da es zur Interaktion durch das abbauende Enzym Xanthinoxidase kommt, wodurch bereits geringe Azathioprin-Dosen eine gefährliche Leukozytopenie provozieren können. Durch eine Dosisreduktion von Azathioprin auf etwa 25 % kann dem Problem abgeholfen werden. Dennoch können hartnäckige gastrointestinale Nebenwirkungen oder Leberwerterhöhungen einen Substanzwechsel ratsam erscheinen lassen. Regelmäßige Kontrollen des Blutbilds und der Leberwerte sind obligat [16, 29, 31] (siehe Patientenfall 1).
Patientenfall 1
E. H., männlich, 77 Jahre
Vor 13 Jahren (im Alter von 64 Jahren) stellte sich spontan eine anhaltende Ptosis des rechten Augenlids ein: morgens mit geringerer Ausprägung, jedoch zunehmend im Tagesablauf. Darauf wurde zunächst wenig geachtet. Schließlich dann doch Konsultation beim Augenarzt, Diagnose: Oculomotoriusparese links.
Erstaunlicherweise verschwand die Ptosis nach einigen Monaten. Im Folgejahr veränderte sich die Sprache: sie war verwaschen, stark näselnd-schnarrend, nicht mehr zu verstehen. Auch traten nun vermehrt Schluckstörungen auf. Dies beunruhigte, sodass eine stationäre Aufnahme zur Abklärung erfolgte.
In der neurologischen Klinik wurden gezielt Myasthenie-spezifische Untersuchungen durchgeführt und die Diagnose Myasthenia gravis Grad II gestellt.
Medikation bei Entlassung:
- Pyridostigmin (Mestinon®) 60 mg 4-mal
- Pyridostigmin (Mestinon® retard) 90 mg zur Nacht
- Prednisolon (Decortin H®) 80 mg
- Azathioprin (Imurek®) 150 mg
- Pantoprazol 20 mg
Regelmäßige monatliche, ärztliche Kontrollbesuche, Differenzialblutbild (insbes. Lymphozyten), Prüfung der Cortison- und Azathioprin-Dosen waren obligatorisch.
Wegen diverser Beschwerden waren in der nachfolgenden Zeitspanne zwei weitere stationäre Aufenthalte notwendig. Herr H. betont ausdrücklich, dass es aus seiner langjährigen Erfahrung wichtig sei, sehr individuell auf die jeweilige Situation einzugehen, da es kein allgemein gültiges Medikationsregime gebe.
Die aktuelle Medikation (100 mg Azathioprin und 120 mg Pyridostigmin täglich) sowie die fachärztliche Betreuung (Myasthenie-Sprechstunde einmal pro Jahr) werden als sehr wichtig beschrieben. Herr H. äußert sich insgesamt sehr zufrieden mit seiner Situation, schätzt den Austausch in der Selbsthilfegruppe und zeigt sich auch sportlich in seiner Boule-Gruppe engagiert.
Obwohl gut etabliert, zeigt sich bei etwa 10 bis 20 % der Patienten kein suffizientes Ergebnis, auch nicht in Kombination mit einem Glucocorticoid, sodass weitere medikamentöse Optionen gefunden werden müssen.
Mycophenolatmofetil ist eine Option, wenn Unverträglichkeiten gegenüber Azathioprin bestehen (positiver Beschluss des G-BA). Es liegt als Prodrug vor, wird in vivo rasch zur Mycophenolsäure aktiviert, die ihrerseits die Inosinmonophosphathydrogenase hemmt, ein Enzym, das in T- und B-Lymphozyten vorkommt. Die Substanz hat ihre eigentliche „Karriere“ bereits in der Transplantationsmedizin gemacht (Unterdrückung der Organabstoßung). Die übliche Dosis beträgt 2 g/Tag, verteilt auf zwei Gaben. Es soll nicht mit Azathioprin kombiniert werden. Die hepatische Verträglichkeit ist besser, dennoch können UAW wie hämolytische Anämie, chronische Diarrhö, opportunistische Infektionen oder sogar ein ZNS-Lymphom seinen Einsatz limitieren. Der Wirkeintritt wird frühestens nach sechs Monaten erwartet [25, 23, 31].
Immunsuppresiva der Reserve (überwiegend Off-Label-Anwendungen)
Methotrexat (MTX) wird schon länger als Zytostatikum bei Leukämie genutzt. Es hemmt die Dihydrofolat-Reduktase und die von der Folsäure abhängige De-novo-Synthese von Purin und Thymidin. Die gestörte Synthese von DNA bzw. RNA führt über den Funktionsverlust zum Zelltod von überwiegend B-Lymphozyten. Obwohl nur wenige Berichte vorliegen, hat MTX wegen langjähriger Erfahrung eine gute Akzeptanz. Die möglichen UAW erscheinen unter Beachtung der Vorsichtsmaßnahmen (Ausschluss von Lebererkrankungen, GI-Ulzera, Blutbildstörungen) gut beherrschbar und mit der wöchentlich zu verabreichenden Dosis von 7,5 bis 15 mg (plus Folsäure-Supplementation nach ein bis zwei Tagen) einfach durchführbar. Ein Wirkeintritt dürfte nach etwa zehn Monaten erwartet werden [31, 23].
Ciclosporin (aus der Transplantationsmedizin bekannt) hemmt die Aktivität von Calcineurin und Interleukin 2 und somit die Aktivität der T-Zellen. Die Dosis beträgt einleitend 3 bis 4 mg/kg KG, es erfolgt ein relativ rascher Wirkeintritt nach vier bis sechs Wochen. Da allerdings zahlreiche Wechselwirkungen bestehen (CYP-Pan-Induktor), wie auch schwere potenzielle UAW-Risiken (Nephrotoxizität, opportunistische Infektionen) vorkommen können, ist eine engmaschige Spiegelkontrolle unerlässlich. Es gilt als ein Mittel der ferneren Reserve. Dem Ciclosporin gleicht Tacrolimus nach Wirkungsprinzip und UAW, hat dosisbezogen jedoch eine deutlich stärkere Wirksamkeit (10- bis 20-fach) [23, 29, 31].
Für alle vorgenannten Stoffe ist eine sorgfältige Nutzen-Risiko-Abwägung für den individuellen Behandlungsfall zu treffen, zudem muss die Thematik Kontrazeption zwingend beachtet werden.
Immunsuppressiva in der Eskalationstherapie
Es bestehen erweiterte Optionen, die überwiegend durch spezialisierte MG-Therapiezentren für dezidierte Fälle vorbehalten sind. Als individueller Heilversuch im Rahmen einer Eskalationstherapie sind sie auch für sonst refraktäre Fälle denkbar. Refraktär bedeutet in diesem Zusammenhang fortbestehende erhebliche MG-Symptomatik trotz adäquater Glucocorticoid- und Azathioprin-Gaben, massive Verschlechterung, Notwendigkeit von IgG-Applikationen sowie schwerwiegende UAW (siehe Patientenfall 2) [26, 29, 31].
Patientenfall 2
D. P., weiblich, 50 Jahre
Vorstellung in neuroimmunologischer Sprechstunde 2019.
Vorbefunde
Generalisierte Acetylcholinrezeptor-Antikörper-positive Myasthenia gravis, ED 1987, MGFA III
- Thymektomie 1988, im Verlauf MRT-Thorax ohne Anhalt für Rezidiv
- Wiederholte krisenhafte Verschlechterungen, zuletzt 2/2019 mit Gabe von intravenösen Immunglobulinen (150 g)
- Bisherige Therapien:
- Azathioprin von 2000 bis 2015 (Hepatopathie)
- Mycophenolatmofetil seit Anfang 2015 (zuletzt 2,5 g tgl.)
- Rituximab 2-mal 1 g Januar und Februar 2017 ohne Effekt
- Wiederholte Immunglobuline in Anbindung an das Universitätsklinikum bis 08/2018
Reanimation im Jahr 2009
Zwischenanamnese
Wiedervorstellung zur Verlaufskontrolle, krisenhafte Verschlechterung. Zunächst guter Effekt durch i. v. Immunglobuline, habe aber in den letzten Wochen wieder nachgelassen. Unlängst Diagnose einer Helicobacter-pylori-positiven Gastritis mit Notwendigkeit einer Antibiose (Tripel-Therapie). Retrospektiv deutliche Verschlechterung der Myasthenie seit dem Jahr 2009 nach Entbindung eines gesunden Kindes. Zuvor habe sie keinerlei körperliche Einschränkungen gehabt, sei Tanzen gewesen und sozial voll integriert. Inzwischen, nach Therapieversagen bzw. der nebenwirkungsbedingten Notwendigkeit des Absetzens von Azathioprin, deutlich eingeschränkte körperliche Belastbarkeit, sie könne den Haushalt notdürftig versorgen, leide unter Kopf- und Rückenschmerzen aufgrund der objektiven Überlastung. Sie sei nicht in der Lage, Treppen zu steigen. Es bestehen bulbäre Symptome mit Schluckbeschwerden, Kauschwäche, einem Kloßgefühl im Hals, mitunter Aspiration von Flüssigkeiten. Sprache nach längerem Sprechen undeutlich, Doppelbilder beim Blick in alle Richtungen, insbesondere beim Blick nach rechts. Seit 2015 Berentung.
Aktuelle Medikation
- Prednison: Decortin® 10 mg
- Mycophenolatmofetil: Cellcept® 2 g
- Pyridostigminbromid: Kalymin® 60 2–2–1 und Mestinon® retard 180 zur Nacht
- Vitamin D3: Dekristol® 20 000 I. E./Woche
- Omeprazol 20 mg
Beurteilung
Aktuell eine Beschwerdezunahme, zwischenzeitlich mit i. v. Immunglobulinen gelindert, jedoch durch die wiederholten und therapeutisch nicht ausreichend effektiven Therapieversuche (Azathioprin, Mycophenolatmofetil, Rituximab) belegt und die erhebliche belastungsinduzierte okulobulbäre sowie Extremitätenschwäche, findet sich eine therapierefraktäre generalisierte Myasthenia gravis.
Eine Therapie mit dem Komplement-Inhibitor Eculizumab wird erwogen. Meningokokken-Impfschutz ist sicherzustellen. Terminierung des Infusionstermins erfolgt über die Ambulanz. Die Medikation wurde aktuell unverändert beibehalten.
Letzter Befund
Augen kräftig, Augenschluss nicht überwindbar, diskrete Ptosis links, gute Mimik, Kopfbeugung kräftig, Abduktion kräftig. Beinhalteversuch 30 Sekunden, Kopfhalteversuch 60 Sekunden, Armhalteversuch 90 Sekunden stabil.
Prozedere
Prednisolon reduziert auf 5 mg, Cellcept® auf 1500 mg täglich, weitere Prednisolon-Reduktion im Verlauf, dann versuchsweise Ausdehnung der Intervalle des Eculizumabs, von der Patientin gewünscht und klinisch vertretbar. Zunächst Wiedervorstellung in 14-tägigem Zyklus für die nächsten 12 Monate.
Rituximab ist ein Anti-CD-20-Antikörper, der zur Depletion CD20-positiver B-Zellen führt und sich als wirksam in der anti-MuSK-seropositiven Myasthenia gravis gezeigt hat [9]. Zweimal pro Jahr werden je zwei Dosen à 1000 mg i. v. (Tag 1 und 15) appliziert, geringere Dosen sind wohl auch möglich. Eine gründliche Nutzen-Risiko-Abwägung ist zu stellen, da schwerwiegende UAW vorkommen können (opportunistische Infektionen). Die zur Therapie genutzten Antikörper sind per se in der Lage, Abwehrprozesse in Gang zu bringen (auf eine sogenannte Desensibilisierung ist dabei zu achten). Von Vorteil kann ein Wirkeintritt nach wenigen Wochen sein [29, 31].
Eculizumab: Da es bei der Besetzung der ACh-Rezeptoren durch die Autoantikörper in der Folge zu einer Beteiligung des körpereigenen Komplementsystems mit Zerstörung der postsynaptischen Strukturen kommt, ist es konsequent, genau in dieses Geschehen einzugreifen. Mit Eculizumab liegt ein IgG2/4k-Antikörper vor, der mit hoher Effizienz an das Komplementprotein C5 bindet und somit die Zerstörung postsynaptischer Strukturen unterbindet [6, 12, 26].
Es wurde unter sehr rigiden Auflagen für die Therapie der refraktären generalisierten Myasthenia gravis zugelassen. Dies bedeutet, dass zuvor zwei erfolglose Therapieversuche mit Immunsuppressiva vorangegangen sein mussten (bei Patienten mit positivem ACh-AK-Nachweis). Die Therapie erfolgt durch ein Eindosieren über vier Wochen mit 900 mg wöchentlich, um dann mit 1200 mg im Intervall von zwei Wochen dauerhaft fortgeführt zu werden [8]. Auf opportunistische Infektionen ist zu achten, gegen Meningokokken ist vorab zu impfen. Mit etwa 400 000 Euro pro Jahr an Medikationskosten handelt es sich um eine extrem kostspielige Option [26]. Weitere Inhibitoren des C5-Komplements befinden sich in der klinischen Erprobung (Ravulizumab, Zilucoplan) [18, 30].
Darüber hinaus finden sich vielversprechende Ansätze mit der Entwicklung von Antikörpern gegen den neonatalen Fc-Rezeptor. Dieser ist membranständig und reguliert die zirkulierenden IgGs. So soll die Bereitstellung von Antikörpern (so auch den autoaggressiven) herabgesetzt werden. Efgartigimod und Rosanoliximab sind Kandidaten mit ersten positiven Ergebnissen [18, 33]. Efgartigimod alfa erhielt Anfang August die europäische Zulassung zur Behandlung erwachsener Patienten mit generalisierter Myasthenia gravis, die ACh-AK-positiv sind [2a].
Myasthene Krise – die Notfallsituation
Bei immerhin bis zu 20 % der MG-Patienten kann es zu einer krisenhaften Zuspitzung mit drastischer Verschlechterung der Lage kommen: Infolge aktueller Zunahme des autoaggressiven Prozesses mit Schwächung der Atem- und Schluckmuskulatur wird eine respiratorische Insuffizienz mit Aspirationsgefahr hervorgerufen. Auslöser können akute (pulmonale) Infektionen, (neu angesetzte) Arzneimittel, Narkosen (post-OP!), Dosisveränderungen oder Adhärenzprobleme der angesetzten MG-Arzneimittel sein [21, 22].
Die myasthene Krise stellt mit einer Mortalität von etwa 3 % trotz intensivmedizinischer Versorgung immer noch eine ernste Komplikation dar. Faktoren wie etwa die Multimorbidität, vorangegangene Krisen, das Lebensalter und das Ausmaß der respiratorischen Beeinträchtigung sind dabei wichtige Risikofaktoren. Die eigentliche Krisenintervention kann durch die Gabe von Pyridostigmin i. v. (maximal 1/30 der p. o. Dosis, üblich als 1 mg langsam injiziert), Anwendung von Immunglobulinen (s. o.), ferner durch die Plasmapherese oder Immunadsorption erfolgen [14, 23].
Eine Reihe von Kliniken haben während der letzten Jahre integrierte Myasthenie-Zentren aufgebaut, die derartige spezialisierte Angebote bereitstellen (Übersicht bei [2]).
Mögliche Unterstützung der Therapie durch klinische Pharmazeuten
Die Myasthenie erfordert Medikationen, die über Jahre oder auf Dauer anzuwenden sind. Dabei kommen überwiegend gut etablierte Arzneimittel zum Einsatz. Deren indizierter und korrekter Gebrauch sichert eine sehr zufriedenstellende Krankheitskontrolle [21]. Es ist somit nur konsequent, wenn die vorhandenen Optionen zur Therapie der Myasthenia gravis möglichst optimiert genutzt werden und mit Bedacht zur Anwendung kommen [28].
Allerdings kann es infolge des Arzneimittelgebrauchs, ob durch Neuansetzung, Komedikation, Wechsel der Präparate, Änderung von Dosen, Anwendungs- und Adhärenzproblemen zu vielfältigen Störungen kommen, wo Unterstützung hilfreich wäre. In der US-Leitlinie gibt es explizit einen Abschnitt („Guidelines for the Pharmacist“), worin die aktive Beteiligung in Umgang mit Komedikationen thematisiert wird [7]. Klinische Pharmazeuten bergen ein Potenzial, da solide Kenntnisse vorliegen bei
- der Identifizierung von Patienten mit erhöhten Risiken,
- der Auswahl geeigneter Arzneimittel bei der Arzneimittelanamnese,
- der Identifizierung von kritischen, die Myasthenie verstärkenden Arzneimitteln,
- der Bewertung von Kontraindikationen,
- der Bewertung von potenziell kritischen Wechselwirkungen und
- der Weitergabe von Anwendungshinweisen,
womit in der Summe zu einer sicheren Medikation und einer besseren Therapie durch Förderung von Adhärenz beigetragen werden kann.
Ein beachtlicher Anteil an Verlaufsverschlechterungen dürfte auf neu angesetzte Arzneimittel zurückführbar sein [23]. Die Liste kritischer Wirkstoffe ist recht lang. Neben einigen wenigen No-Gos (D-Penicillamin, Botulinumtoxin, Interferone, Lebendimpfstoffe, Suxamethonium), die eine Myasthenia gravis triggern bzw. zur Exazerbation bringen können, gibt es eine große Gruppe potenziell riskanter Medikamente, die möglichst vermieden werden sollten, wie etwa Magnesiumsalze, i. v. Glucocorticoide, Betablocker und Calciumantagonisten, einige Antibiotika (Gyrasehemmer, Makrolide, Aminoglykoside) sowie Checkpoint-Inhibitoren. Hier gilt es, rechtzeitig nach weniger riskanten Alternativen zu schauen. Die dritte Gruppe – umfangreich genug – umfasst problematische Substanzen, die unter guter (klinischer) Beobachtung (und Dosisanpassung) anzuwenden sind. In jedem Falle sollte nach Dosisänderung oder Neuansetzen eines Arzneimittels stets sehr aufmerksam auf eine eventuelle Veränderung der Myasthenie-Symptomatik geachtet werden (Tab. 4, aktuelle Übersichten bei DMG, MGFA, Neurologienetz, [7, 20, 23, 31]).
Tab. 4. Auflistung von für Myasthenia-gravis-(MG-)Patienten kritischen Medikationen (auszugsweise nach DMG, MGFA, Neurologienetz)
|
Kritische Medikationen – Betrachtung der Risiken |
||
|
Stoffklasse |
Typ, Beispiel |
Bemerkungen und Hinweise |
|
Antibiotika |
Aminoglykoside (Gentamicin u. a.) |
Können MG verschlechtern, Alternativen erwägen |
|
Gyrasehemmer (Ciprofloxazin u. a.) |
Können MG verschlechtern, Alternativen erwägen |
|
|
Makrolide (Clarithromycin u. a.) |
Können MG verschlechtern, Alternativen erwägen |
|
|
Betablocker |
Sympatholytika (Metoprolol u. a.) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
Antiepileptika |
Antikonvulsiva (Phenytoin u. a.) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
Botulinumtoxine |
Anti-Spastik (Botox u. a.) |
Unterbindet neuromuskuläre Aktivierung; sehr strenge Indikationsstellung!! |
|
Calcium-Blocker |
Antihypertensiva (Amlodipin u. a.) |
Können MG verschlechtern (Transmission/Muskel), Alternativen erwägen |
|
Antiarrhythmika (Verapamil u. a.) |
Können MG verschlechtern (Transmission/Muskel), Alternativen erwägen |
|
|
Checkpoint-Inhibitoren |
Krebstherapie (Nivolumab u. a.) |
CAVE: de novo MG möglich; nur unter strengster Abwägung |
|
Chinin |
Malaria-Mittel (= Curare-artig) |
CAVE: kann MG verschlechtern |
|
Chloroquin |
Rheuma-Reserve, Resochin u. a.; (= immunsuppressiv) |
Kann MG verschlechtern; falls indiziert: eng überwachen |
|
Corticosteroide |
Immunsuppressiva (Prednisolon u. a.) |
Eine wichtige Option bei MG, die die Erkrankung jedoch während der ersten beiden Therapie-Wochen verschlechtern kann! |
|
Deferoxamin |
Hämatotherapie |
Chelatbildner, kann MG verschlechtern, überwachen |
|
Diuretika |
Antihypertensiva (Torasemid u. a.) |
Können MG verschlechtern (Elektrolyte), Dosis anpassen |
|
D-Penicillamin |
Rheuma-Reserve, Resochin u. a. (= immunsuppressiv) |
CAVE: kann MG hervorrufen, streng vermeiden! |
|
Hypnotika |
Benzodiazepine (Oxazepam u. a.) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
Z-Substanzen (Zopiclon u. a.) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
|
Antihistaminika (Doxylamin u. a.) |
Können MG verschlechtern, vermeiden! |
|
|
Impfstoffe |
Lebendvakzine (Varizella-Zoster u. a.) |
CAVE: streng kontraindiziert |
|
Totvakzine |
Gefahr eines unzuverlässigen Impfschutzes (Titer prüfen!) |
|
|
Lokalanästhetika |
Procainamid |
Können MG verschlechtern, Vorsicht, Lidocain-Typ bevorzugen |
|
Magnesium-Salze |
Tokolytikum (Mg-Varia) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
Narkosemittel |
Muskelrelaxanzien (Atacuronium u. a.) |
CAVE: Curare-Typ drastische Dosisreduktion, Succinycholin-Typ wählen |
|
Opioid-Analgetika |
Analgesie (Morphin u. a.) |
Können MG verschlechtern, unter Vorsicht nutzbar |
|
Parkinson-Medikamente |
Anticholinergika (Budipin u. a.) |
Können MG deutlich verschlechtern, meiden, Ausweichpräparate! |
|
Psychopharmaka |
Trizyklische Antidepressiva (Doxepin u. a.) |
Können MG deutlich verschlechtern, meiden, Alternativen! |
|
Anticholinerge Neuroleptika (Promethazin) |
Können MG deutlich verschlechtern, Ausweichpräparate |
|
|
Bipolar-Medikation (Lithium u. a.) |
Können MG verschlechtern, enges Spiegelmonitoring |
|
|
Sexualhormone |
Estrogen/Gestagen |
Stör-Effekte auf MG im Zyklus-Verlauf möglich |
|
Statine |
Lipidsenker (Atorvastatin u. a.) |
Auf Muskelwirkungen achten, unter Vorsicht nutzbar |
|
Urologika |
Spasmolytika (Atropin, Oxybutynin u. a.) |
Auf Muskelwirkungen achten, unter Vorsicht nutzbar |
In Zusammenhang mit der Myasthenia gravis sollten Herausforderungen durch besondere Problemlagen angesprochen werden, die sich beispielsweise durch eine Operation, Schwangerschaft oder das Impfen ergeben.
OP und Narkose: Nach Möglichkeit soll eine Lokalanästhesie angestrebt werden (mit einem Anästhetikum vom Amid-Typ). Für eine OP sollen die Curare-ähnlichen Stoffe zur Anwendung kommen (z. B. Alcuronium), die jedoch mit nur 10 bis 50 % der üblichen Dosis und damit deutlich geringer dosiert werden müssen. Bedingt durch die Myasthenia gravis kann bereits eine respiratorische Schwäche mit Schluckstörungen (Aspirationsgefahr) erschwerend vorhanden sein. Auch die Auswahl an Stoffen zur üblichen Prämedikation ist zu reflektieren (zentral dämpfende vermeiden, keine Benzodiazepine). Die vorhandene MG-Dauermedikation (Azathioprin oder Pyridostigmin) wird üblich fortgeführt. Die Umrechnung bei der postoperativen Umstellung der oralen Pyridostigmin-Dosis auf eine erforderliche i. v. Dosis beträgt 1 zu 30. Auch während dieser Phase muss noch auf eventuell krankheitsbedingte Schluckstörungen geachtet werden (Aspirationsgefahr durch postoperatives Erbrechen) [11, 23, 31].
Schwangerschaft: Das Vorliegen einer Myasthenia gravis steht einer möglichen Schwangerschaft bzw. einem Kinderwunsch nicht entgegen. Vor Eintritt der Schwangerschaft sollte allerdings ein stabiler Remissionszustand erreicht werden, und dies durch eine – nach aktuellem Stand – sichere Medikation. Mit den ACh-Inhibitoren und den Glucocorticoiden stehen bewährte Standardmedikationen zur Verfügung. Kritisch gesehen werden allerdings etwaige Vorbehandlungen mit Mycophenolat, MTX oder Tacrolimus, die spätestens bereits drei Monate vor einer geplanten Schwangerschaft abgesetzt und auf ein unproblematisches Mittel umgestellt werden müssen. Hilfestellung bietet das Pharmakovigilanz- und Beratungszentrum für Embryonaltoxikologie Embryotox [35].
Die hormonellen Umstellungsvorgänge während der Schwangerschaft können per se insbesondere im ersten Trimenon zu leichteren Verschlechterungen der Myasthenia gravis führen. Die Therapie mit Azathioprin und Ciclosporin kann nach derzeitigem Kenntnisstand während der Schwangerschaft fortgeführt werden, es sollte jedoch unter Behandlung mit diesen oder den vorgenannten kritischen Stoffen nicht gestillt werden. Zum Zeitpunkt der Geburt muss auf die sogenannte neonatale Myasthenie geachtet werden, die bei etwa 10 % der Neugeborenen vorkommt. Die Kinder zeigen transient eine Muskelschwäche, sind unfähig zu trinken und sollten bis zum Abklingen der Symptome für einige Tage Acetylcholinesterase-Inhibitoren erhalten. Im Falle von MG-Exazerbationen während der Schwangerschaft gelten IgGs oder gegebenenfalls die Plasmapherese als sichere Verfahren [10, 23, 13a].
Impfungen: Sie sind ein gezielt gewähltes Training für das Immunsystem, um den Organismus für einen späteren Kontakt mit einem Erreger zu ertüchtigen. Infektionen, die mit einer Aktivierung immunogener Prozesse einhergehen, gelten an sich als ein Trigger für myasthene Krisen. Daher ist beim kalkulierten Einsatz von Impfstoffen stets Vorsicht geboten. Die Impfstoffe (Antigene) werden weit überwiegend als Toxoid/Tot- oder Lebend-Impfstoffe angeboten. Die Totimpfstoffe werden relativ gut vertragen. Dennoch sollte der Titer geprüft werden, da eine immunsuppressive MG-Medikation die Immunantwort behindern und somit den intendierten Impfschutz abschwächen kann. Die Lebendvakzine sind hingegen bei Patienten unter Immunsuppression absolut kontraindiziert und sollten bei MG-Patienten ohne immunsuppressive Medikation nur unter strengster Indikationsstellung erwogen werden [31, 23].
Aktuelle Empfehlungen für MG-Patienten der STIKO stellen den sicheren Nutzen der Corona- und Influenza-Impfstoffe heraus.
Anwendungshinweise – Myasthenie im Alltag
Betroffene sollten für etwaige Symptomverschlechterungen (muskuläre Beschwerden) sensibilisiert werden, insbesondere, wenn diese in Zusammenhang mit Infekten oder Fieber auftreten. Auch neu oder vermehrt auftretende Schluck- und Atemstörungen sollten Anlass für eine ärztliche/neurologische Abklärung sein. Die MG-Medikation muss regelmäßig und gewissenhaft eingenommen werden. Dosisänderungen oder situativ hinzugefügte Arzneimittel können schwerwiegende Folgen nach sich ziehen. Wichtige Aktivitäten sollten vormittags erfolgen, über den Tag hinweg ist zunehmend – trotz Erholungspausen – mit Leistungsabfall und Müdigkeit zu rechnen. Patienten sollten einen Notfallausweis und auch Notfallmedikamente insbesondere auf Reisen mit sich führen [23].
Fazit
Die Autoimmunkrankheit Myasthenia gravis zeigt sich klinisch als sehr heterogen, die Verläufe und der Ausprägungsgrad sind höchst individuell. Gut etablierte Medikationen bestehen: Die symptomatisch wirksamen Acetylcholinesterase-Inhibitoren kommen zumeist gleich nach Diagnosestellung zum Einsatz, bei Symptomverschlechterung muss auf Glucocorticoide erweitert werden. Im Verlauf der Langzeittherapie kommen weitere, immunsuppressive Arzneimittel hinzu, einmal um Glucocorticoide einzusparen, aber auch um therapierefraktäre Behandlungen zu meistern. Krisenartige Entwicklungen und Exazerbationen, die eine intensivmedizinische Behandlung erfordern, können durch besondere Situationen (z. B. Infektionen, Stress, Operationen, Medikamente) auftreten. Insgesamt kann heute ein gutes und zufriedenstellendes Outcome erreicht werden.
Danksagung
Für die Unterstützung wird gedankt: A. Hünerfauth, H. Henckel, C. Sprenger und K. Green (HD).
Interessenkonflikterklärung
Die Autoren erklären, dass kein Interessenkonflikt vorliegt.
Literatur
1. Conti-Fine BM, et al. Myasthenia Gravis – past, present, and future. J Clin Invest 2006;116:2843–54.
2. Deutsche Myasthenie Gesellschaft (DMG). Informationsplattform, Fortbildungen und Schriften, Verzeichnis der Myasthenie-Zentren, Selbsthilfegruppen-Angebote. www.dmg.de.
2a. European Medicines Agency. Vyvgart. https://www.ema.europa.eu/en/medicines/human/EPAR/vyvgart (Zugriff am 26.08.2022).
3. Farmakidis C, et al. Treatment of myasthenia gravis. Neurol Clin 2018;36:311–37.
4. Heibel S, de Groot K. Aphereseverfahren bei Autoimmunerkrankungen. Einsatz in der Primär- und Eskalationstherapie. Klinikarzt 2019;48:360–9.
5. Herdegen T. Pharmakologisch: Glucocorticoide. Dtsch Apo Ztg 2012;152:5372–402.
6. Hoffmann S, et al. Complement deposition of the neuromuscular junction in seronegative myasthenia gravis. Acta Neuropathol 2020;139:1119–22.
7. Howard JF. Myasthenia gravis – A manual for the health care provider. Myasthenia Gravis Foundation of America, 2009.
8. Howard JF, et al. Safety and efficacy of eculizumab in acetylcholine-receptor antibody-positive refractory generalized myasthenia gravis. Lancet Neurol 2017;16:976–86.
9. Ioro R, et al. Efficacy and safety of rituximab for the treatment of myasthenia gravis. J Neurol 2015;262:1115–9.
10. Klehmet J, et al. Myasthenia gravis und Schwangerschaft. Akt Neurol 2014;41:447–53.
11. Köhler W, Sieb JP. Myasthenie. 4. Auflage. Bremen: Uni med Verlag, 2012.
12. Koneczney I, Herbst R. Myasthenia gravis – Pathogenetic effects of autoantibodies on neuromuscular architecture. Cells 2019;8:1–35.
13. Kottlors M, Glocker F. Myasthenia gravis. In: Hufschmidt A, et al. (Hrsg.). Neurologie compact. 8. Auflage. Stuttgart: Thieme; 2020. doi: 10.1055/b-007-170972.
13a. Kühnert M, et al. Recommendations of the AGG (Section Maternal Disease) for Myasthenia Gravis in Pregnancy. Geburtshilfe Frauenheilkd 2021;81:1301–6.
14. Mäurer M. Therapie der Myasthenia gravis – Myasthene Krisen unbedingt verhindern. DNP Der Neurologe & Psychiater 2019;20:36–41.
15. Mantegazza R, et al. Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiatr Disease Treatment 2011;7:151–60.
16. Melms A, Hohlfeld R. Myasthenia gravis und myasthene Syndrome. In: Brandt T, et al. (Hrsg.). Therapie und Verlauf neurologischer Erkrankungen. 4. Auflage. Stuttgart: Kohlhammer Verlag, 2003:1259–83.
17. Menghesha H, et al. Der Stellenwert der Thymektomie ohne Thymom in der Therapie der Myasthenia gravis. Chirurg 2021; https://doi.org/10.1007/s00104-021-01436-3.
18. Menon D, Barnett C, Bril V. Novel treatments in myasthenia gravis. Front Neurol 2020;11:538.
19. Müllges W, Stoll G. Myasthenia gravis. Nervenarzt 2019;10:1055–66.
20. Narayanaswami P, et al. International consensus guidance for management of myasthenia gravis 2020 update. Neurology 2021;96:114–22.
20a. Schneider-Gold C. Abnorme Muskelermüdbarkeit: Was steckt dahinter? DNP Der Neurologe & Psychiater 2022;23:47–53.
21. Schneider-Gold C, Toyka KV. Myasthenia gravis – Pathogenese und Immuntherapie. Dtsch Ärztebl 2007;104:420–6.
22. Schroeter M, Thayssen G, Kaiser J. Myasthenie – Exazerbation und Krise. Akt Neurol 2018;45:271–7.
23. Schumm F. Leitfaden für Myasthenie und das Lambert-Eaton-Syndrom. 13. Auflage. Deutsche Myasthenie Gesellschaft, 2021.
24. Sieb JP. Myasthenia gravis – Neues zur Pathogenese, Diagnose und Therapie. Internistische Praxis 2017;57:293–303.
25. Sieb JP. Myasthenia gravis – Überblick über die immunsuppressiven Therapiemöglichkeiten unter besonderer Berücksichtigung von Mycophenolatmofetil. Arzneimitteltherapie 2010;28:151–7.
26. Sieb JP. Therapierefraktäre Myasthenia gravis – Eculizumab als neue Therapie-Option. Psychopharmakotherapie 2018;25:227–33.
27. Silvestri NJ, Wolfe GI. Myasthenia gravis. Sem Neurol 2012;32:215–26.
28. Sussman J, et al. Myasthenia gravis – Association of British Neurologist’s management guidelines. Pract Neurol 2015;15:199–206.
29. Urban PP, Jacobi C. Immunsuppressiva bei Myasthenia gravis. Bremen: Deutsche Myasthenie Gesellschaft e. V. (DMG), 2021.
30. Vissing J, et al. Minimal symptom expression in patients with acetylcholine receptor antibody-positive refractory generalized myasthenia gravis treated with eculizumab. J Neurol 2020;267:1991–2001.
31. Wiendl H. Diagnostik und Therapie der Myasthenia gravis und des Lambert-Eaton-Syndroms. Leitlinie-AWMF-Reg-Nr. 030/87. DGN 2017:1–60. (Hinweis: Die Leitlinie befindet sich derzeit in Überarbeitung. Die Auswahl der medikamentösen Optionen wird sich zukünftig nach dem klinischen Erscheinungsbild der Myasthenia gravis und dem (Auto-)Antikörperstatus richten.)
32. Wolfe GI, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med 2016;375:511–22.
33. Wolfe G, et al. IgG regulation through FcRn blocking: A novel mechanism for the treatment of myasthenia gravis. J Neurol Sci 2021;430:118074.
34. www.dmg-online.de (reevaluiert am 10.08.2022).
35. www.embryotox.de (reevaluiert am 10.08.2022).
36. www.myasthenia.org (reevaluiert am 10.08.2022).
37. www.neurologienetz.de (reevaluiert am 10.08.2022).
38. www.youtube.com/watch?v=uRoRsmvkhTl (Prostigmin Tutorial, Fa. Roche 1935) (reevaluiert am 10.08.2022).
Modifizierter Nachdruck aus Krankenhauspharmazie 2022;43:43–55
Dr. Werner Kropf, Dorothée Möller, Pfalzklinikum für Neurologie und Psychiatrie, Weinstraße 100, 76889 Klingenmünster, E-Mail: werner.kropf@pfalzklinikum.de
Myasthenia – What is the reason for weak and exhausted muscles? An overview of the disease and its medications
Myasthenia gravis (MG) is known for being hardly represented in neurology. Patients show some kind of abnormous fluctuating weakness of their muscles which increases when put under strain and becomes less when the muscles relax. MG is an autoimmune process characterised by antibodies which are directed against the acetylcholine receptors of the neuromuscular junction. These acetylcholine receptors are blocked and destroyed and the innervation of the functioning of the skeletal muscles are, as a consequence, affected negatively. Apart from smaller muscles (e. g. ocular and bulbar muscles), the controllers of extremities such as muscles which support breathing can be seriously affected and may require intensive medical care. In the 30ies of the last century, inhibitors of the enzyme acetylcholinesterase proved to be an effective medication for a symptomatic therapy. These inhibitors provide more of the neurotransmitter acetylcholin and still count among the most important options in therapy. Advances in knowledge of the pathologic and immunologic processes have led to a more rational use of immunosuppressant drugs which play an important part in the safe therapy guidance and control of the disease, nowadays. Infections, stress and hormonal changes or co-medications can lead to a spontaneous worsening of the disease (myasthenic crisis) which result in the activation of an autoimmune process as a hazardous situation. Involvement of clinical pharmacists can be helpful in optimising suitable medication. In fact, as up to now a complete curative therapy is still unavailable but considering essential precautions, satisfactory treatment results with a high quality of life and few impairments can be achieved for the patients.
Key words: myasthenia, medications, acetylcholinesterase inhibitors, immunosuppressants
Psychopharmakotherapie 2022; 29(05):169-180