Martina Hahn und Sibylle C. Roll, Frankfurt am Main
Arzneimittelinteraktionen können zu unerwünschten, zum Teil schwerwiegenden Arzneimittelereignissen führen [15, 28, 29]. Insbesondere bei Polypharmazie (Multimedikation) ist dies der Fall, da die Anzahl der möglichen Interaktionen exponentiell mit der Anzahl eingenommener/verordneter Medikamente steigt und somit das Risiko einer potenziell relevanten Interaktion. Arzneimittelinteraktions-Datenbanken können dabei helfen, Interaktionen zu erkennen und, wenn möglich, zu vermeiden [11, 17, 18]. Allerdings ergibt sich in der klinischen Beobachtung das Phänomen, dass selbst relevante Interaktionen nicht zwingend zu einem unerwünschten Ereignis führen. Das tatsächliche Eintreten bleibt (erfreulicherweise) oft aus. Dies wiederum führt manchmal zu Konflikten zwischen Apothekern, die auf theoretische Interaktionsrisiken hinweisen, und Ärzten, die eine andere klinische Beobachtung machen. Es entsteht eine „alert fatigue“ auf beiden Seiten. Der Apotheker „mag“ nicht mehr anrufen – der Arzt „mag“ die Hinweise des Apothekers nicht mehr hören [4]. Dabei wird geschätzt, dass allein in den USA jährlich 100 000 Patienten infolge von Interaktionen versterben, eine Warnung also dringend angezeigt wäre. 10 % der stationären Patienten erleiden während des Aufenthalts im Krankenhaus unerwünschte Arzneimittelereignisse, 3,6 % aller Krankenhausaufnahmen in Europa sind Folge von unerwünschten Arzneimittelwirkungen [6]. Wie kann erklärt werden, dass nicht alle Patienten Anzeichen für eine pharmakokinetische Interaktion zeigen, obwohl ausreichende Evidenz für eine relevante Interaktion (z. B. starke Hemmeffekte aus In-vivo-Daten) vorliegen?
Etliche Studien haben den Zusammenhang zwischen Genotyp und Arzneimittelkonzentration im Serum ermittelt. In diesen Studien gab es jedoch starke Streuungen. Mittlerweile ist bekannt, dass die Komedikation, aber auch weitere Faktoren (Inflammation, Schwangerschaft, Alter, Nieren- und Leberfunktion) zu einer sogenannten Phänokonversion – also einem passager veränderteren Phänotyp – führen können [19, 23, 24]. In vielen Studien ist dieses Phänomen weitgehend missachtet worden: Es erfolgte eine Gleichsetzung des genetischen Phänotyps mit dem klinisch beobachteten Phänotyp. Auch wurde zum Teil nur auf wenige damals bekannte Polymorphismen hin untersucht. Heute kennen wir beispielsweise für CYP2D6 149 Polymorphismen, wie auf der Homepage des Pharmacogene Variation Consortium (https://www.pharmvar.org/) dargestellt. Auf welche dieser Polymorphismen in einem konkreten Kollektiv geprüft wird, ist für einen korrekten Befund entscheidend. Nach Publikationen zur Häufigkeit in bestimmten geographischen Regionen kann heute ein möglichst schmales und damit kosteneffizientes Panel mit möglichst hoher „Trefferquote“ angewendet werden [13]. Dadurch, dass mögliche Einflüsse auf den Phänotyp in den Studien der 1990er- und 2000er-Jahre unberücksichtigt blieben, wurde die Pharmakogenetik-Implementierung ausgebremst und nimmt erst in letzter Zeit deutlich an Fahrt auf. Heute kann man feststellen, dass die Arzneimittelinteraktion allein und auch der Genotyp allein nicht ausreichen, um die Wirkstoffkonzentration eines Arzneimittels vorherzusagen – erst in der Kombination (und gegebenenfalls auch in Kombination mit weiteren Faktoren wie Nierenfunktionsstörung) ist eine verlässlichere Aussage zur Pharmakokinetik des betroffenen Wirkstoffs möglich, wie im Weiteren gezeigt wird.
Pharmakokinetische Arzneimittelinteraktionen
Arzneimittel können sich gegenseitig in der Aufnahme, Distribution, Metabolismus und Elimination beeinflussen. In diesem Fall spricht man von pharmakokinetischen Interaktionen. Davon abzugrenzen sind pharmakodynamische Interaktionen, zum Beispiel auf Rezeptorebene, bei denen Arzneistoffe sich in ihrer pharmakologischen Wirkung beeinflussen.
Besonders bedeutsam sind Interaktionen, die die Enzyme des Arzneimittelmetabolismus betreffen. Man unterscheidet Substrate von Inhibitoren und Induktoren der Enzyme. Entsprechende Tabellen liefern Aufschluss, ob ein Arzneimittel ein Inhibitor oder Induktor ist, den Substrat-Spiegel also anhebt oder senkt: Die Inhibition eines Enzyms durch Arzneistoff A kann zu erhöhten Serumkonzentrationen eines Wirkstoffs B führen. Bei der Induktion führt Arzneistoff A („perpetrator drug“) dazu, dass Arzneistoff B („victim drug“) schneller abgebaut wird.
Nach FDA-Definition gibt es drei Klassen von Inhibitoren und Induktoren: stark, mittelstark und schwach. „Mittelstark“ definiert dabei eine klinisch relevante Interaktion, da hier von einem 2- bis 5-fachen Anstieg der Area under the curve (AUC) ausgegangen werden muss. Nicht bei jedem Patienten ist jedoch eine relevante Interaktion zu beobachten. Verantwortlich dafür sind unter Anderem genetische Polymorphismen.
Pharmakogenetik
Pharmakogenetische Untersuchungen haben zunehmend an Bedeutung gewonnen, werden aber außer in der Onkologie noch nicht in der Routineversorgung angewendet. Um Material für eine Genotypisierung zu gewinnen, genügt ein Mundschleimhautabstrich, sie ist also schnell und einfach durchzuführen. Leitlinien können dem Behandler helfen, die Genotypisierungsbefunde in klinisches Handeln zu übersetzen. Das Clinical Pharmacogenetics Implementation Consortium (CPIC) hat bereits Guidelines mit konkreten Handlungsempfehlungen für über 100 Arzneimittel veröffentlicht. Die Dutch Pharmacogenetics Working Group (DPWG), das Canadian Pharmacogenomics Network for Drug Safety (CPNDS), und das French National Network (Réseau) of Pharmacogenetics (RNPGx) haben ebenfalls Phänotyp-basierte Leitlinien entwickelt und publiziert [1]. Für die Psychiatrie hat die Pharmakogenetik (PGx) eine besondere Bedeutung, da das klinische Outcome nur subjektiv mittels Fragebogen und aufgrund der Wirklatenz erst nach Wochen eingeschätzt werden kann. Verlässliche Marker wie der Genotyp können daher hier von besonderer Bedeutung sein und dazu führen, dass der Patient eine schnellere Response und hoffentlich später auch eine Remission erlangt [3]. Das CPIC hat Leitlinien für selektive Serotonin-Wiederaufnahmehemmer (SSRI) und Trizyklika publiziert, die dem Behandler konkrete Handlungsanweisungen beim Vorliegen bestimmter Genotyp-Konstellationen liefern [21, 22].
In verschiedenen Untersuchungen konnte gezeigt werden, dass eine Testung schon vor der Verordnung der Arzneimittel hilfreich sein könnte. So wurde in einer US-amerikanischen Kohortenstudie mit Daten aus den Jahren 2011 bis 2017 gefunden, dass bei Kindern jährlich rund 10 000/100 000 Verordnungen Arzneimittel betrafen, für die es eine sehr gute Evidenz gibt, dass die pharmakogenetische Testung sinnvoll ist (davon jeweils rund 250/100 000 Verordnungen für Citalopram, Escitalopram bzw. Amitriptylin) [32]. In mindestens 1300/100 000 Fällen würde die Kenntnis des Genotyps eine Empfehlung zur Veränderung der Verordnung ausgelöst haben („actionable genotype“) [32].
Turner et al. zeigten an Patienten (n = 1456) mit Non-ST-Myokardinfarkten, dass bei 98,7 % der Patienten ein „actionable genotype“ vorlag. Weiterhin fand man, dass bei 77,1 % der Patienten Interaktionen vorlagen. 38,7 % der Interaktionen wurde als relevant eingestuft. Dabei handelte es sich bei 59,1 % um Arzneimittelinteraktionen, 11,6 % Arzneimittel-Gen-Interaktionen, 26,3 % Arzneimittel-Arzneimittel-Gen-Interaktionen und zu 2,9 % Arzneimittel-Gen-Gen-Interaktionen [35].
Was genau ist darunter zu verstehen und wie kommen diese Interaktionen zustande?
Definitionen
|
Begriff |
Definition |
|
Drug-Drug-Interaction (DDI) |
Liegt vor, wenn ein Arzneimittel den Abbau eines weiteren gleichzeitig eingenommenen Arzneimittels des Patienten beeinflusst |
|
Drug-Gene-Interaction (DGI) |
Liegt vor, wenn der genetische Phänotyp die Abbaugeschwindigkeit eines Arzneimittels verändert |
|
Drug-Drug-Gene-Interaction (DDGI) |
Liegt vor, wenn der genetische Phänotyp und ein weiteres Arzneimittel die Abbaugeschwindigkeit eines Arzneimittels verändern |
|
Phänokonversion |
Liegt vor, wenn eine Diskrepanz zwischen dem ermittelten genetischen Phänotyp und der wahren Abbaugeschwindigkeit des Arzneimittels besteht (z. B. durch Interaktionen, Schwangerschaft, Inflammation, Alter, Geschlecht, Leber- und Nierenfunktion) |
|
Drug-Gene-Gene-Interaction (DGGI) |
Liegt vor, wenn die erwartete Kapazität des Abbaus eines Arzneimittels durch den genetischen Phänotyp eines zweiten Enzyms im Abbauweg verändert wird |
*Gemeint sind im vorliegenden Kontext pharmakokinetische Arzneimittelinteraktionen
Genetische Polymorphismen
Ein genetischer Polymorphismus ist eine Variation in der DNA-Sequenz. Man unterscheidet drei Arten von Polymorphismen: a) Single-Nucleotide-Polymorphismus (SNP), bei dem ein einziges Basenpaar verändert ist, b) Deletion oder Insertion von sogenannten Tandem-Repeats (VNTR [variable number tandem repeats]) und c) Copy number variants (CNV; Kopienzahlvariationen), bei denen die Anzahl der Genkopien zwischen Individuen variiert (z. B. bei CYP2D6).
Diese Polymorphismen führen dazu, dass
- das Codon verändert wird und damit eine andere Aminosäure transkribiert wird, wodurch eine veränderte Aktivität des CYP-Enzyms resultieren kann,
- ein vorzeitiges STOP-Codon entsteht, das zum Abbruch der Transkription führt (dadurch entsteht kein Protein),
- Veränderungen an den Exon- und Intron-Splicing-Junctions auftreten (dadurch entsteht ein nicht-funktionstüchtiges Protein),
- die Stabilität der mRNA verändert wird (dadurch entsteht ein nicht-funktionstüchtiges Protein),
- die Enhancer-Aktivität verändert wird (Genablesung häufiger oder seltener, z. B. bei CYP2C19-Ultrarapid-Metabolizern),
- es keine Konsequenz hat.
Die Konsequenz richtet sich ferner danach, in welcher Region der Polymorphismus auftritt:
- In der Kodier-Region können synonyme Mutationen (stille Mutationen, die die gleiche Aminosäure kodieren wie das ursprüngliche Gen), Mis-Sense-Mutationen und Non-Sense-Mutationen entstehen
- In der Intron-Region entstehen Veränderungen im Splicing und der Bindung des Transkriptions-Faktors
- In der Promotor- oder Enhancer-Region verändert ein SNP die Transkriptions-Faktor-Bindung oder die RNA-Polymerase-Bindung
Der Genotyp wird durch die Sequenz der DNA an einer bestimmten Stelle determiniert. Für jedes Gen existieren zwei einander entsprechende Genorte auf zwei Chromosomen; dies bezeichnet man als Diplotyp (Allel 1 und Allel 2). Aus diesem Genotyp resultiert ein genetischer Phänotyp.
Veranschaulicht sei dies am Beispiel von CYP2D6 (Abb. 1). CYP2D6 ist, wie viele Phase-I-Enzyme, sehr polymorph. Aus der biochemischen Aktivitätsprüfung sind vier Varianten bekannt: normale Funktion, verminderte Funktion, keine Funktion, erhöhte Funktion. Noch genauer werden die Aktivitäten durch einen Punktewert, den sogenannten Activity-Score ausgedrückt (Tab. 1) [7]. Für jedes Stern-Allel wird ein Punktewert definiert, je nachdem, wie funktionstüchtig das Enzym ist (Abb. 1) [12, 14]. Daraus resultieren fünf genetische Phänotypen: Normal (NM), Intermediate Metabolizer (IM), Rapid Metabolizer (RM), Ultrarapid Metabolizer (UM) und Poor Metabolizer (PM).
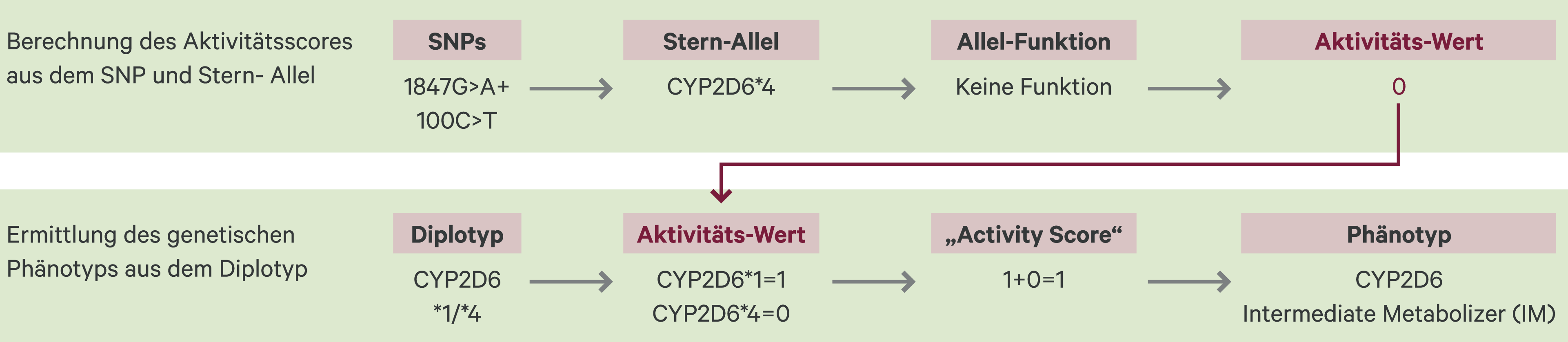
Abb. 1. Berechnung des Phänotyps aus den vorliegenden Single-Nucleotide-Polymorphismen (SNPs). Der Genotypisierungsbefund zeigt zunächst an, welche Base ausgetauscht wurde (SNP). Die daraus resultierenden Genvarianten werden mit Stern-Allel (* und Nummer des Allels) bezeichnet. Für viele Stern-Allele ist eine Funktionsveränderung bekannt, zum Beispiel gelten für CYP2D6 die Aktivitäts-Werte null für *4 und *3, eins für *1, *5, *17. Daraus ergibt sich unter Berücksichtigung des Diplotyps ein Activity-Score, aus dessen Wert ein genetischer Phänotyp, im Beispiel Intermediate Metabolizer (vgl. Tab. 1), ermittelt wird.
Tab. 1. Beispiel für Activity-Scores für CYP2D6
|
Activity-Score |
Allele (Beispiele) |
Genetischer Phänotyp |
|
> 2,25 |
*1/*1xN, *1/*2xNa, *2 /*2xNa, *1 × 2/*9 |
Erhöhte Aktivität, Ultrarapid Metabolizer (UM) |
|
≤ 2,25 bis ≥ 1,25 |
*1/*10, *1/*41, *1/*9, *1/*1, *1/*2, *2 × 2/*10 |
Wildtyp, Normal Metabolizer (NM) |
|
> 0 bis < 1,25 |
*4/*10, *4/*41, *10/*10, *10/*41, *41/*41, *1/*5, *1/*4 |
Reduzierte Aktivität, Intermediate Metabolizer (IM) |
|
0 |
*3/*4, *4/*4, *5/*5, *5/*6 |
Nicht funktionstüchtig, Poor Metabolizer (PM) |
a N: Anzahl der Duplikate bei einer Copy Number Variation, z. B. *1 × 2, *1 × 3
Das Vorliegen bestimmter Polymorphismen variiert regional stark, wie in Tabelle 2 dargestellt [16].
Tab. 2. Häufigkeiten genetischer Polymorphismen für CYP2D6 und CYP2C19 im Vergleich
|
Enzym |
Phänotyp |
Europa |
Afrika |
Ost-Asien |
Mittlerer Osten |
Psychiatrische Patienten in Deutschland (vollstationär, mit MDD) (n = 108) [16] |
|
CYP2D6 |
UM |
1–5 % |
Bis zu 40 % |
1 % |
10–15 % |
1 % (+3 % UM/PM*) |
|
NM |
80–85 % |
50 % |
70–75 % |
75–80 % |
53 % |
|
|
IM |
5–10 % |
5–10 % |
25 % |
10 % |
35 % |
|
|
PM |
7–10 % |
< 1 % |
< 1 % |
1–2 % |
7 % |
|
|
CYP2C19 |
UM |
5 % |
3–5 % |
1–2 % |
5–10 % |
39 % (31 % heterozygot = Rapid Metabolizer) |
|
NM |
70 % |
65–70 % |
30–35 % |
75–80 % |
32 % |
|
|
IM |
20 % |
15–20 % |
40–45 % |
10–15 % |
27 % |
|
|
PM |
3–5 % |
5–10 % |
Bis zu 25 % |
< 1 % |
2 % |
* 3 % der psychiatrischen Patienten hatten eine Genduplikation, wobei durch das Verfahren nicht determiniert werden konnte, ob das funktionstüchtige oder das nicht-funktionstüchtige Gen dupliziert vorliegt. MDD: Depression (Major depressive disorder); IM: Intermediate Metabolizer; NM: Normal Metabolizer; PM: Poor Metabolizer; UM: Ultrarapid Metabolizer
Arzneimittel-Gen-Interaktionen (Drug-Gene-Interactions)
Im Fokus der pharmakogenetischen Forschung standen speziell Arzneimittel mit engem therapeutischem Bereich wie Warfarin (CYP2C9- und VKORC1-Genotyp und Risiko für Hirnblutungen oder Schlaganfall) und Clopidogrel (thrombotische Ereignisse oder Blutungen), prognoseverändernde Arzneimittel wie Tamoxifen (Rezidivrisiko aufgrund CYP2D6-Genotyp), Irinotecan (UGT1A1-Genotyp und Risiko für Myelosuppression) und Thiopurine (TPMT-Genotyp und Risiko für Myelosuppression).
Die Leitlinien für die Psychopharmakotherapie beziehen sich in ihren Empfehlungen auf CYP2D6 und CYP2C19. Diese beiden Enzyme sind diejenigen mit den meisten bekannten Polymorphismen. Sowohl das CPIC, aber auch die DPWG haben bereits viele Leitlinien entwickelt, die eine Dosierungsempfehlung für jeden genetischen Phänotyp (IM, PM etc.) geben, und folgen damit Publikationen von Kirchheiner et al. vor nunmehr 20 Jahren, die den Zusammenhang zwischen Genotyp und Serumkonzentration für Antidepressiva und Antipsychotika gezeigt haben [26]. So wurde auch publiziert, dass die Serumspiegel von Escitalopram bei CYP2C19-PMs 95 % und bei IMs 30 % höher liegen als bei NMs [9]. Ein Abfall der Spiegel um 13 % und 36 % wurde für RMs (heterozygot *1/*17) beziehungsweise UMs (homozygot *17/*17) beschrieben. Das CPIC formulierte aufgrund solcher Daten bereits vor fünf Jahren Leitlinien zur Dosierung von SSRI und Trizyklika (TZA) [21, 22]. Es wird geschätzt, dass die Hälfte aller Patienten > 65 Jahre in einem Zeitraum von vier Jahren ein Arzneimittel verordnet bekommt, für das bereits jetzt Pharmakogenetik-Leitlinien vorliegen [29]. Dosierungsempfehlungen aufgrund des Genotyps existieren für fast alle Antidepressiva und viele Antipsychotika, eine Antipsychotika-Leitlinie des CPIC ist bereits in Vorbereitung. Die PharmGKB-Datenbank (www.pharmgkb.org) liefert Auskunft über die Evidenz der einzelnen Empfehlungen und eignet sich daher gut als Nachschlagewerk. Datenbanken wie die MediQ-Datenbank (www.mediq.ch) ermöglichen bereits die Eingabe des Genotyps zur Analyse und Berücksichtigung von Arzneimittel-Gen-Interaktionen.
Arzneimittel-Arzneimittel-Gen-Interaktionen (Drug-Drug-Gene-Interactions)
Die FDA teilt die CYP-Inhibitoren und die CYP-Induktoren in drei Kategorien:
- Schwach (< 2-facher Anstieg der AUC bzw. < 50%-Anstieg der Clearance)
- Mittelstark (> 2- und < 5-facher Anstieg der AUC bzw. 50- bis 80%-Anstieg der Clearance)
- Stark (> 5-facher Anstieg der AUC bzw. > 80%-Anstieg der Clearance)
Starke und mittelstarke Inhibitoren sowie Induktoren führen definitionsgemäß zu relevanten Interaktionen, da hier Empfehlungen für Dosisanpassungen bestehen bzw. Kontraindikationen für eine gleichzeitige Einnahme vorliegen. Einige Beispiele sind in Tabelle 3 dargestellt.
Tab. 3. Überblick über die definierten genetischen Phänotypen der unterschiedlichen CYP-Enzyme, der Substrate und Arzneistoffe, die als Inhibitoren oder Induktoren wirken und eine Phänokonversion verursachen können
|
CYP |
Genetische Phänotypen |
Substrate |
Inhibitoren und Induktoren |
|
1A2 |
Erhöhte Aktivität, normale Aktivität, unbekannte Aktivität |
Duloxetin, Olanzapin, Clozapin, Theophyllin, Coffein |
Ciprofloxacin, Fluvoxamin, Enoxacin, |
|
2A6 |
PM, IM, NM, UM |
Nicotin |
|
|
2B6 |
NM, IM, PM, RM, UM |
Bupropion, Cyclophospamid, Efavirenz, Methadon |
Clopidogrel, Ticlopidin, Carbamazepin, Efavirenz, Rifampicin |
|
2C8 |
Erhöhte Aktivität, normale Aktivität, verminderte Aktivität |
Glitazone, Paclitaxel, Repaglinid |
Gemfibrozil, Trimethoprim, Rifampicin, Johanniskraut |
|
2C9 |
NM, IM, PM |
Celecoxib, Losartan, NSAR, Phenytoin, Warfarin, Phenprocoumon, Glibenclamid |
Amiodaron, Fluconazol, Rifampicin |
|
2C19 |
NM, IM, PM, RM, UM |
Clopidogrel, Diazepam, Protonenpumpeninhibitoren (PPI), Escitalopram, Citalopram, Sertralin, Trimipramin, Amitriptylin, Clomipramin, Agomelatin |
Fluvoxamin, Fluconazol, Esomeprazol, Omeprazol, Rifampicin |
|
2D6 |
NM, IM, PM, UM |
Fluoxetin, Paroxetin, Sertralin, Amitriptylin, Clomipramin, Venlafaxin, Atomoxetin, Betablocker, Codein, Tramadol, Tamoxifen, Hydrocodon, Oxycodon, Dextromethorphan |
Fluvoxamin, Fluoxetin, Paroxetin, Bupropion, Duloxetin, Melperon, Terbinafin Beachte: es gibt keine CYP2D6-Induktoren |
|
3A4 |
Erhöhte Aktivität, normale Aktivität, verminderte Aktivität |
Aprepitant, Calciumkanalblocker, Dronedaron, Makrolide, Protease-Inhibitoren, Statine, Quetiapin, Triazolam |
Azole, Clarithromycin, Fluvoxamin Ritonavir, Verapamil, Grapefruit, Rifampicin, Phenytoin, Johanniskraut |
|
3A5 |
NM, IM, PM |
Tacrolimus |
Azole, Verapamil, Diltiazem, Phenobarbital, Carbamazepin, Rifampicin |
Kursiv = Induktoren
Allerdings beobachtet man in der klinischen Praxis bei Kombination dieser Inhibitoren oder Induktoren mit entsprechenden Substraten nicht immer den erwarteten definierten Anstieg oder Abfall der AUC. Hier kommt nun die Phänokonversion ins Spiel. Der genetische Phänotyp wird durch Einnahme eines Induktors oder Inhibitors verändert. Es entsteht eine Genotyp-Phänotyp-Diskrepanz, die besonders für CYP2C19 und CYP2D6 beschrieben wird und damit besonders in der Psychiatrie eine große Rolle spielt, da dies die Abbauwege der meisten psychotropen Substanzen betrifft. Dabei kommen als exogene Faktoren auch nicht benannte, vom Patienten aber eingenommene Medikamente infrage, die Interaktionen verursachen können. Darüber hinaus beeinflussen andere, endogene Faktoren den Phänotyp, beispielsweise Nierenfunktionsstörungen, Leberfunktionsstörungen oder Inflammation. Solche Faktoren können bei Vorliegen des genetischen Phänotyps in Kombination mit dem Befund eines therapeutischen Drug-Monitorings (TDM) leichter identifiziert werden.
Das Phänomen „Phänokonversion“ ist in der klinischen Praxis häufig, wie von einigen Autoren bereits untersucht:
In einer großen australischen Kohorte konnte gezeigt werden, dass der Anteil von CYP2D6-Poor-Metabolizern durch Phänokonversion (z. B. durch Gabe eines CYP2D6-Inhibitors) von 5,4 % auf 24,7 % steigt [31]. Für CYP2C19 erhöhte sich der Anteil von CYP2C19-Poor-Metabolizern von 2,7 % auf 17 % im untersuchen Kollektiv von über 5000 Patienten. Durch Phänokonversion erhöht sich also die Anzahl von Poor Metabolizern deutlich über die, die anhand von Populationswerten zu erwarten wäre.
Darüber hinaus konnten Storelli et al. zeigen, dass Menschen mit einem nicht-funktionstüchtigen CYP2D6-Allel ein erhöhtes Risiko für Phänokonversion zeigen: Sie verglichen 17 homozygote NM (NM/NM) mit 17 heterozygoten NM (NM/PM). Die Probanden erhielten 5 mg Dextromethorphan und 10 mg Tramadol zu drei Sitzungen. Bei der zweiten Sitzung wurde zusätzlich Duloxetin, bei Sitzung 3 zusätzlich Paroxetin verabreicht. Eine Phänokonversion zu IM (71 % vs. 25 %; p = 0,009) bei Gabe von Duloxetin und zu PM (94 % vs. 56 %; p = 0,011) bei Gabe von Paroxetin trat insbesondere in den heterozygoten Probanden auf. Der Serumspiegel von Dextromethorphan stieg besonders bei homozygoten Probanden bei Gabe von Paroxetin. Diese Interaktion war also stärker als bei heterozygoten Probanden, wenn man den relativen Anstieg des Serumspiegels betrachtet. Also auch innerhalb von Phänotyp-Gruppen (hier NM) gibt es noch Unterschiede, die bei der Interpretation Berücksichtigung finden müssen [34]. Ähnliches gilt für CYP2C19: In einer experimentellen klinischen Studie mit 34 Probanden wurde nach 28 Tagen Protonenpumpenhemmer-Gabe (Omeprazol oder Esomeprazol) bei 93 % der Probanden einer Phänokonversion gefunden. Während alle IMs zu PMs konvertiert waren, war es in der heterogenen Gruppe der NMs bei einem Teil zu IM- und bei einem anderen Teil zu PM-Konvertierung gekommen [27].
Solche Daten finden in den oben beschriebenen Activity-Scores bereits Ausdruck (Tab. 1). Spezielle Faktoren, die den Einfluss von Inhibitoren und Induktoren definieren (z. B. 0,5 oder 2), wurden publiziert und können zur Abschätzung der Phänokonversion genutzt werden [10] (Tab. 4 und 5).
Tab. 4. Phänokonversions-Berechnung bei CYP2D6
|
Activity-Score CYP2D6 |
Genetischer Phänotyp |
Schwacher oder mittelstarker Inhibitor |
Starker Inhibitor |
|
0 |
PM |
Activity-Score x 0,5 = PM |
Activity-Score x 0 = PM |
|
> 0 < 1,25 |
IM |
Activity-Score x 0,5 = IM |
Activity-Score x 0 = PM |
|
> 1,25 < 2,25 |
NM |
Activity-Score x 0,5 = IM |
Activity-Score x 0 = PM |
|
> 2,25 |
UM |
Activity-Score x 0,5 = NM |
Activity-Score x 0 = PM |
Tab. 5. Phänokonversion bei CYP2C19
|
Genetischer Phänotyp CYP2C19 |
Komedikation mit moderatem oder starkem Inhibitor Prädiktiver Phänotyp |
|
NM, IM |
PM |
|
RM, UM |
IM |
|
PM |
PM |
|
Komedikation mit moderatem oder starkem Induktor; Prädiktiver Phänotyp |
|
|
NM, RM |
UM |
|
IM |
NM |
|
PM |
PM |
|
UM |
UM |
Dies ist besonders in der Psychiatrie relevant: In einer deutschen Kohorte von 27 396 stationären psychiatrischen Patienten erhielten 14,4 % aller Patienten einen CYP-Inhibitor oder -Induktor – also jeder sechste Patient. Bei 43,6 % der Patienten kam es potenziell zu einer relevanten Interaktion bei Verordnung von Inhibitoren und/oder Induktoren, das heißt, der Patient hat ein entsprechendes Substrat dieser durch Interaktion in der Aktivität veränderten CYP-Enzyme eingenommen [20]. In dieser Untersuchung wurde jedoch nicht genotypisiert, sodass über den Grad der Phänokonversion und der Phänotypen keine Aussage getroffen werden konnte.
Monte et al. zeigten zudem, dass auch die Gabe weiterer Substrate eine Phänokonversion auslösen können. Sie nutzten dazu ebenfalls Dextromethorphan und analysierten die Dextromethorphan-Dextrorphan-Quotienten, um eine Phänokonversion nach Gabe eines Substrats festzustellen. Die gleichzeitige Gabe eines weiteren CYP2D6-Substrats verursachte ein 9,49-fach höheres Risiko für Phänokonversion, also eine Genotyp-Phänotyp-Diskrepanz [30].
Jedoch muss andererseits auch festgestellt werden, dass die Zugabe eines Inhibitors oder Induktors nicht immer in einer Phänokonversion resultiert. Durch gegenläufige Effekte des genetischen Polymorphismus und der Interaktion kommt es zu einem Shift des resultierenden Phänotyps, der Phänokonversion. Es können mehrere Phänomene auftreten, wie in Tabelle 4, 5 und 6 dargestellt.
Tab. 6. Einfluss der Gabe eines Inhibitors auf den Metabolismus eines Medikaments bei Patienten mit unterschiedlichem Genotyp (gültig für alle Enzyme)
|
Patient ist |
Erhält |
Resultat |
|
Poor Metabolizer |
Starken Inhibitor |
Keine Veränderung |
|
Intermediate Metabolizer |
Starken Inhibitor |
Verlangsamter Abbau |
|
Normal Metabolizer |
Starken Inhibitor |
Deutlich verlangsamter Abbau |
|
Ultrarapid Metabolizer |
Starken Inhibitor |
Ganz stark verlangsamter Abbau |
Dies zeigt, wie komplex das Zusammenspiel aus Interaktion und Genotyp ist.
Erhält ein CYP2C19-UM beispielsweise einen CYP2C19-Induktor, so verändert sich der Phänotyp nicht (Tab. 4 und 5). Genauso verändert sich der Phänotyp durch Inhibitor-Gabe bei einem Poor Metabolizer nicht, wie am Beispiel der Gabe von Fluvoxamin und Lansoprazol bei unterschiedlichen genetischen Phänotypen gezeigt werden konnte [39]. Zur Abschätzung der Relevanz einer Interaktion ist die Kenntnis des genetischen Phänotyps daher wichtig. Ein internetbasiertes Tool liefert Hinweise auf den Einfluss des Genotyps und einer Arzneimittelinteraktion auf die AUC eines Substrats: https://www.ddi-predictor.org.
Ein Beispiel: Citalopram (CYP2C19-Substrat) und Omeprazol (CYP2C19-Inhibitor) werden kombiniert und man würde grundsätzlich einen Anstieg des Citalopram-Serumspiegels erwarten. Bei einem Normal-Metabolizer wäre dies auch der Fall (AUC-Quotient 1,27, also eine um 27 % erhöhte Bioverfügbarkeit), bei einem Poor Metabolizer bleibt der AUC-Quotient jedoch 1. Diese Prädiktion der Bioverfügbarkeit beruht jedoch auch wiederum nur auf dem Hauptabbauweg des Arzneimittels und berücksichtigt weder weitere genetische Polymorphismen noch Nebenabbauwege, wie in den folgenden Abschnitten dargestellt.
Arzneimittel-Gen-Gen-Interaktionen (Drug-Gene-Gene-Interactions)
Arzneimittel-Gen-Gen-Interaktionen können erklären, warum manchmal eine Diskrepanz zwischen gemessenem erwartetem Serumspiegel bei Kenntnis eines Genotyps vorliegt: Viele Arzneimittel werden nämlich durch mehrere CYP-Enzyme abgebaut, beispielsweise wird Amitriptylin durch CYP2C19 und CYP2D6 abgebaut. Dies ist in Abbildung 2 unter V dargestellt.
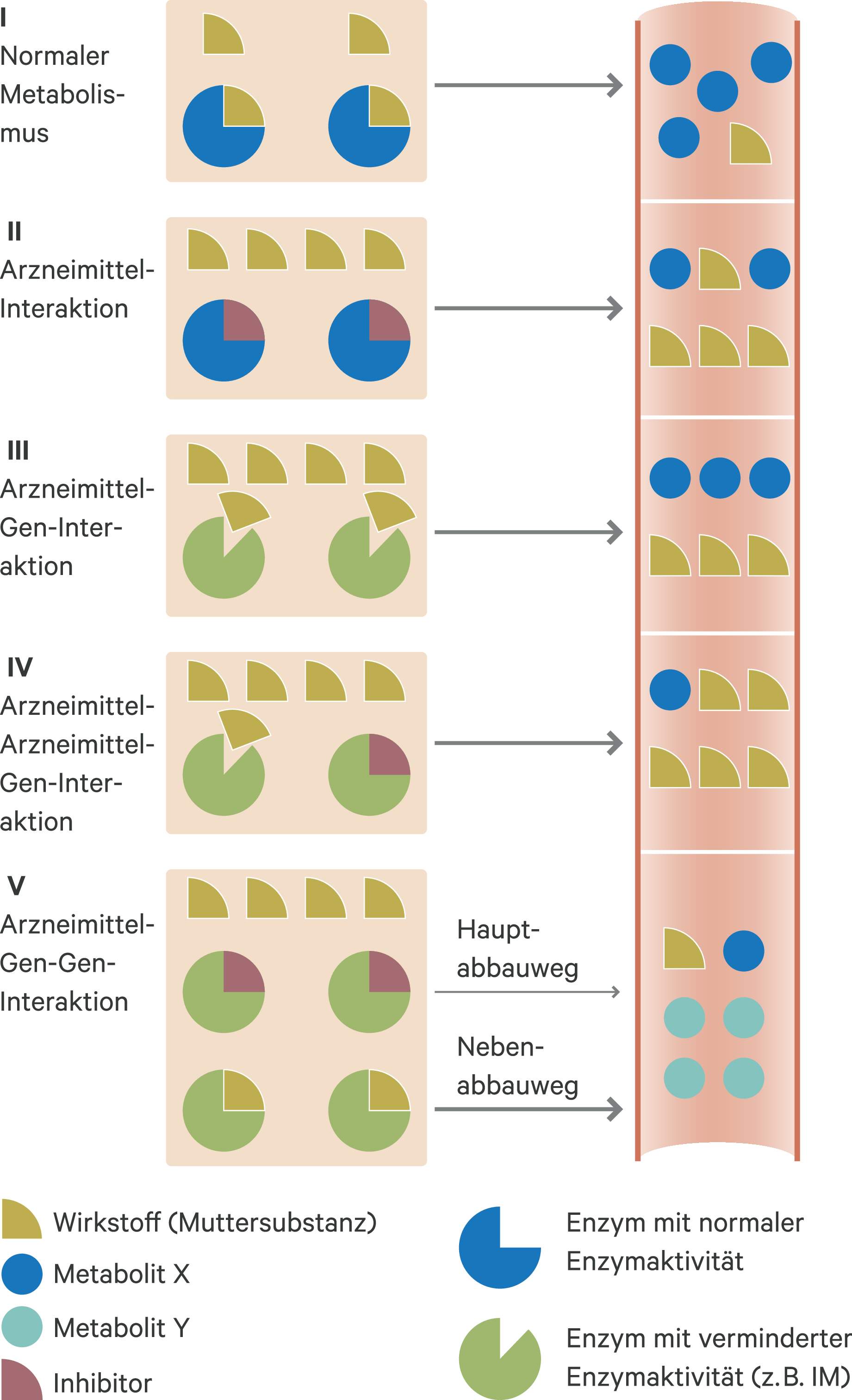
Abb. 2. DDIs, DGIs, DDGIs und DGGIs im Vergleich. I) Normaler Abbau, es entstehen Metabolite durch das abbauende Enzym. II) Bei Arzneimittelinteraktionen kommt es z. B. zum Anstieg durch Inhibition des abbauenden Enzyms, die Muttersubstanz akkumuliert. III) Bei Polymorphismen, z. B. Intermediate-Metabolizer-Status, kommt es zum verminderten Abbau und somit zur Akkumulation der Muttersubstanz. IV) DDGI: Es kommt zur Phänokonversion. Die Abbaugeschwindigkeit wird durch eine Inhibition des Enzyms weiter verlangsamt. Die Muttersubstanz akkumuliert mehr, als dies aufgrund des genetischen Phänotyps zu erwarten wäre. V) DGGI: Durch den genetischen Phänotyp eines zweiten abbauenden Enzyms eines Arzneimittels kommt es trotz Inhibition des hauptsächlich abbauenden Enzyms zu einem schnellen Metabolismus des Arzneimittels. Es bilden sich jedoch andere Metabolite. Dies ist auch bei genetischen Polymorphismen auf beiden Enzymen der Fall, z. B. PM auf dem Hauptweg, NM auf dem Nebenweg. (Modifiziert nach [17a])
Beide Enzyme können Polymorphismen aufweisen. Der Phänotyp beider Enzyme, beispielsweise PM CYP2D6 und UM CYP2C19, beeinflusst den Serumspiegel von Amitriptylin (Abb. 2). Würde man nur den CYP2D6-Genotyp berücksichtigen, so würde man extrem hohe Plasmaspiegel erwarten. Durch den Ultrarapid-Metabolizer-Status im zweiten abbauenden Enzym wird der Spiegel jedoch deutlich niedriger sein. Die Leitlinie von Hicks et al. empfiehlt in diesem Falle den Einsatz von TDM [20]. So brauchen Patienten mit IM/IM-Status eine deutlich geringere Amitriptylin-Dosis als NM/IM- oder UM/IM-Patienten. Entsprechend wurden für einige Medikamente bereits Kreuztabellen publiziert, die eine Dosisempfehlung aufgrund beider Phänotypen abgeben, so beispielsweise für Trizyklika (CYP2D6 und CYP2C19) [20], aber auch Statine (SLCO1B1 und ABCG2) [31], Thiopurine (TMTP und NUDT15) [30] und Warfarin (CYP2C9 und VKORC1). Statt der Genotypisierung eines Gens (z. B. nur CYP2D6) empfiehlt sich daher ein Panel, damit auch die hier vorgestellten Arzneimittel-Gen-Gen-Interaktionen berücksichtigt werden können. Gerade bei PM-Status des Hauptabbauwegs werden der zweite Abbauweg und dessen Enzymaktivität besonders relevant. In der PharmLine-Studie wurde untersucht, wie oft mehrere Abbauwege eines Arzneimittels durch Interaktionen und/oder genetische Polymorphismen beeinflusst wurden. Bei 55,1 % der Patienten lag ein genetischer Phänotyp vor, der eine Dosisanpassung gemäß den Leitlinien erforderte. Bei 44,9 % lag eine Arzneimittel-Arzneimittel-Gen-Interaktion vor, durch Phänokonversion veränderte sich also der Phänotyp. Für Escitalopram konnte gezeigt werden, dass Patienten mit CYP2C19-IM/PM- und CYP3A4-IM/PM-Genotyp ein höheres Risiko für Umstellung oder Dosisreduktion aufwiesen als Patienten, bei denen eine Kombination von CYP2C19-IM/PM und CYP3A4-NM vorlag (aOR 4,38; 95%-Konfidenzintervall [KI] 1,22–15,69 und aOR 2,75; 95%-KI 1,03–7,29) [2].
Solche Arzneimittel-Gen-Gen-Interaktionen scheinen also eine hohe Relevanz zu besitzen.
Die detaillierten Abbauwege sind auf www.pharmgkb.org für fast alle Antidepressiva dargestellt. Für die Psychiatrie haben Bousman et al. ein Panel aus CYP2D6, CYP2C19, CYP2C9, HLA-A und HLA-B vorgeschlagen [5]. Die DWPG empfiehlt einen Gen-Pass, der 58 Genvarianten von 14 sogenannten „Pharmakogenen“ (CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, F5, HLA-B, NUDT15, SLCO1B1, TPMT, UGT1A1 und VKORC1) beschreibt und damit 49 Arzneistoffe abdeckt [36]. Dieser Pass könnte dann jedem Behandler vorgelegt werden, so wie aktuell bereits in den Niederlanden, wo Patienten diesen Pass in Form einer Plastikkarte mitführen und überall vorzeigen können bzw. mit QR-Code in die Verordnungssoftware/Apothekensoftware einlesen lassen können.
Bedeutung für die Praxis
TDM – der Goldstandard in der Therapiesteuerung – liefert stark variable Werte bei gleicher Dosis und erlaubt die Messung des tatsächlichen Phänotyps [23]. Die Interpretation der Befunde ohne Genotypisierungsbefund bleibt jedoch teilweise vage. Falsche Vermutungen zum Genotyp können eine Problemlösung arzneimittelbezogener Probleme verzögern. Bei Bekanntsein des Genotyps kann der Serumspiegel daher präziser interpretiert werden. Daraus ergeben sich auch andere Therapiealgorithmen in der klinischen Praxis, beispielsweise Adhärenzförderung oder Suche nach Inhibitoren oder Induktoren. Nichtgenetische Faktoren, die den Phänotyp beeinflussen, können so aufgedeckt werden und entsprechende Maßnahmen (Behandlung der Entzündung, Vermeidung einer Arzneimittelinteraktion, Anpassung an eine veränderte Leber- oder Nierenfunktion) abgeleitet werden [19].
Der Vorteil bei einer Genotypisierung vor Beginn einer Pharmakotherapie liegt in der Vermeidung von unerwünschten Arzneimittelwirkungen aufgrund von Überdosierungen zu Therapiebeginn. Da TDM erst nach fünf Halbwertszeiten des Wirkstoffs durchgeführt werden kann, um valide Talspiegel zu ermitteln, kann es für die Aufdeckung eines stark erhöhten Plasmaspiegels bereits zu spät sein – die unerwünschte Arzneimittelwirkung ist eventuell schon früher eingetreten. TDM und Pharmakogenetik ergänzen sich gut und können beide auf ihre Weise zu einer erhöhten Arzneimitteltherapiesicherheit führen. Auch hinsichtlich Nichtansprechens bietet die Genotypisierung Vorteile. Patienten wollen eine schnelle Linderung und sind daher offen für pharmakogenetische Untersuchungen, wenn dadurch ein wochenlanges An- und wieder Absetzen eines Antidepressivums entfallen kann, wie es beispielsweise bei CYP2C19-Ultrarapid-Metabolizern und Citalopram zu erwarten wäre. Durch ein Zusammenspiel aus Gentypisierung und therapeutischem Drug-Monitoring kann die Arzneimitteltherapiesicherheit deutlich erhöht werden.
Ausblick
Pharmakogenetische Untersuchungen sind den meisten aktuellen Studien zufolge bereits kosteneffizient bzw. kosteneinsparend [24]. Testungen werden zudem immer günstiger, sodass eine Kosteneffizienz-Steigerung zu erwarten ist.
Erste Verträge zur integrierten Versorgung (IV) mit einer Krankenkasse konnten für 2022 in Deutschland geschlossen werden, sodass erstmalig auch im ambulanten Sektor eine Genotypisierung ohne Kosten für den Patienten angeboten werden kann.
Immer mehr Studien belegen den Zusammenhang zwischen Metabolismus und Polymorphismen und so nimmt die Evidenz dieser Arzneimittel-Gen-Interaktion stetig zu. Einige Interaktionsdatenbanken haben sich dem Thema Genotypisierung bereits geöffnet und erlauben die Eingabe eines genetischen Phänotyps zur Prüfung auf Arzneimittel-Gen-Interaktionen. Eine Analyse von Arzneimittel-Gen-Gen- oder Arzneimittel-Arzneimittel-Gen-Interaktionen ist aufgrund der Komplexität jedoch noch nicht möglich und bedarf auch weiterhin der fachkundigen Interpretation, beispielsweise durch klinische Pharmazeuten. Auch die Leitlinien empfehlen derzeit das Hinzuziehen von klinischen Pharmazeuten bei bestimmten Genkonstellationen, da Pharmazeuten in der Regel ein besseres Wissen zur Pharmakokinetik und Pharmakogenetik aufweisen [21, 22]. In den USA und Großbritannien gibt es bereits verschiedene Kurse (Diplom, Master) an den Universitäten zu „precision medicine“ für Pharmazeuten. In Deutschland müssen diese Fortbildungsmöglichkeiten dringend geschaffen werden, damit klinische Pharmazeuten oder andere Berufsgruppen diese Aufgabe in Zukunft erfüllen können, um die behandelnden Ärzte zu unterstützen.
Die Kosteneffizienz ist zwar durch einige, jedoch insgesamt nur durch wenige Studien belegt. Eine große Frage bleibt auch aufgrund der vorliegenden Ergebnisse noch: Wann „lohnt“ sich der Einsatz von Pharmakogenetik? – Vor Einnahme des Medikaments oder erst bei Auftreten von Nebenwirkungen bzw. Nicht-Ansprechen? Ergebnisse aus der multizentrischen PREPARE-Studie werden Evidenz für den Nutzen von Genotypisierungen vor der Einnahme von Medikamenten – darunter auch viele Psychopharmaka – zur Vermeidung von unerwünschten Arzneimittelereignissen liefern [37] und den Klinikern eine Entscheidungshilfe an die Hand geben, wann oder bei welchen Arzneimitteln Genotypisierungen vorgenommen werden sollten.
Einige Studien weisen jedoch schon darauf hin, dass eine Verbesserung der Symptome bei jedem vierten Patienten durch pharmakogenetische Testungen erzielt werden kann [38]. Besonders für Medikamente mit engem therapeutischem Bereich, zu denen auch Trizyklika gehören, konnte gezeigt werden, dass die unerwünschte Arzneimittelwirkung vom Genotyp abhängt: Für CYP2D6-Intermediate-Metabolizer, die Amitriptylin einnehmen, war das Risiko für unerwünschte Arzneimittelwirkungen mit 76,5 % deutlich höher als das von Normal-Metabolizern mit 12,1 % (p = 0,00001). Dabei korrelierte die Nortriptylin- und nicht die Amitriptylin-Serumonzentration mit dem Auftreten der unerwünschten Arzneimittelwirkung [33].
Fazit
Durch immer aussagekräftigere Daten zur Bedeutung eines Genotyps und genetischen Phänotyps für die Wirksamkeit und Verträglichkeit von Arzneimitteln erscheint eine Genotypisierung vor der Verordnung („preemptive testing“) bestimmter Medikamente schon jetzt durchaus sinnvoll. Da die pharmakogenetischen Befunde lebenslang gültig sind, dürfte dies Vorgehen auch kosteneffizient sein. Unter Berücksichtigung von DDI, DGI, DGGI, und DDGI kann die Arzneimitteltherapiesicherheit für Patienten deutlich erhöht werden und Risikopatienten für unerwünschte Arzneimittelwirkungen können besser identifiziert werden. Eine Kostenerstattung durch alle Krankenkassen ist wünschenswert, jedoch sollte jeder Patient für sich selbst entscheiden dürfen, ob er diese Diagnostik nutzen möchte, auch wenn die Kosten nicht erstattet werden. Eine Vorenthaltung dieser Diagnostik erscheint nicht mehr zeitgemäß.
Abkürzungen
|
AUC |
Area under the curve, Maß der Bioverfügbarkeit |
|
CPIC |
Clinical Pharmacogenetics Implementation Consortium |
|
CYP |
Cytochrom P450 |
|
DDI |
Drug-Drug-Interaction = Arzneimittelinteraktion |
|
DGI |
Drug-Gene-Interaction = Arzneimittel-Gen-Interaktion |
|
DDGI |
Drug-Drug-Gene-Interaction = Arzneimittel-Arzneimittel-Gen-Interaktion |
|
DGGI |
Drug-Gene-Gene-Interaction = Arzneimittel-Gen-Gen-Interaktion |
|
DPGW |
Dutch Pharmacogenetics Working Group |
|
FDA |
Food and Drug Administration |
|
IM |
Intermediate Metabolizer |
|
NM |
Normal Metabolizer |
|
PM |
Poor Metabolizer |
|
RM |
Rapid Metabolizer |
|
UM |
Ultrarapid Metabolizer |
Interessenkonflikterklärung
Die Autorinnen geben an, keine Interessenkonflikte zu haben.
Literatur
1. Abdullah-Koolmees H, van Keulen AM, Nijenhuis M, Deneer VHM. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front Pharmacol 2021;11:595219. Published online 2021 Jan 25. doi: 10.3389/fphar.2020.595219
2. Bahar MA, Lanting P, Bos JHJ, Sijmons RH, et al. B. Impact of drug-gene-interaction, drug-drug-interaction, and drug-drug-gene-interaction on (es)citalopram therapy: the PharmLines Initiative. J Pers Med 2020;10:256. Published online 2020 Nov 28. doi: 10.3390/jpm10040256
3. Bättig VAD, Roll SC, Hahn M. Pharmacogenetic testing in depressed patients and interdisciplinary exchange between a pharmacist and psychiatrists results in reduced hospitalization times. Pharmacopsychiatry 2020;53:185–92.
4. Baumann P, Hahn M, Roll SC, Stämpfli D. Psychiatrische Patienten fragen ihren Arzt und Apotheker – wie kann die ambulante Versorgung optimiert werden? Psychopharmakotherapie 2020;27:270–7.
5. Bousman CA, Al Maruf A, Mueller DJ. Towards integration of pharmacogenetics in psychiatry: a minimum, evidence-based genetic testing panel. Curr Opin Psychiatry 2019;32:7–15.
6. Bouvy J, De Bruin M, Koopmanschap M. Epidemiology of adverse drug reactions in Europe: a review of recent observational studies. Drug Saf 2015;38:437–53.
7. Caudle KE, Sangkuhl K, Whirl‐Carrillo M, Swen JJ, et al. Standardizing CYP2D6 genotype to phenotype translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci 2020;13:116–24.
8. Chan SL, Ng HY, Sung C, Chan A, et al. Economic burden of adverse drug reactions and potential for pharmacogenomic testing in Singaporean adults. Pharmacogenomics J 2019;19:401–10.
9. Chang M, Tybring G, Dahl ML, Lindh JD. Impact of cytochrome P4502C19 polymorphisms on citalopram/escitalopram exposure: a systematic review and meta-analysis. Clin Pharmacokinet 2014;53:801–11.
10. Cicali EJ, Elchynski AL, Cook KJ, Houder JT, et al. How to integrate CYP2D6 phenoconversion into clinical pharmacogenetics: a tutorial. Clin Pharmacol Ther 2021;110:677–87.
11. Fritz D, Ceschi A, Curkovic I, Huber M, et al. Comparative evaluation of three clinical decision support systems: prospective screening for medication errors in 100 medical inpatients. Eur J Clin Pharmacol 2012;68:1209–19.
12. Gaedigk A, Ndjountché L, Divakaran K, Bradford D, et al. Cytochrome P4502D6 (CYP2D6) gene locus heterogeneity: characterization of gene duplication events. Clin Pharmacol Ther 2007;81:242–51.
13. Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, et al. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med 2017;19:69–76.
14. Gaedigk A, Simon SD, Pearce RE, Bradford LD, et al. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 2008;83:234–42.
15. Gurwitz JH, Field TS, Avorn J, et al. Incidence and preventability of adverse drug events in nursing homes. Am J Med 2000;109:87–94.
16. Hahn M, et al. Frequencies of genetic polymorphisms of clinically relevant gene-drug pairs in a german psychiatric inpatient population. Pharmacopsychiatry 2020;53:1–10.
17. Hahn M, Roll SC. Validierung von Interaktionsdatenbanken in der Psychopharmakotherapie. Nervenarzt 2018;89:319–26.
17a. Hahn M, Roll SC. The influence of pharmacogenetics on the clinical relevance of pharmacokinetic drug-drug interactions: drug-gene, drug-gene-gene and drug-drug-gene interactions. Pharmaceuticals 2021;14:487.
18. Hedna K, Andersson ML, Gyllensten H, Hagg S, et al. Clinical relevance of alerts from a decision support system, PHARAO, for drug safety assessment in the older adults. BMC Geriatr 2019;19:164.
19. Hefner G, Shams ME, Unterecker S, Falter T, et al. Inflammation and psychotropic drugs: the relationship between C-reactive protein and antipsychotic drug levels. Psychopharmacology (Berl) 2016;233:1695–705.
20. Hefner G, Wolff J, Hahn M, et al. Prevalence and sort of pharmacokinetic drug-drug interactions in hospitalized psychiatric patients. J Neural Transm (Vienna) 2020;127:1185–98.
21. Hicks JK, Bishop JR, Sangkuhl K, Müller DJ, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther 2015;98:127–34.
22. Hicks JK, Swen JJ, Thorn CF, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther 2013;93:402–8.
23. Hiemke C, et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017. Pharmacopsychiatry 2018;51:9–62.
24. Jaquenoud Sirot E, van der Velden JW, Rentsch K, et al. Therapeutic drug monitoring and pharmacogenetic tests as tools in pharmacovigilance. Drug Saf 2006;29:735–68.
25. Karamperis K, Koromina M, Papantoniou P, Skokou M, et al. Economic evaluation in psychiatric pharmacogenomics: a systematic review. Pharmacogenomics J 2021;21:533–41.
26. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173–92.
27. Klieber M, Oberacher H, Hofstaetter S, Beer B, et al. CYP2C19 phenoconversion by routinely prescribed proton pump inhibitors omeprazole and esomeprazole: clinical implications for personalized medicine. J Pharmacol Exp Ther 2015;354:426–30.
28. Lazarou J, Pomeranz B, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200–5.
29. Leone R, Magro L, Moretti U, et al. Identifying adverse drug reactions associated with drug-drug interactions data mining of a spontaneous reporting database in Italy. Drug Saf 2010;33:667–75.
30. Monte AA, West K, McDaniel KT, et al. CYP2D6 genotype phenotype discordance due to drug-drug interaction. Clin Pharmacol Ther 2018;104:933–9.
31. Mostafa S, Kirkpatrick CMJ, Byron K, Sheffield L. An analysis of allele, genotype and phenotype frequencies, actionable pharmacogenomic (PGx) variants and phenoconversion in 5,408 Australian patients genotyped for CYP2D6, CYP2C19, CYP2C9 and VKORC1 genes. J Neural Transm (Vienna). 2019;126:5–18.
32. Ramsey LB, Ong HH, Schildcrout JS, et al. Prescribing prevalence of medications with potential genotype-guided dosing in pediatric patients. JAMA Netw Open 2020;3:e2029411. Published 2020 Dec 1. doi: 10.1001/jamanetworkopen.2020.29411.
33. Steimer W, Zöpf K, von Amelunxen S, Pfeiffer H, et al. Amitriptyline or not, that is the question: pharmacogenetic testing of CYP2D6 and CYP2C19 identifies patients with low or high risk for side effects in amitriptyline therapy. Clin Chem 2005;51:376–85.
34. Storelli F, Matthey A, Lenglet S, Thomas A, et al. Impact of CYP2D6 functional allelic variations on phenoconversion and drug-drug interactions. Clin Pharmacol Ther 2018;104:148–57.
35. Turner RM, de Koning EM, Fontana V, Thompson A, et al. Multimorbidity, polypharmacy, and drug-drug-gene interactions following a non-ST elevation acute coronary syndrome: analysis of a multicentre observational study. BMC Med 2020;18:367. Published 2020 Nov 25. doi: 10.1186/s12916-020-01827-z.
36. van der Wouden CH, van Rhenen MH, Jama WOM, Ingelman-Sundberg M, et al. Development of the PGx-passport: a panel of actionable germline genetic variants for pre-emptive pharmacogenetic testing. Clin Pharmacol Ther 2019;106:866–73.
37. van der Wouden CH, Böhringer S, Cecchin E, Cheung KC, et al.; Ubiquitous Pharmacogenomics Consortium. Generating evidence for precision medicine: considerations made by the Ubiquitous Pharmacogenomics Consortium when designing and operationalizing the PREPARE study. Pharmacogenet Genomics 2020;30:131–44.
38. Walden LM, Brandl EJ, Tiwari AK, Cheema S, et al. Genetic testing for CYP2D6 and CYP2C19 suggests improved outcome for antidepressant and antipsychotic medication. Psychiatry Res 2019;279:111–5.
39. Yasui-Furukori N, Saito M, Uno T, Takahata T, et al. Effects of fluvoxamine on lansoprazole pharmacokinetics in relation to CYP2C19 genotypes. J Clin Pharmacol 2004;44:1223–9.
Prof. Dr. Martina Hahn, Klinik für Psychiatrie, Psychosomatik und Psychotherapie des Universitätsklinikums Frankfurt, Heinrich-Hoffmann-Straße 10, 60528 Frankfurt am Main, E-Mail: Martina.Hahn@kgu.de
Prof. Dr. Sibylle C. Roll, Klinik für psychische Gesundheit, varisano Klinikum Frankfurt Höchst, Gotenstraße 6–8, 65929 Frankfurt am Main
Pharmacokinetic drug interaction in the field of psychopharmacotherapy – the relevance is depending on the genotype
Drug interactions are a well-known cause of adverse drug events. Drug interaction databases can help the clinician to recognize and at the same time avoid such interactions and adverse events. However, not every interaction leads to an adverse drug event. The concentration of the drug is depending on other factors like inflammation, age, gender, pregnancy, renal or liver dysfunction AND pharmacogenetic polymorphisms. Also, if inhibitors or inducers of drug metabolizing enzymes (e. g. CYP, UGT) are added to drug therapy, phenoconversion can occur and lead to a genetic phenotype that mismatches the observable phenotype. Drug-drug-gene and drug-gene-gene interactions influence the concentration of the drug and its metabolites. To date there have been few studies published on the impact of genetic variations on drug-drug interactions. This article shows examples and evidence of drug-drug-gene interactions, as well as drug-gene-gene interactions. Phenoconversion is explained and methods to calculate the phenotype are described. Clinical recommendations are given regarding the integration of pharmacogenetic results in the assessment of the relevance of drug interactions in the future.
Key words: pharmacogenetic testing, drug-drug-gene-interactions, drug-gene-gene interactions
Psychopharmakotherapie 2022; 29(01):17-26