Holger Petri, Bad Wildungen*
Die Behandlung der Human-immunodeficiency-virus(HIV)-Infektion hat durch Einführung hochwirksamer Arzneistoffkombinationen in den letzten Jahren große Fortschritte gemacht. Menschen mit einer HIV-Infektion wird bei konsequent durchgeführter antiretroviraler Therapie (ART) ein Leben ohne wesentliche Einschränkungen ermöglicht bei annähernd normaler Lebenserwartung [38].
Antiretrovirale HIV-Therapeutika haben unterschiedliche Angriffspunkte im HIV-Replikationszyklus (Abb. 1). Nukleosidische bzw. nukleotidische und nichtnukleosidische Reverse-Transkriptase-Inhibitoren (NRTI und NNRTI) verhindern die Transkription von einzelsträngiger viraler RNA in eine doppelsträngige DNA durch Hemmung des HIV-Enzyms Reverse Transkriptase. Proteaseinhibitoren (PI) hemmen die HIV-Protease. Dieses Enzym spaltet das sogenannte gag-pol-Protein in seine Untereinheiten. Unterbleibt dieser Schritt, entstehen Viruspartikel, die nicht infektiös sind. Ein weiteres Schlüsselenzym im HIV-Replikationszyklus ist die Integrase, die dazu beiträgt, die provirale DNA in das Wirtsgenom zu integrieren. Integraseinhibitoren (INI) verhindern die Insertion proviraler DNA in das humane Genom. Entry-Inhibitoren unterbinden den Eintritt von HI-Viren in die Zellen. Fusionsinhibitoren greifen am HIV-Hüllprotein gp120 an und verhindern so den Eintritt in die Zielzelle. Der Korezeptorantagonist Maraviroc blockiert CCR5-Rezeptoren, an denen CCR5-trope HI-Viren an die Zielzelle andocken [38].
Als Pharmakoenhancer werden niedrig dosiertes Ritonavir und Cobicistat eingesetzt. Diese sogenannten Booster hemmen das Cytochrom-P450(CYP)-Isoenzym 3A4 in therapeutischer Intention. Hierdurch wird das pharmakokinetische Profil der Proteaseinhibitoren und des Integrase-Inhibitors Elvitegravir verbessert mit dem Ziel, eine anwendungsfreundliche Einnahme zu fördern.
Vor dem Hintergrund des Resistenzrisikos und einer möglichst guten Verträglichkeit werden antiretrovirale Therapeutika mit unterschiedlichem Wirkprofil kombiniert [11, 27, 38].
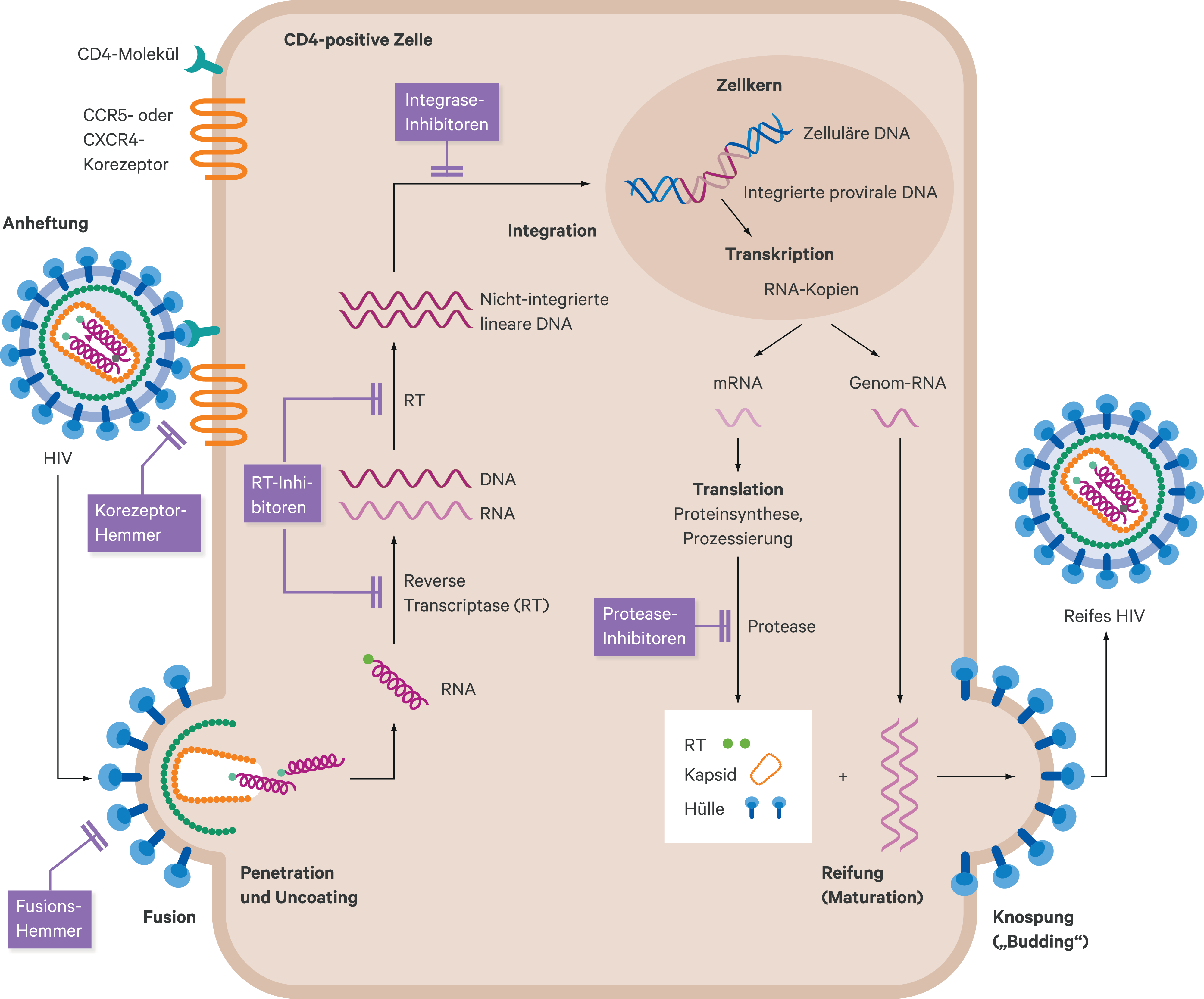
Abb. 1. Schematische Darstellung der Angriffspunkte der verschiedenen HIV-Therapeutika
Nukleosidische und nukleotidische Reverse-Transkriptase-Inhibitoren (NRTI)
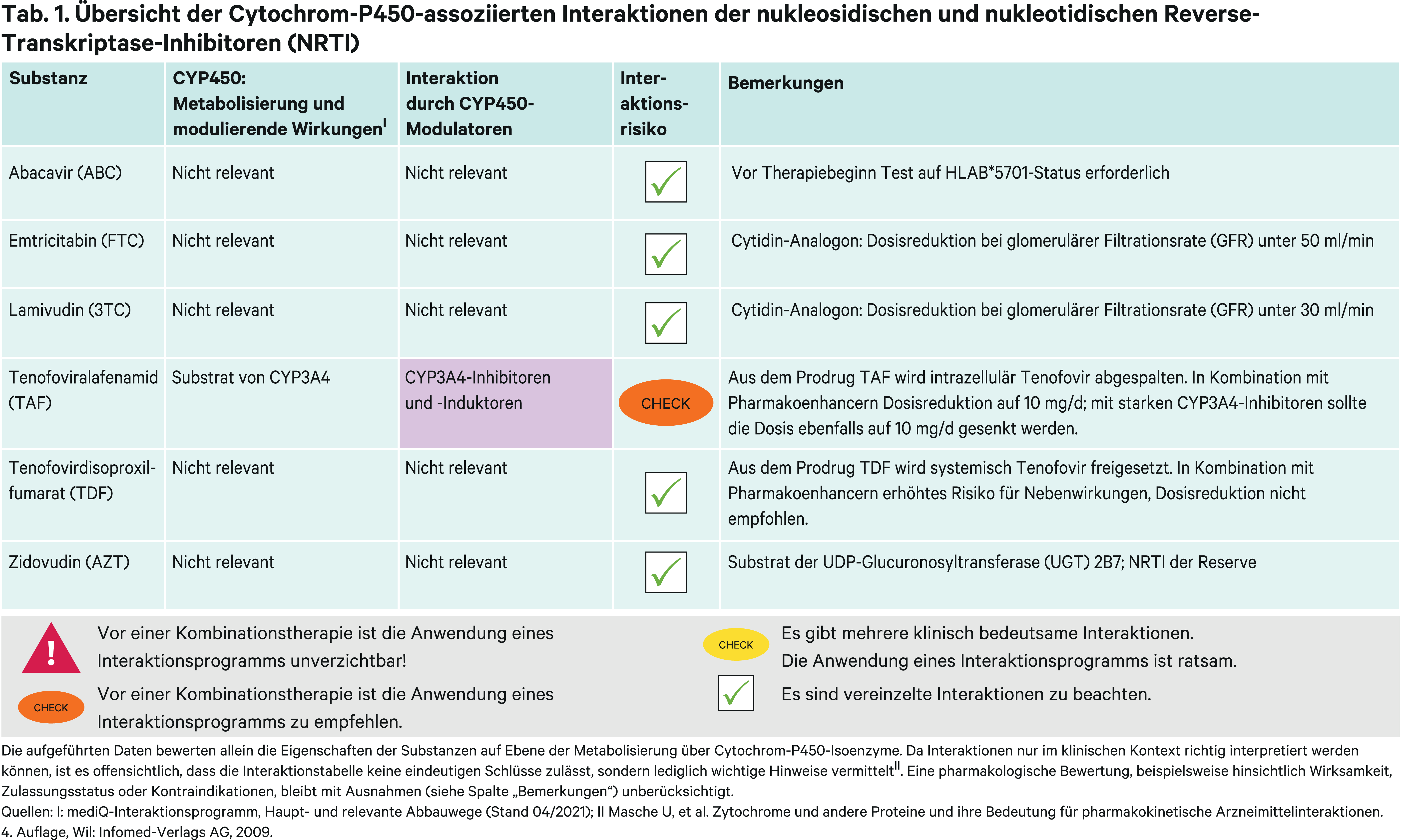
Gemäß nationalen wie internationalen Leitlinien enthält die initiale antiretrovirale Therapie (ART) ein bis zwei Nukleosid-/Nukleotidanaloga (NRTI) („Backbone-Nukes“) [11, 38]. Als nukleosidische RTI stehen die beiden Cytidin-Analoga Emtricitabin und Lamivudin und das Guanosin-Analogon Abacavir primär zur Auswahl, wobei Emtricitabin und Lamivudin nicht miteinander kombiniert werden [38]. Als nukleotidischer RTI wird Tenofovir eingesetzt. Der Einsatz von Zidovudin bleibt nur noch bestimmten Resistenzsituationen vorbehalten [27].
Abacavir
Abacavir (ABC) unterliegt keinen relevanten, metabolischen Interaktionen. Das Guanosin-Analogon ist mit dem Risiko für Überempfindlichkeitsreaktionen (Hypersensitivitätsreaktionen, HSR) besonders bei Trägern des HLAB*5701-Allels assoziiert. Vor der Verordnung ist ein Test auf einen HLAB*5701-Status erforderlich [67]. Somit ist Abacavir nicht für die postexpositionelle Prophylaxe der HIV-Infektion zum Beispiel nach Nadelstichverletzungen geeignet [13].
Emtricitabin und Lamivudin
Für die beiden Cytidin-Analoga Emtricitabin (FTC) und Lamivudin (3TC) besteht ebenfalls kein Potenzial für pharmakokinetische Wechselwirkungen [27, 67]. Beide sind vergleichbar wirksam und verträglich. Von praktischer Relevanz ist, dass die Lamivudin-Dosis schon bei einer glomerulären Filtrationsrate (GFR) von < 50 ml/min zu reduzieren ist und 3TC-haltige Fixkombinationen aufgebrochen werden müssen. Bei FTC-haltigen Fixkombination ist das erst ab einer GFR < 30 ml/min notwendig [27, 67].
Zidovudin
Zidovudin (AZT) war 1987 unter dem Handelsnamen Retrovir® das erste gegen das HI-Virus gerichtete, antiretrovirale Medikament auf dem Markt. Es hat an Bedeutung verloren, weil es schlechter verträglich ist als die neueren NRTI. Gastrointestinale, aber auch myelotoxische Nebenwirkungen limitieren den Einsatz. Auch die Lipoatrophie, die bei den älteren NRTI eine belastende Nebenwirkung ist und für den Vertriebsstopp von Didanosin und Stavudin geführt hat, bessert sich bei Wechsel von AZT auf andere Therapien. Zudem muss es zweimal täglich eingenommen werden und kommt für die heute gängigen Once-Daily-Kombinationen folglich nicht infrage.
Zidovudin hat somit als NRTI nur noch Reservestatus. Sein Einsatz ist bestimmten Resistenzkonstellationen vorbehalten. Bei der Mutation K65R, die zu einer Tenofovir-Resistenz führt, kann eine Überempfindlichkeit der Viren für AZT genutzt werden, möglicherweise reichen dafür auch niedrigere Dosen [27]. Pharmakokinetische Interaktionen können über Phase-II-Reaktionen zustande kommen, da Zidovudin primär über die UDP-Glucuronosyltransferase (UGT) 2B7 metabolisiert wird. Die Anwendung der UGT-Inhibitoren Fluconazol und Valproinsäure führte bei Patienten zu einer 74- bzw. 80%igen Erhöhung der Zidovudin-Exposition [51, 67]. Eine erhöhte Toxizität kann nicht ausgeschlossen werden [67]. Es sind niedrige Zidovudin-Stärken verfügbar, wodurch Dosisanpassungen möglich sind. Durch den UGT-Induktor Rifampicin kann sich der AZT-Plasmaspiegel halbieren [21]. Zidovudin wird renal tubulär unter Beteiligung von organischen Anionentransportern (Organic anion transporter, OAT) aktiv sezerniert. Durch den OAT-Inhibitor Probenecid verdoppelt sich die AUC des NRTI [67].
Tenofovir
Tenofovir (TFV) liegt als Tenofovirdisoproxilfumarat (TDF) und Tenofoviralafenamid (TAF) in zwei Prodrugformen vor. Intrazellulär wirksam wird Tenofovir durch Phosphorylierung zu Tenofovirdiphosphat (TFV-DP).
Tenofovirdisoproxil wird nach systemischer Aufnahme über Esterasen zu Tenofovir gespalten. Für Tenofovir ist das Potenzial für CYP-bedingte Wechselwirkungen gemäß In-vitro-Daten gering [67]. Die Kombination mit Induktoren wie Rifampicin führte zu keiner relevanten pharmakokinetischen Wechselwirkung [67]. Mit Ritonavir geboosterte Proteaseinhibitoren können die Tenofovir-AUC um 22 bis 37 % erhöhen [26]. Eine Dosisanpassung wird aber nicht empfohlen [67].
Aus Tenofoviralafenamid wird intrazellulär durch eine lysosomale Carboxypeptidase Tenofovir abgespalten. Die Tenofovirplasmaspiegel sind unter TAF-Einnahme 91 % niedriger als unter TDF-Einnahme, wohingegen die intrazellulären Tenofovirdiphosphatspiegel 6,5-fach höher sind [26]. Tenofoviralafenamid ist ein Substrat der Arzneimitteltransporter P-Glykoprotein (P-gp) und Breast cancer resistance protein (BCRP). Es wird geringfügig über CYP3A4 metabolisiert [67]. Mit Ritonavir und Cobicistat geboosterte ART-Kombinationsregime erfordern eine TAF-Dosisreduktion von 25 mg auf 10 mg [26, 67]. Die AUC von Tenofovir kann um das 2,1-bis 4,2-Fache ansteigen. Tenofovir erhöht das Risiko renaler und ossärer Komplikationen bei Langzeitanwendung. Im direkten Vergleich sind ungeboosterte TAF- und TDF-Regime vergleichbar wirksam und verträglich. Enthält die Therapie einen Pharmakoenhancer, ist die Verträglichkeit von Tenofoviralafenamid in reduzierter Dosis besser als die von Tenofovirdisoproxil, das in der Regel nur in einer Fixdosis bei Erwachsenen angewendet wird [26].
Auch bei Ketoconazol- und Itraconazol-Einnahme wird eine Dosisreduktion von TAF empfohlen [67]. Begründet wird dies mit einer starken P-gp-Hemmung. Metabolische Gründe scheinen aber eine größere Bedeutung zu spielen. Zum einen ist Ritonavir kein starker P-gp-Inhibitor und zum anderen steigert die Dreifachkombination direkter antiviraler Agenzien (DAA) zur Behandlung der chronischen Hepatitis C aus Sofosbuvir/Velpatasvir/Voxilaprevir trotz der starken P-gp-Hemmung die AUC von TAF nur um 52 % und erfordert keine Dosisreduktion [22, 67].
CYP3A4-Induktoren (Abb. 2) sind auch P-gp-Induktoren, wenn auch weniger stark ausgeprägt [18]. Carbamazepin senkt die TAF-AUC um 55 % und die Tenofovir-Exposition um 23 %. Wegen des möglichen Wirkungsverlusts werden Induktoren nicht in Kombination mit Tenofoviralafenamid empfohlen [67]. In einer klinischen Studie wurde in Kombination mit dem P-gp-Induktor Rifampicin die TAF-Dosis in einem ungeboosterten Therapieregime von 1 × 25 mg auf zweimal täglich erhöht. Hierdurch waren sowohl die Plasmaspiegel von TAF als auch die intrazelluläre TFV-DP-Exposition vergleichbar mit der einmal täglichen Einnahme von 25 mg TAF ohne Induktor [9]. Da Rifampicin keinen Einfluss auf die Pharmakokinetik von Tenofovirdisoproxil hat, sollte TDF bevorzugt mit potenten Induktoren angewendet werden.
Integrase-Inhibitoren (INI)
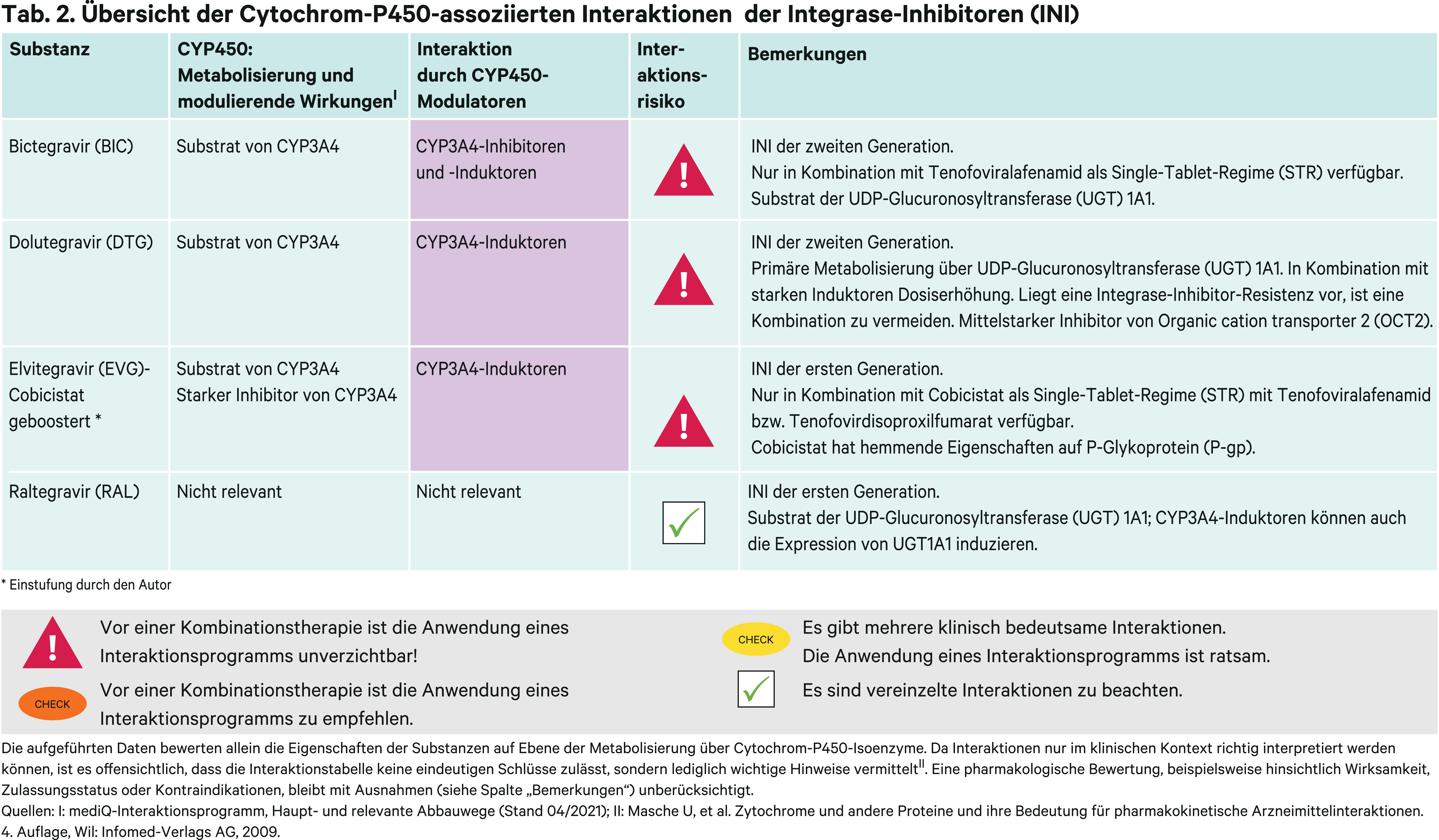
Integrase-Inhibitoren, syn. Integrase-Strangtransfer-Inhibitoren (INSTI), sind erkennbar an der Endung „gravir“ und werden gemäß nationalen sowie internationalen Leitlinien als erste Wirkstoffgruppe zur Kombination mit den „NRTI-Backbone-Nukes“ empfohlen. Sie sind kurz- und mittelfristig ausreichend wirksam und verträglich. Im Jahr 2007 kam mit Raltegravir der erste INSTI auf den Markt, sodass Daten zur Langzeittoxizität naturgemäß limitiert sind. Raltegravir und Elvitegravir bilden die Vertreter der ersten Generation. Nachteil dieser beiden Integrase-Inhibitoren ist die relativ geringe Resistenzbarriere. Bictegravir ist wie Elvitegravir nur als Single-Tablet-Regime (STR) verfügbar. Mit Dolutegravir ist ein flexibler Einsatz in Ein- und Mehrtablettenregimen möglich [27].
Erstgenerations-INI
Raltegravir
Raltegravir (RAL) wird CYP-unabhängig über UGT1A1 metabolisiert.Es wird unverändert oder als Raltegravir-Glucuronid renal und biliär eliminiert. In einer Untersuchung mit radioaktiv markiertem Raltegravir fielen ungefähr 70 % der gesamten Radioaktivität auf Raltegravir als wichtigste zirkulierende Substanz. Die übrige Radioaktivität im Plasma fiel auf Raltegravir-Glucuronid. Probanden des *28/*28-Genotyps, die Enzyme mit reduzierter Aktivität exprimieren, hatten verglichen mit Probanden des Wildtyps im geometrischen Mittel eine 41%ige Erhöhung der AUC, die aber klinisch vernachlässigbar ist [65]. Die gute Verträglichkeit bestätigte sich in einer weiteren Studie mit HIV-Patienten. Hier war zwar die AUC bei *28-Trägern über das Doppelte erhöht, die unerwünschten Arzneimittelwirkungen traten aber nicht klinisch relevant häufiger oder stärker zutage [5]. Raltegravir kann bei Erwachsenen zweimal täglich mit 400 mg oder einmal täglich mit 1200 mg dosiert werden.
Atazanavir als starker UGT1A1-Inhibitor steigert die AUC von Raltegravir um 68 %, mit Ritonavir geboostert hingegen nur um 41 % wegen gegenläufiger UGT-induzierender Effekte des Wirkverstärkers. Ritonavir induziert UGT1A1 und kompensiert damit die Hemmung des Enzyms durch Atazanavir. Rifampicin als starker Induktor senkt die Exposition von Raltegravir in der 400-mg-Einzeldosis um 40 % und die 12h-Plasmakonzentration um 61 %. Auch bei Verdopplung der Dosis auf 800 mg sind die 12h-Plasmaspiegel vergleichbar niedrig [64]. Es wird daher unter gleichzeitiger Anwendung mit Rifampicin nur die zweimal tägliche Einnahme zu je 400 mg empfohlen und die 1200-mg-Einzeldosis sollte wegen möglicher subtherapeutischer Talspiegel gemieden werden. Das gleiche Dosierungsschema gilt mit anderen UGT1A1-Induktoren wie Phenytoin, Phenobarbital oder Tipranavir/Ritonavir [67].
Elvitegravir
Elvitegravir (EVG) wird über CYP3A4 metabolisiert und ist nur geboostert mit Cobicistat in Anwendung. Hierdurch ist eine einmal tägliche Einnahme in Fixkombination mit Emtricitabin und TDF/TAF als Single-Tablet-Regime möglich. Mit weiteren Inhibitoren wie Ketoconazol steigt die Elvitegravir-Exposition um 48 %, was nicht klinisch relevant ist für die antiretrovirale Therapie mit Elvitegravir [48]. Bei Komedikation mit CYP3A4-Induktoren drohen Therapieversagen und Resistenzentwicklung. Carbamazepin senkt die AUC von EVG um 69 %, die Talspiegel sinken um 97 %. Starke CYP3A4-Induktoren sind unter Elvitegravir-Einnahme kontraindiziert. Auch von der Anwendung mittelstarker Induktoren wie Bosentan sollte Abstand genommen werden, weil diese auch die Plasmaspiegel des Pharmakoenhancers in den subtherapeutischen Bereich fallen lassen könnten. Mit Cobicistat als starker CYP3A4- und P-gp-Hemmer ergeben sich zahlreiche, weitere pharmakokinetische Interaktionsrisiken [67].
Zweitgenerations-INI
Bictegravir
Bictegravir (BIC) ist wie Elvitegravir nur als Single-Tablet-Regime verfügbar, muss aber nicht geboostert werden. BIC ist ein Substrat von CYP3A4 und UGT1A1. Der starke CYP3A4-Hemmer Voriconazol erhöht die AUC von Bictegravir um 61 %. Stärker ist die Wirkung durch Atazanavir, das sowohl CYP3A4 und UGT1A1 hemmt. Der Proteaseinhibitor erhöht die Bictegravir-Exposition um das 4-Fache, ähnlich hoch wie bei Cobicistat-geboostertem Atazanavir [69]. Somit ist der metabolische Abbau von Bictegravir über UGT von größerer Bedeutung. Zu beachten ist zudem, dass Bictegravir nur in Fixkombination mit Tenofoviralafenamid in der Stärke 25 mg verfügbar ist. Durch die TAF-betreffenden Interaktionen ist die Anwendung von Bictegravir somit zusätzlich eingeschränkt. Durch Induktion von CYP3A4 und UGT1A1 kann die Exposition von Bictegravir deutlich sinken. Rifampicin reduziert die AUC um 75 % [69]. Das Antibiotikum ist wie Johanniskraut-haltige Präparate unter Bictegravir-Therapie kontraindiziert. Andere starke Induktoren wie Carbamazepin sollten gemieden werden, weil auch sie gleichzeitig die TAF-Exposition senken [67].
Dolutegravir
Dolutegravir (DTG) wird hauptsächlich über Metabolisierung via UGT1A1 eliminiert. Es ist aber auch ein Substrat von CYP3A4. Durch Atazanavir stieg die Exposition bei gesunden Probanden um das Doppelte [55]. Dies erfordert aber keine Dosisanpassung. Vielmehr sind Wechselwirkungen mit Induktoren zu beachten. Durch Carbamazepin, Efavirenz, Rifampicin und Tipranavir/Ritonavir sanken in pharmakokinetischen Studien die AUC-Werte um etwas mehr als die Hälfte [56, 58]. Durch eine zweimal tägliche statt der gewöhnlich einmal täglichen Einnahme kann die Plasmaspiegelsenkung ausgeglichen werden [15]. Bei Einnahme anderer, vermutlich die Exposition senkender, Induktoren wie Johanniskraut, Oxcarbazepin und Phenytoin beträgt die empfohlene Dosis bei Erwachsenen folglich zweimal täglich 50 mg [67]. Besonders im Falle einer Integrase-Inhibitor-Resistenz soll aber eine Reduktion der Dolutegravir-Plasmaspiegel vermieden werden [67].
Dolutegravir kann die renale Elimination von Substraten des organischen Kationentransporters 2 (Organic cation transporter 2, OCT2) mindern. Die AUC von Metformin steigt durch Dolutegravir um 79 % bei einmal täglich 50 mg Dolutegravir und um 145 % bei zweimal täglich Dolutegravir. Eine Dosisanpassung, besonders bei nachlassender Nierenfunktion, ist zur Vermeidung von Laktazidosen in Betracht zu ziehen. Fampridin ist als OCT2-Substrat unter Dolutegravir-Einnahme kontraindiziert [67].
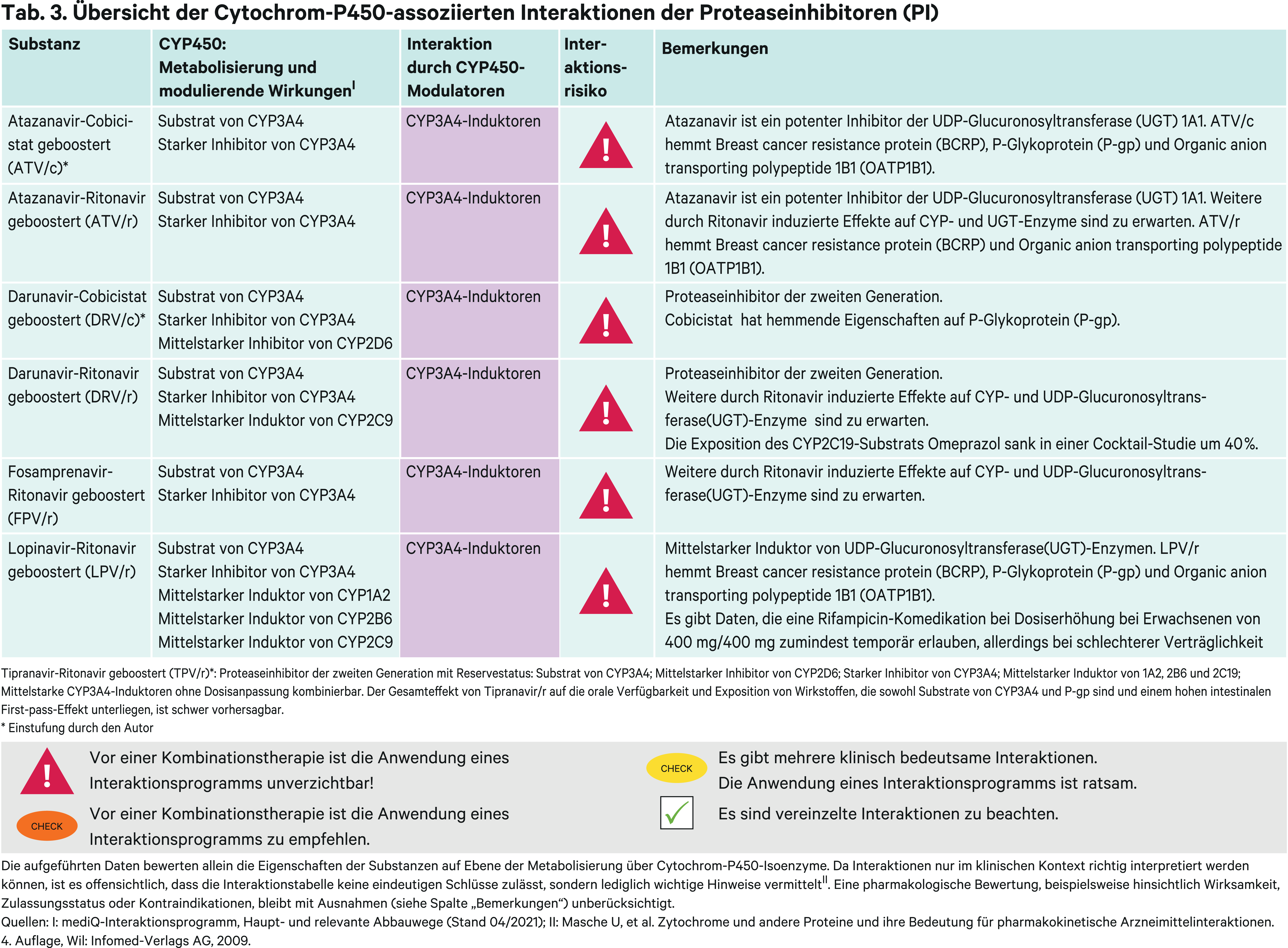
Proteaseinhibitoren (PI)
Die verfügbaren Proteaseinhibitoren sind an der Endung „-navir“ zu erkennen. Als Substrate von CYP3A4 werden sie obligat mit Pharmakoenhancern kombiniert. Für Atazanavir und Darunavir kann das Boostern wahlweise mit Cobicistat erfolgen, für alle anderen ist nur niedrig dosiertes Ritonavir zugelassen. Gewöhnlich wird die Boosterung durch ein „/r“ oder „/c“ im Anschluss an den Substanznamen kenntlich gemacht. Abhängig vom Wirkverstärker unterscheidet sich das pharmakokinetische Interaktionsprofil über die starke Hemmung von CYP3A4-Substraten hinaus [27]. Für die Abschätzung des Interaktionsrisikos einer noch nicht untersuchten Arzneimittelkombination kann es empfehlenswert sein zu prüfen, ob es Daten aus Untersuchungen mit einem anderen geboosterten Proteasehemmer mit dem komedizierten Wirkstoff gibt, wodurch analoge Rückschlüsse gezogen werden könnten. Beispielgebend sind die induktiven Effekte auf CYP1A2 und 2B6 zu nennen sowie die Phänotyp-abhängigen Wechselwirkungen von Voriconazol mit Ritonavir geboosterten Therapien. Protaseaseinhibitoren werden nach ihrer Struktur in zwei Generationen unterteilt. Darunavir und Tipranavir haben als Vertreter der zweiten Generation mit ihrer nichtpeptidischen Struktur eine höhere Resistenzbarriere als die Vertreter der ersten Generation [27].
Erstgenerations-PI
Atazanavir
Atazanavir (ATV) wird in einigen Ländern wegen seiner CYP3A4-hemmenden Eigenschaften auch ohne Booster angewandt. Jedoch ist ohne Boosterung die Resistenzbarriere vermutlich niedriger. Durch den Induktor Rifampicin wird eine 72%ige Abnahme der Ritonavir-geboosterten Atazanavir-AUC verursacht [8]. Die verminderte Exposition sollte nicht mit einer Atazanavir-Dosissteigerung kompensiert werden, da diese sehr häufig zu Leberreaktionen führt [24, 67]. Von der gleichzeitigen Einnahme mit Rifampicin wird abgeraten. Andere Induktoren wie Carbamazepin oder Phenytoin können unter engmaschiger Überwachung des virologischen Ansprechens der Patienten komediziert werden. Im Gegensatz dazu sollen die beiden NNRTI Efavirenz und Nevirapin nicht mit Atazanavir/r gleichzeitig angewendet werden. Hintergrund dieser Empfehlung ist dabei nicht eine verringerte ATV-Exposition, sondern um 42 bzw. 59 % reduzierte Talspiegel des Proteaseinhibitors und damit möglicherweise temporäre subtherapeutische Plasmaspiegel [67]. Dieser Einwand kann bei Anwendung anderer starker CYP3A4-Induktoren nicht ausgeschlossen werden. Bei einer notwendigen Efavirenz-Komedikation sollte die Ritonavir-Dosis verdoppelt werden.
Therapielimitierend ist bei Atazanavir seine UGT1A1-hemmende Wirkung. UGT1A1 ist das wesentliche Enzym für die Elimination von Bilirubin. Patienten mit Morbus Meulengracht (Gilbert-Syndrom) sind Träger des UGT1A1*28-Allels, das mit einer um 70 % reduzierten Transkription verbunden ist [39]. Durch die Hyperbilirubinämie können die Patienten einen Ikterus entwickeln [7]. Besondere Vorsicht ist geboten, wenn Atazanavir gleichzeitig mit UGT1A1-Substraten mit enger therapeutischer Breite verabreicht wird. Zum Beispiel wird der aktive Metabolit des Chemotherapeutikums Irinotecan, SN38, über diese Transferase eliminiert.
Weitere pharmakokinetische Wechselwirkungen sind durch den jeweils gewählten Pharmakoenhancer zu erwarten.
Die komplexen Wechselwirkungen mit Voriconazol sind in einer CYP2C19-genotypbasierten Studie mit gesunden Probanden untersucht worden und sind prinzipiell auf alle anderen PI/r-Kombinationen übertragbar. Das Azol-Antimykotikum Voriconazol wird primär über CYP2C19 verstoffwechselt und nachgeordnet über CYP3A4. Bei Probanden mit mindestens einem funktionellen CYP2C19-Allel sinken die Voriconazol-Spiegel um ein Drittel, begründet durch die CYP2C19-induktiven Effekte von Ritonavir. Bei Probanden ohne funktionales CYP2C19-Allel steigt die Voriconazol-AUC dagegen um das 5,6-Fache, weil in dieser Situation auch der metabolische Abbau des Nebenwegs durch die CYP3A4-Blockade ausfällt [71]. Vor dem Hintergrund dieser dramatischen Änderungen pharmakokinetischer Eckdaten des Reserve-Antimykotikums sollte bei Therapieeinleitung eine CYP2C19-Geno- oder Phänotypisierung in Betracht gezogen werden.
Fosamprenavir
Fosamprenavir (FPV) ist das Phosphatester-Prodrug von Amprenavir. Es wird rasch in-vivo zu Amprenavir hydrolysiert. Fosamprenavir wird mit Ritonavir geboostert. Rifampicin hat in einer klinischen Studie die AUC von Amprenavir um 82 % gesenkt [47]. Jedoch wurde es ohne den Pharmakoenhancer eingesetzt. Bedingt durch den Boostereffekt des Wirkverstärkers fällt die Expositionsänderung bei Kombination mit dem Induktor Phenytoin moderat aus [67]. Weitere pharmakokinetische Interaktionen von FPV/r können durch den Wirkverstärker Ritonavir verursacht werden. Eine Besonderheit ist die Plasmaspiegelabsenkung von Paroxetin durch FPV/r. Der vermutete Mechanismus ist die Verdrängung des Antidepressivums von seiner Plasmabindung am Alpha-1-acid Glykoprotein (AGP), einem Akut-Phase-Protein. Dafür spricht auch, dass in der Studie mehr freies, ungebundenes Paroxetin im Plasma gefunden wurde [62].
Lopinavir
Lopinavir (LPV) ist nur mit Ritonavir in Fixkombination verfügbar. Die Standarddosis bei Erwachsenen beträgt 400 mg/100 mg zweimal täglich. Optional kann Lopinavir/r auch einmal täglich mit 800 mg/200 mg eingenommen werden [67]. Die Rifampicin-induzierte Abnahme der Lopinavir-Konzentration kann durch Super-Boostern kompensiert werden. Hierzu wird LPV/r in der Standarddosis 400mg/100mg zusätzlich mit 300 mg Ritonavir zweimal täglich kombiniert [35]. Dies sollte aber nur Situationen vorbehalten bleiben, die einer gleichzeitigen Anwendung mit Rifampicin bedürfen. Die erhöhte Ritonavir-Dosis ist mit Leberwerterhöhungen und einer Zunahme gastrointestinaler Beschwerden assoziiert [43, 45]. Jedoch gibt es auch Daten, die eine akzeptable Verträglichkeit bei HIV/Tb-koinfizierten Patienten zeigen [6, 10]. Hierzulande wird das Antibiotikum primär in der Behandlung temporärer Nicht-Tuberkulose-Infektionen wie einer Spondylodiszitis eingesetzt. Bei einer gleichzeitigen Tuberkulose-Erkrankung sollte das schwächer CYP3A4-induzierend wirkende Rifabutin bevorzugt werden [67]. Wie bei Atazanavir kommt es durch Komedikation mit Efavirenz und Nevirapin zu einer Abnahme der Lopinavir-Talspiegel um 39 %. Wird die PI/r-Dosis um 25 % erhöht, sind die Lopinavir-Talspiegel vergleichbar mit den Lopinavir-Werten ohne Kombination mit den beiden NNRTI [54]. Eine einmal tägliche Einnahme ist dann aber nicht mehr möglich [67]. Bei Anwendung anderer CYP3A4-Induktoren wie Carbamazepin oder Phenobarbital sollte eine Dosiserhöhung in Erwägung gezogen werden, obwohl es hierfür aus klinischen Studien keine Daten gibt.
Die Durchführung von Arzneimittelinteraktionsstudien wurde durch die Möglichkeit, mehr als eine potenzielle Arzneimittelinteraktion (DDI) innerhalb einer einzigen Studie zu bewerten, revolutioniert. Cocktail-Studien bieten eine Möglichkeit, innerhalb einer einzigen Studie nach DDI über mehrere Stoffwechselwege zu suchen. Diese Studien werden in der Regel an gesunden Probanden durchgeführt und verwenden die gleichzeitige Verabreichung von Indexsubstraten und die Bestimmung von Biomarkern, um die Aktivitäten der Arzneimittel-metabolisierenden Enzyme (DME) vor und während der Arzneimittelbehandlung gleichzeitig zu bewerten. Zu den Vorteilen der Verwendung von Cocktail-Studien in der Arzneistoffentwicklung gehören eine reduzierte Probandenvariabilität, erhöhte Effizienz und geringere Kosten. Potenzielle Einschränkungen können durch ein geeignetes Studiendesign berücksichtigt werden. Da Cocktail-Studien das potenzielle Ausmaß von DDI bewerten, können Rückschlüsse auf die Medikamentendosierung und -anwendung gezogen werden [44]. Die metabolische Aktivität eines Perpetrators wird bestimmt durch die Veränderung des metabolischen Quotienten. Dieser setzt sich zusammen aus dem Verhältnis Muttersubstanz zum Metaboliten, der spezifisch über das CYP-Isoenzym gebildet wird, beispielsweise bei CYP2D6 Dextromethorphan zu Dextrorphan [66].
In einer Cocktail-Studie konnte ein induktiver Effekt von Lopinavir/r auf die CYP1A2-, 2C9- und 2C19-Testsubstrate Coffein, S-Warfarin und Omeprazol gezeigt werden. Die Enzymaktivität von CYP1A2 stieg um 43 %, die von CYP2C9 um 29 % und die von CYP2C19 um das Doppelte [68]. Bupropion wird primär über CYP2B6 verstoffwechselt [34]. Die Plasmaspiegelsenkung von Bupropion um 57 % durch LPV/r deutet auf eine Induktion von CYP2B6 hin [28].
Zweitgenerations-PI
Darunavir
Darunavir (DRV) ist ein nichtpeptidischer Proteaseinhibitor. DRV kann bei ART-naiven Patienten oder vorbehandelten Patienten ohne Virusmutationen, die mit einer DRV-Resistenz assoziiert sind, wahlweise mit Ritonavir oder Cobicistat angewendet werden. Unter diesen Voraussetzungen wird Darunavir mit 800 mg einmal täglich dosiert, wahlweise mit 100 mg Ritonavir oder 150 mg Cobicistat geboostert. Für beide Möglichkeiten ist ein Single-Tablet-Regime verfügbar [27, 67]. Bei mehrfach vorbehandelten Patienten kann Darunavir auch bei PI-resistenten Viren noch eine antivirale Wirkung entfalten [27, 67]. Hierzu muss Darunavir jedoch mit 2 × 600 mg dosiert werden. Wegen der zweimal täglichen Einnahme kann der Proteaseinhibitor nur mit Ritonavir geboostert werden, weil Cobicistat nur für die einmal tägliche Einnahme zur Verfügung steht. In Abhängigkeit des verwendeten Pharmakoenhancers zeigt Darunavir ein unterschiedliches Interaktionsprofil, das berücksichtigt werden muss, wenn von einem Pharmakoenhancer zum anderen gewechselt wird. Beiden gemein sind die Interaktionen mit CYP3A4-Substraten. In Kombination mit dem Induktor Rifampicin sind DRV/r sowie DRV/c kontraindiziert [67]. Das Superboostern bei DRV/r wurde in einer Studie untersucht und musste aufgrund von schwerwiegenden hepatotoxischen Nebenwirkungen abgebrochen werden [17]. Es bleibt zu erwägen, ob bei einer zwingend notwendigen Kombination mit dem Antibiotikum in diesem Fall ein vorübergehender Wechsel auf Lopinavir/r in der Dosis von 400 mg/400 mg möglich ist. Der Metabolismus von Cobicistat ist in stärkerem Umfang abhängig von CYP3A4 und sein Boostereffekt kann bei gleichzeitiger Anwendung mit CYP3A4-Induktoren vermindert sein [61, 67]. Andere starke CYP3A4-Induktoren als Rifampicin sind unter einer DRV/c-Therapie kontraindiziert und moderat starke CYP3A4-Induktoren wie Bosentan sollten gemieden werden. Das verfügbare Single-Tablet-Regime ist dann nicht mehr einsetzbar. Dagegen bleiben die pharmakokinetischen Parameter von DRV/r durch den Induktor Carbamazepin unberührt [67].
DRV/r in der Dosis zweimal täglich 600 mg/100 mg ist mit Efavirenz kombinierbar. Bei der einmal täglichen Einnahme sinken die Darunavir-Talspiegel in einen subtherapeutischen Bereich [59]. In Kombination mit Nevirapin als NNRTI ist keine Dosiserhöhung notwendig. Unter DRV/r fallen die AUC-Werte der SSRI-Antidepressiva Paroxetin und Sertralin um 39 % bzw. 49 %. Für die Plasmaspiegelsenkung von Paroxetin kann, wie bei Fosamprenavir/r, eine Verdrängung am Bindungsprotein angenommen werden. Für Sertralin ist ein beschleunigter metabolischer Abbau zu vermuten. In einer Cocktail-Studie stieg die AUC von Dextromethorphan als CYP2D6-Testsubstrat um das 2,7-Fache. Die Exposition von Omeprazol reduzierte sich um 42 % und von S-Warfarin um 21 %. Die Enzymaktivität nahm für CYP2C9 um 55 % und für CYP2C19 um 31 % zu [53].
Tipranavir
Tipranavir (TPV) war der erste Vertreter der nichtpeptidischen Proteaseinhibitoren und wird mit 200 mg Ritonavir zweimal täglich kombiniert, damit ausreichend hohe Plasmaspiegel erreicht werden [27]. So müssen bei der Standarddosis von 500 mg/200 mg TPV/r in den verfügbaren Stärken Tipranavir mit 250 mg als Kapseln und Ritonavir mit 100 mg als Tabletten bereits acht feste Einheiten täglich eingenommen werden [67]. Dieses Einnahmeschema und das komplexe Interaktionsprofil führten dazu, dass Tipranavir/r nur noch bei Therapieresistenz auf mehrere Proteaseinhibitoren eine Reserveoption darstellt. In einer Cocktail-Studie zeigten sich CYP-induzierende Effekte im Steady-State von TPV/r. Die AUC von Coffein sank um 47 % und die von Omeprazol um 70 %. Der Effekt auf S-Warfarin war mit 12 % im Steady-State gering. Die AUC von Dextromethorphan stieg um das 5,5-Fache [16]. Mit der peroralen und i. v.- Gabe von Midazolam als CYP3A4-Indexsubstrat wurde der Effekt von TPV/r auf hepatische und intestinale CYP3A4-Enzyme in Bezug auf die Erstdosis und im Steady-State untersucht. Nach parenteraler Gabe stieg Midazolam im Steady-State um das 3-Fache und nach peroraler Einnahme um das 10,4-Fache. Folglich wird die intestinale CYP3A4-Aktivität stärker gehemmt als die hepatische. Nach Ersteinnahme war die AUC von Midazolam um jeweils das 5-Fache bzw. das 36-Fache erhöht, was auf einen induktiven Effekt im Steady-State hinweist. Ähnlich verhält es sich mit dem P-gp-Testsubstrat Digoxin. Auch hier zeigte sich bei peroraler Einnahme ein höherer Effekt auf die Exposition als nach parenteraler Gabe. Im Steady-State zeigte sich eine induktive Wirkung, während nach einmaliger Gabe die Hemmung von P-gp festgestellt wurde [16]. Ähnlich verhielt es sich in einer Studie mit Loperamid. Die Exposition des peripher wirkenden Opioidagonisten wurde durch Tipranavir bei gesunden Probanden um über die Hälfte gesenkt [42]. Somit ist es schwierig den Gesamteffekt von Tipranavir/r auf die orale Bioverfügbarkeit und Exposition von Wirkstoffen vorherzusagen, die sowohl Substrate von CYP3A4 als auch P-gp sind und einem hohen intestinalen First-pass-Metabolismus unterliegen [67].
Eine AUC-Reduktion von 56 % bei Bupropion weist auf einen induktiven Effekt von Tipranavir/r auf CYP2B6 hin. Dazu passt auch das Absinken der AUC von Methadon um die Hälfte, ebenfalls ein Substrat von CYP2B6 [67].
Carbamazepin als CYP3A4-Induktor senkt den Talspiegel von Tipranavir um 61 % und sollte, wie andere starke CYP3A4-Induktoren, gemieden werden [67]. Efavirenz als mittelstarker Induktor hatte keinen Einfluss auf die pharmakokinetischen Parameter [36]. Mittelstarke Induktoren sind mit Tipranavir kombinierbar, Rifampicin und Johanniskraut-haltige Arzneimittel sind dagegen kontraindiziert [67].
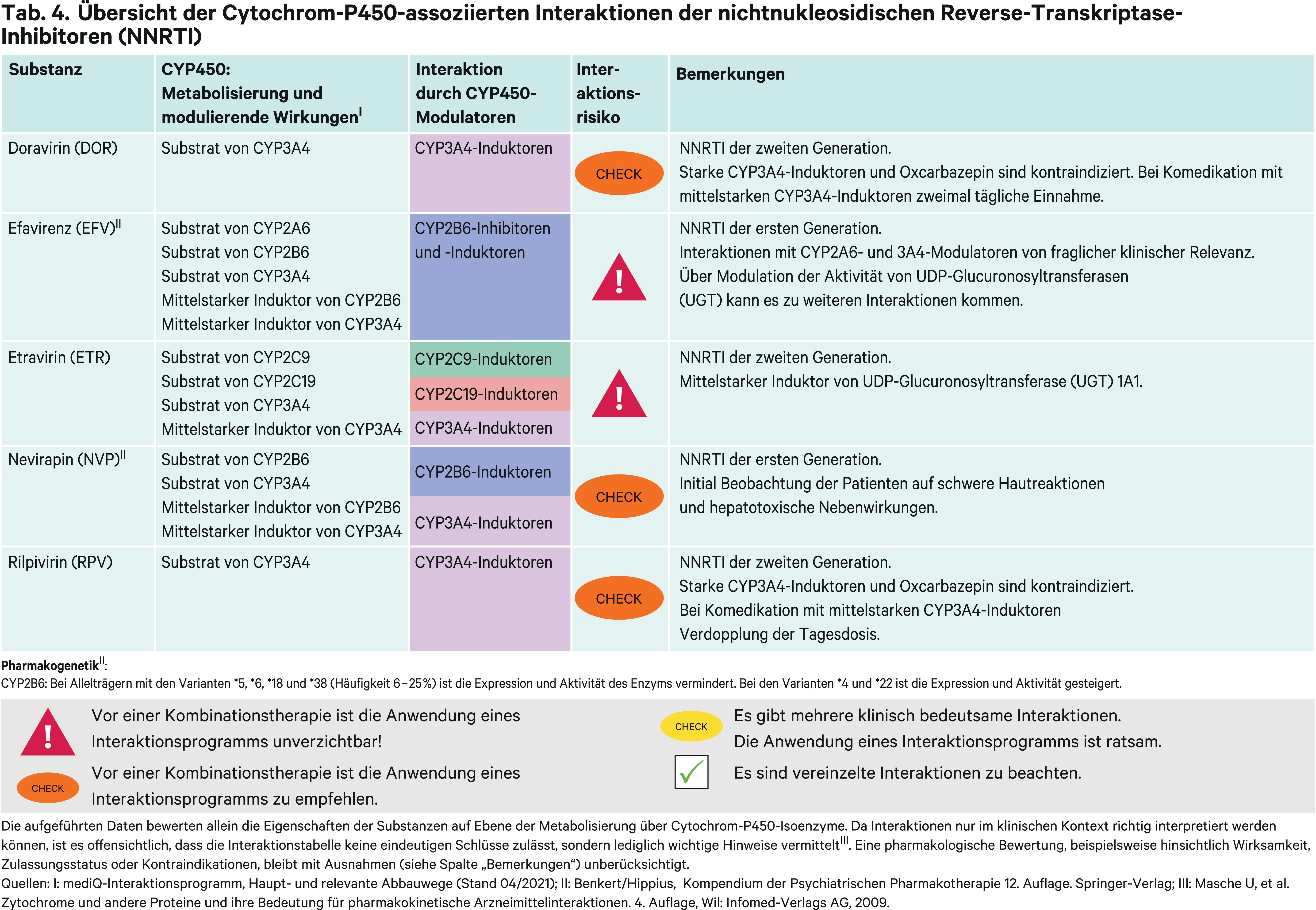
Nichtnukleosidische Reverse-Transkriptase-Inhibitoren (NNRTI)
Die Erstgenerations-NNRTI Nevirapin und Efavirenz spielen bei Erwachsenen in der antiretroviralen Therapie derzeit keine Rolle mehr. Nevirapin als erster Vertreter dieser neuen Wirkstoffklasse hat eine vergleichsweise geringe Resistenzbarriere [27]. Zudem besteht initial die Gefahr von schwerwiegenden, lebensbedrohlichen Auswirkungen auf die Haut einschließlich Stevens-Johnson-Syndrom (SJS) und toxischer epidermaler Nekrolyse sowie hepatotoxischer Reaktionen [67]. Somit kommt es nicht für die postexpositionelle Prophylaxe der HIV-Infektion in Frage [13]. Efavirenz zeigt eine schlechte Verträglichkeit. Besonders zentralnervöse Störungen wie Schwindel und Benommenheit sowie lebhafte Träume oder Alpträume limitieren seinen Einsatz [27].
Die Zweitgenerations-NNRTI sind erkennbar an der Endung „-virin“. Doravirin, Etravirin und Rilpivirin sind deren Vertreter, wobei Etravirin und Rilpivirin nicht ohne Einschränkungen anwendbar sind. Etravirin war der erste Zweitgenerations-NNRTI und ist in Kombination mit geboosterten Proteaseinhibitoren zugelassen bei antiretroviral vorbehandelten Patienten. Rilpivirin darf grundsätzlich nicht bei hochvirämischen Patienten verschrieben werden [27]. Somit bleibt als Mittel der Wahl für die Erstlinientherapie mit einem NNRTI Doravirin.
Erstgenerations-NNRTI
Efavirenz
Efavirenz (EFV) wird hauptsächlich über CYP2B6 verstoffwechselt. Bei Kombination mit dem CYP2B6-Inhibitor Voriconazol (Abb. 2) steigt die Exposition um 44 % und die Efavirenz-Tagesdosis muss um die Hälfte reduziert werden [14, 67]. Die Einnahme von Efavirenz ist verbunden mit neuropsychiatrischen Nebenwirkungen. Deutliche Erhöhungen der Bioverfügbarkeit können zu schwerwiegenden Nebenwirkungen führen, wobei der genetische Polymorphismus, unabhängig der Komedikation, von klinischer Relevanz ist. CYP2B6 wird polymorph exprimiert. Ein Patient entwickelte schwere neuropsychiatrische Nebenwirkungen, nachdem eine Efavirenz-enthaltende Single-Tablet-Regime-Einnahme begonnen wurde. Der gemessene Plasmaspiegel des NNRTI lag 40-fach höher als zu erwarten war. Gemäß einer genetischen Analyse war der Patient Träger des Haplotyps CYP2B6*6/*38, womit eine deutlich eingeschränkte Metabolisierungskapazität von CYP2B6-Substraten verbunden ist. Nach Beendigung der Efavirenz-Therapie sistierten die Symptome über einen mehrwöchigen Verlauf vollständig [3]. Interaktionen mit Induktoren sind von geringer Bedeutung für die Efavirenz-Exposition. Mit dem Induktor Rifampicin fällt die EFV-AUC bei gesunden Probanden um 26 % und kann durch moderate Dosissteigerung ausgeglichen werden [67]. Dieses Vorgehen bestätigte sich bei HIV-TB-koinfizierten Patienten, bei denen die AUC bei der höchsten Efavirenz-Dosis mit Rifampicin nur um wenige Prozentpunkte niedriger lag im Vergleich zur niedrigeren Dosis ohne Rifampicin [4]. Dieser pharmakokinetische Vorteil kann bei der Behandlung der Tuberkulose von HIV-infizierten Kindern und Jugendlichen genutzt werden [12].
Als „Perpetrator“ kann Efavirenz zahlreiche Wechselwirkungen auslösen. Im Sinne einer bidirektionalen Interaktion senkt EFV die Voriconazol-AUC über eine CYP2C19-Induktion um 77 % [14]. Die Exposition von Bupropion fällt um 55 %, die der CYP3A4-Substrate Diltiazem und Simvastatin wird durch EFV um 69 % reduziert [23, 32, 49]. In einer klinischen Studie zeigte Efavirenz eine 41%ige Steigerung der metabolischen Aktivität auf CYP2C19 [41]. Durch UGT1A1-Induktion kommt es zum Absinken der AUC von Raltegravir um 36 % [29]. Dies ist aber klinisch nicht relevant [67].
Nevirapin
Nevirapin (NVP) ist ein Substrat von CYP2B6 und 3A4. Wechselwirkungen mit starken CYP3A4-Hemmern sind von geringer Bedeutung. Ketoconazol und Itraconazol führen zu keiner signifikanten Veränderung der pharmakokinetischen NVP-Parameter [30, 37]. Eine klinische Studie mit Voriconazol liegt nicht vor, die Hinweise auf einen möglichen klinischen Effekt durch CYP2B6-Inhibitoren geben könnte. Nach Anwendung von Rifampicin sinkt die AUC um 58 % [50]. Ein Einfluss des mittelstarken Induktors Rifabutin konnte nicht festgestellt werden. Nevirapin induziert seinen eigenen Metabolismus. Somit führen starke Induktoren wie Rifampicin zu einer signifikanten Senkung der AUC. Eine gleichzeitige Anwendung mit Rifampicin wird daher nicht empfohlen und andere starke Induktoren sollten ebenfalls gemieden werden, obwohl es hierzu keine Empfehlungen in den Herstellerangaben gibt. Der Einfluss von moderaten Induktoren auf die Nevirapin-Exposition erscheint vernachlässigbar.
Da Nevirapin ein CYP3A4-Induktor ist, kann Cobicistat nicht als Pharmakoenhancer genutzt werden. Die AUC-Werte der CYP3A4-Substrate Itraconazol und Ketoconazol fallen um 61 und 72 % [30, 37]. In einer Studie zeigte sich ein AUC-Abfall des Substitutionsmittels Methadon um 40 %; hierfür wird eine CYP2B6-Induktion verantwortlich gemacht [60].
Zweitgenerations-NNRTI
Doravirin
Doravirin (DOR) wird hauptsächlich über CYP3A4 metabolisiert. Rifampicin senkte in einer Studie die AUC von Doravirin um 88 % und der Talspiegel fiel auf 3 % [33]. Wie Rifampicin sind auch andere starke CYP3A4-Induktoren sowie Oxcarbazepin kontraindiziert. Mit dem mittelstarken Induktor Rifabutin halbiert sich die Doravirin-Exposition [33]. Sollte eine Begleitmedikation mit mittelstarken CYP3A4-Induktoren nicht vermieden werden können, so ist Doravirin zweimal täglich zu dosieren [67]. Zwar können starke CYP3A4-Hemmer wie Ketoconazol die Doravirin-AUC um das Dreifache erhöhen, eine Dosisanpassung ist aber wegen der allgemein guten Verträglichkeit des NNRTI nicht notwendig [33]. Ein leichter induktiver Effekt auf das CYP3A4-Testsubstrat Midazolam sollte bei einer Therapie mit den Calcineurin-Inhibitoren Ciclosporin und Tacrolimus beachtet werden, die über dieses CYP-Isoenzym abgebaut werden [33, 67].
Etravirin
Etravirin (ETR) wird über CYP2C9, 2C19 und 3A4 verstoffwechselt. Wechselwirkungen mit CYP-Inhibitoren (Abb. 2) sind von geringem Ausmaß und erfordern keine Dosisanpassung. Fluconazol als potenter Hemmer aller drei CYP-Enzyme vermochte die AUC von Etravirin um 86 % zu erhöhen. Starke CYP3A4-Inhibitoren wie Atazanavir/r oder Voriconazol erhöhen die ETR-Exposition um etwa ein Drittel [25]. Etravirin ist nur zugelassen in Kombination mit geboosterten Proteaseinhibitoren [67]. Jedoch kommt hierfür Tipranavir/r nicht infrage, weil die ETR-Exposition bei HIV-Infizierten aufgrund von induktiven Effekten von TPV/r auf CYP2C9 und 2C19 um 76 % reduziert wurde [52]. Andere starke Induktoren können zu verminderten Plasmakonzentrationen von Etravirin führen und sollten gemieden werden. Mittelstarke Induktoren wie Rifabutin und Lopinavir/r vermindern die Etravirin-Exposition um etwa ein Drittel. Dies erfordert jedoch keine Etravirin-Dosisanpassung.
Etravirin kann durch seine CYP3A4-Induktion zu klinisch relevanten Plasmaspiegelsenkungen von CYP3A4-Substraten führen. So sank die Exposition von Maraviroc um 53 % und von Sildenafil um 57 % [25]. Dadurch bedingt wird eine gleichzeitige Anwendung mit Cobicistat-geboosterten Proteaseinhibitoren nicht empfohlen, sodass letztendlich nur die Kombination mit Ritonavir-geboosterten PI-Regimen zur Verfügung steht.
Bemerkenswert ist die Interaktion von Etravirin mit Dolutegravir. Begründet über eine UGT1A1-Induktion sank in einer klinischen Studie die AUC des Integrase-Inhibitors um 71 %. Die Plasmakonzentration von Raltegravir sinkt durch Etravirin nur um 10 %. Wird Etravirin mit einem geboosterten Proteaseinhibitor wie Lopinavir/r oder Darunavir/r kombiniert, verliert sich der induktive Effekt auf Dolutegravir. Die pharmakokinetischen Parameter von Dolutegravir waren bei Anwendung von Lopinavir/r nur unwesentlich verändert, unter Darunavir/r war die AUC um 25 % reduziert, was aber klinisch nicht bedeutsam ist [57].
Dies ist ein Beispiel dafür, dass Ritonavir-geboosterte ART-Regime zuweilen unvorhersagbare und mechanistisch nicht erklärbare Wechselwirkungen zeigen können. Es zeigt aber auch, dass die in der Regel mit gesunden Probanden gewonnenen Ergebnisse aus pharmakokinetischen Wechselwirkungsstudien bei HIV-Patienten nicht zwingend übertragbar sind. So wurde in vier Fallbeispielen dokumentiert, dass nach Initiierung einer Etravirin-Therapie die Raltegravir-Talspiegel in einen subtherapeutischen Bereich fielen [40].
Rilpivirin
Rilpivirin (RPV) ist ein Substrat von CYP3A4. Darunavir/r erhöhte in einer klinischen Studie bei HIV-seronegativen Probanden die AUC von Rilpivirin um das 2,3-Fache [20]. Dies ist ohne Konsequenz für die Dosierung von Rilpivirin. Rifampicin reduzierte in einer weiteren Untersuchung die Exposition von Rilpivirin um 80 % [20]. Starke CYP3A4-Induktoren sind unter Rilpivirin-Therapie kontraindiziert [67]. Eine 42%ige Senkung der Exposition durch Rifabutin kann wie bei gleichzeitiger Anwendung anderer mittelstarker CYP3A4-Induktoren durch Verdopplung der Rilpivirin-Dosis ausgeglichen werden [67]. Gemäß Herstellerangaben darf Oxcarbazepin nicht angewendet werden [67].
Pharmakoenhancer
Als pharmakokinetische Booster über eine starke CYP3A4-Hemmung werden niedrig dosiertes Ritonavir in variabler Dosis und Cobicistat in Fixdosis genutzt. Ziel ist es, durch Verbesserung pharmakokinetischer Parameter die Einnahmefrequenz und Medikamentenzahl zu senken, gleichmäßigere Wirkstoffspiegel zu erhalten, die Anwendungsfreundlichkeit zu verbessern und so im Ergebnis den Therapieerfolg zu steigern [61]. Ritonavir kann mit allen Proteaseinhibitoren kombiniert werden, Cobicistat nur mit Atazanavir und Darunavir sowie dem Integrase-Inhibitor Elvitegravir.
Da über 60 % der Arzneimittel über CYP3A4 verstoffwechselt werden, ist eine Vielzahl unerwünschter Wechselwirkungen möglich. Dies veranschaulicht sich unter anderem am Beispiel von topisch und intraartikulär applizierten Glucocorticoiden, die Substrate von CYP3A4 sind. Aufgrund der erhöhten systemischen Verfügbarkeit ist in zahlreichen Kasuistiken beschrieben, wie Fluticason und Triamcinolon eine adrenale Suppression und konsekutiv ein Cushing-Syndrom induzierten [2, 19].
Ritonavir
Ritonavir wurde als Proteaseinhibitor in höherer Dosierung als antiretrovirale Substanz eingeführt. Hierfür ist es noch zugelassen, aber wegen der vergleichsweise schlechten Verträglichkeit wird von einer therapeutischen Anwendung abgeraten. Für den Boostereffekt reicht eine niedrigere und besser verträgliche Tagesdosis. Pharmakokinetische Wechselwirkungen von Ritonavir können durch Inhibition von CYP2D6 und P-gp zustande kommen. Die Daten zum Ausmaß einer CYP2D6-Hemmung sind nicht eindeutig und abhängig vom jeweils verwendeten Proteaseinhibitor. Während gemäß Herstellerangaben von Lopinavir/r diesbezüglich kein Interaktionsrisiko ausgeht, sank die CYP2D6-Aktivität durch LPV/r um zwei Drittel, was Ergebnisse aus Cocktail-Studien mit Dextromethorphan und Darunavir/r bzw. Tipranavir/r bestätigen [66]. Mit Desipramin als CYP2D6-Indexsubstrat wurde eine geringe Erhöhung der Exposition von 26 % ermittelt [61]. Ritonavir in Booster-Dosis führte in einer klinischen Studie bei simultaner Einnahme zu keiner signifikanten Änderung der AUC des P-gp-Substrates Dabigatranetexilat [46].
Induktive Eigenschaften auf CYP1A2, 2B6, 2C9 und 2C19 sowie auf UGT erweitern das Spektrum unerwünschter Interaktionen mit Ritonavir. Über eine UGT1A1-Induktion kann es beispielsweise zu einem Abfall der Ethinylestradiol-Plasmaspiegel kommen [61]. Lamotrigin wird primär über UGT1A4 metabolisiert. Lopinavir/r reduzierte in einer Studie die Lamotrigin-Spiegel um die Hälfte [63].
Rosuvastatin ist ein Substrat des organic anion transporting polypeptide 1B1 (OATP1B1) und von BCRP. Durch ATV/r und LPV/r ist die AUC von Rosuvastatin um das 3,13- bzw. 2-Fache erhöht. In Kombination mit den anderen Proteaseinhibitoren fällt die AUC-Erhöhung geringer aus [61, 67].
Cobicistat
Cobicistat wird über CYP3A4 metabolisiert. Carbamazepin senkt die AUC des Pharmakoenhancers um 84 %. Starke CYP3A4-Induktoren sind unter Cobicistat-haltigen ART-Regimen kontraindiziert, da es nur in einer Dosis verfügbar ist. Auch von einer gleichzeitigen Anwendung mit mittelstarken Induktoren wie Bosentan wird abgeraten [67].
Cobicistat ist ein schwacher CYP2D6-Hemmer und im Vergleich zu Ritonavir stärkerer P-gp-Hemmer. Die AUC von Desipramin steigt um etwa 60 % [61]. Die AUC von Dabigatranetexilat wurde in einer klinischen Studie um das 2,3-Fache erhöht, was sich auch nicht durch eine zeitversetzte Einnahme vermindern ließ [46]. Die AUC-Erhöhung von Rosuvastatin ist durch ATV/c mit 242% deutlich höher als durch DRV/c mit 93% bzw. durch EVG/c mit 38% [61, 67].
Entry-Inhibitoren
Maraviroc ist der erste und bislang einzige CCR5(CC-Motiv-Chemokin-Rezeptor-5)-Antagonist. Der metabolische Abbau erfolgt primär über CYP3A4. Die empfohlene Maraviroc-Dosis richtet sich nach der Begleitmedikation. Die Standarddosis wird halbiert, wenn der Patient gleichzeitig starke CYP3A4-Hemmer mit oder ohne CYP3A4-Induktoren einnimmt (Abb. 2). Eine Ausnahme bildet Tipranavir/r. Bei Kombination mit starken CYP3A4-Induktoren soll die Dosis verdoppelt werden. Hintergrund dieser Empfehlungen ist die Steigerung der Exposition von Maraviroc durch starke CYP3A4-Inhibitoren wie Ketoconazol, Atazanavir und Darunavir/r um das Vier- bis Fünffache, zum Teil trotz der Einnahme von CYP3A4-Induktoren [1]. Der Induktor Rifampicin reduziert die Maraviroc-AUC um 63 %. Unterschiedliche Auswirkungen haben die NNRTI Efavirenz und Nevirapin auf die Plasmaspiegel des CCR5-Antagonisten. Während Efavirenz die 12h-AUC von Maraviroc um 53 % reduziert, ist Nevirapin ohne Einfluss [31]. Bei Kombination mit Efavirenz wird daher eine Dosiserhöhung von Maraviroc empfohlen. Somit ist auch Vorsicht geboten bei Anwendung mittelstarker CYP3A4-Induktoren [67].
Für die parenteral anzuwendenden Entry-Inhibitoren Fostemsavir und Ibalizumab sind keine pharmakokinetischen Interaktionen zu erwarten [67].
Fazit
Die antiretrovirale Therapie (ART) der HIV-1-Infektion hat über Jahrzehnte einen enormen Fortschritt erfahren. Die ART ist nach derzeitigem Wissensstand eine lebenslange Therapiestrategie, die ohne Unterbrechungen durchzuführen ist. Eine Therapiekombination soll anwendungsfreundlich sein, um die Adhärenz zu fördern. Eine geringe Einnahmefrequenz und niedrige Medikamentenzahl wird durch die Pharmakoenhancer befördert – ein Kunstgriff, bei dem die üblicherweise unerwünschten Wechselwirkungen therapeutisch genutzt werden. Andererseits bergen sie aber das Potenzial schier unübersichtlicher Interaktionen, da über 60 % aller Arzneimittel über CYP3A4 metabolisiert werden [70]. Vor der Einleitung als auch unter der Therapie mit antiretroviralen Arzneistoffen bei Änderung der Begleitmedikation ist eine Medikationsanalyse dringend empfehlenswert. Hierzu sollten neben den Herstellerangaben auch Interaktionsdatenbanken sowie die Internetseite der University of Liverpool www.hiv-druginteractions.org herangezogen werden. Bei der Interpretation der Datenbankergebnisse sowie bei der Wahl möglicher Alternativ-Arzneimittel sollte die Expertise in der HIV-Therapie erfahrener Ärzte oder Apotheker zu Rate gezogen werden. Eine suboptimale Therapie durch zu geringe Wirkstoffspiegel gefährdet das internationale 90–90–90-Ziel der UNAIDS (United Nations Programme on HIV/AIDS), mit dem versucht wird, die HIV-Epidemie erfolgreich einzugrenzen oder gar zu beenden. Diese Initiative beinhaltet, dass mindestens 90 % aller Menschen weltweit mit AIDS diagnostiziert sein sollen, von diesen mindestens 90 % medikamentös behandelt und von bei diesen wiederum mindestens 90 % der Therapien erfolgreich sein sollten. Da neben pharmakokinetischen Wechselwirkungsrisiken bei einzelnen Arzneimitteln auch die Art der Anwendung und die Verwendung von magensäureblockierenden Mitteln von Bedeutung sein kann, bietet sich darüber hinaus ein Medikationsmanagement an. Dies befördert eine interdisziplinäre Zusammenarbeit zwischen Arzt und Apotheker zur Sicherstellung einer möglichst höchsteffektiven und risikoarmen antiretroviralen Therapie.
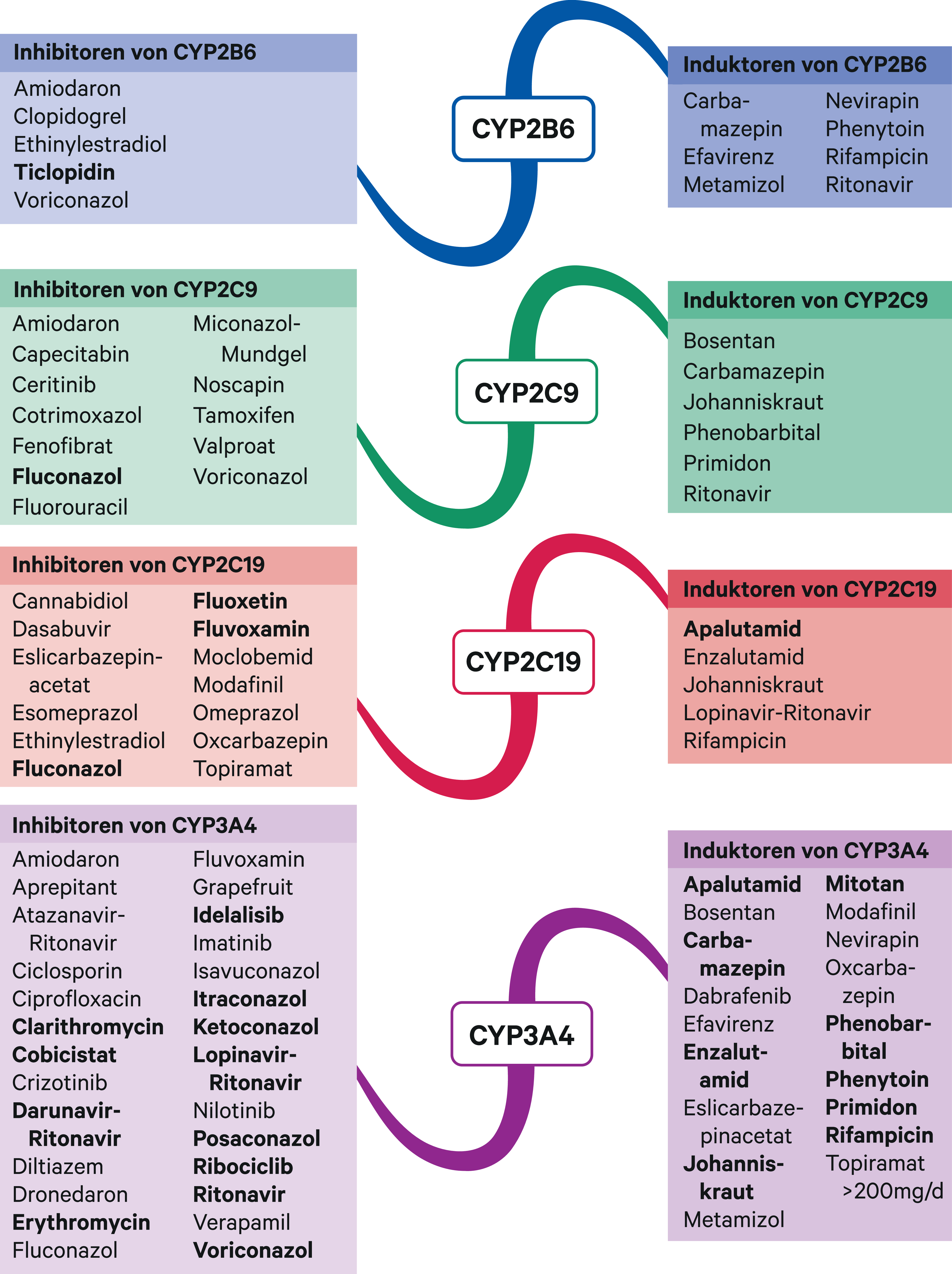
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2B6, 2C9, 2C19 und 3A4 (Stand 04/2021) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Abel S, Russell D, Taylor-Worth RJ, et al. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol 2008;65 Suppl 1:27–37.
2. Alidoost M, Conte GA, Agarwal K, et al. Iatrogenic Cushing’s syndrome following intra-articular triamcinolone injection in an HIV-infected patient on cobicistat presenting as a pulmonary embolism: Case Report and Literature Review.Int Med Case Rep J 2020;13:229–35.
3. Anagnostopoulos A, Rotger M, Aouri M, Kuster SP, Telenti A, Décosterd LA, Günthard HF. Efavirenz intoxication due to a new CYP2B6 constellation. Antivir Ther 2013;18:739–43.
4. Atwine D, Baudin E, Gelé T, et al. Effect of high-dose rifampicin on efavirenz pharmacokinetics: drug-drug interaction randomized trial. J Antimicrob Chemother 2020;75:1250–8.
5. Belkhir L, Seguin-Devaux C, Elens L, et al. Impact of UGT1A1 polymorphisms on Raltegravir and its glucuronide plasma concentrations in a cohort of HIV-1 infected patients. Sci Rep 2018;8:7359.
6. Boulanger C, Rolla V, Al-Shaer MH, et al. Evaluation of super-boosted lopinavir/ritonavir in combination with rifampicin in HIV-1-infected patients with tuberculosis. Int J Antimicrob Agents 2020;55:105840.
7. Burchell B, Hume R. Molecular genetic basis of Gilbert’s syndrome. J Gastroenterol Hepatol 1999;14:960–6.
8. Burger DM, Agarwala S, Child M, et al. Effect of rifampin on steady-state pharmacokinetics of atazanavir with ritonavir in healthy volunteers. Antimicrob Agents Chemother. 2006;50:3336–42.
9. Custodio JM, West S, Lutz J. Twice-daily administration of tenofovir alafenamide in combination with rifampin: potential for tenofovir alafenamide use in HIV-TB coinfection. In: Program and abstracts of the 16th European AIDS Conference; October 25–27, 2017; Milan, Italy. Abstract PS13/4.
10. Decloedt EH, Maartens G, Smith P, et al. The safety, effectiveness and concentrations of adjusted lopinavir/ritonavir in HIV-infected adults on rifampicin-based antitubercular therapy. PLoS One 2012;7:e32173.
11. Deutsche AIDS-Gesellschaft e. V. Deutsch-Österreichische Leitlinien zur antiretroviralen Therapie der HIV-1-Infektion. Version 9 vom 03.09.2020, AWMF-Register-Nr.: 055–001.
12. Deutsche AIDS-Gesellschaft e. V. Deutsch-Österreichische Leitlinien zur antiretroviralen Therapie der HIV-1-Infektion bei Kindern und Jugendlichen. Version 9 vom 03.09.2020, AWMF-Register-Nr.: 048–011.
13. Deutsche AIDS-Gesellschaft e. V. Deutsch-Österreichische Leitlinien zur Postexpositionellen Prophylaxe der HIV-Infektion (update 2018). AWMF-Register-Nr.: 055–004.
14. Desta Z, Metzger IF, Thong N, et al. Inhibition of Cytochrome P450 2B6 Activity by Voriconazole Profiled Using Efavirenz Disposition in Healthy Volunteers. Antimicrob Agents Chemother 2016;60:6813–22.
15. Dooley KE, Sayre P, Borland J, et al. Safety, tolerability, and pharmacokinetics of the HIV integrase inhibitor dolutegravir given twice daily with rifampin or once daily with rifabutin: results of a phase 1 study among healthy subjects. J Acquir Immune Defic Syndr 2013;62:21–7.
16. Dumond JB, Vourvahis M, Rezk NL, et al. A phenotype-genotype approach to predicting CYP450 and P-glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin Pharmacol Ther 2010;87:735–42.
17. Ebrahim I, Maartens G, Wiesner L, et al. Pharmacokinetic profile and safety of adjusted doses of darunavir/ritonavir with rifampicin in people living with HIV. J Antimicrob Chemother 2020;75:1019–25.
18. Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: Review of clinical drug-drug interaction studies. Clin Pharmacokinet 2020;59:699–714.
19. Foisy MM, Yakiwchuk EM, Chiu I, et al. Adrenal suppression and Cushing’s syndrome secondary to an interaction between ritonavir and fluticasone: a review of the literature. HIV Med 2008;9:389–96.
20. Ford N, Lee J, Andrieux-Meyer I, et al. Safety, efficacy, and pharmacokinetics of rilpivirine: systematic review with an emphasis on resource-limited settings. HIV AIDS (Auckl) 2011;3:35–44.
21. Gallicano KD, Sahai J, Shukla VK, et al. Induction of zidovudine glucuronidation and amination pathways by rifampicin in HIV-infected patients. Br J Clin Pharmacol 1999;48:168–79.
22. Garrison KL, Kirby B, Stamm LM, et al. Drug-drug interaction profile of Sofosbuvir/Velpatasvir/Voxilaprevir fixed-dose combination. Presented at the International Liver Congress, 19–23 April 2017, Amsterdam, the Netherlands.
23. Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005;39:307–12.
24. Haas DW, Koletar SL, Laughlin L, et al. Hepatotoxicity and gastrointestinal intolerance when healthy volunteers taking rifampin add twice-daily atazanavir and ritonavir. J Acquir Immune Defic Syndr 2009;50:290–3.
25. Havens JP, Podany AT, Scarsi KK, et al. Clinical Pharmacokinetics and Pharmacodynamics of Etravirine: An Updated Review. Clin Pharmacokinet 2020;59:137–54.
26. Hill A, Hughes SL, Gotham D, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate: is there a true difference in efficacy and safety? J Virus Erad 2018;4:72–9.
27. Hoffmann C, Rockstroh JK, HIV 2020/2021. Medizin Fokus Verlag.
28. Hogeland GW, Swindells S, McNabb JC, et al. Lopinavir/ritonavir reduces bupropion plasma concentrations in healthy subjects. Clin Pharmacol Ther 2007;81:69–75.
29. Iwamoto M, Wenning LA, Petry AS, et al. Minimal effects of ritonavir and efavirenz on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother 2008;52:4338–43.
30. Jaruratanasirikul S, Sriwiriyajan S. Pharmacokinetic study of the interaction between itraconazole and nevirapine. Eur J Clin Pharmacol 2007;63:451–6.
31. Jenkins T, et al. The effect of P450 inducers on the pharmacokinetics of CCR5 antagonist UK-427,857 in healthy volunteers. 5th International Workshop on Clinical Pharmacology of HIV Therapy, Rome, April 2004, abstract 37.
32. Kaul S, Ji P, Dudley J, et al. Pharmacokinetic (PK) interaction between efavirenz (EFV) and diltiazem (DTZ) or itraconazole (ITR) after multiple-dose administration in adult healthy subjects. 14th Conference on Retroviruses and Opportunistic Infections (CROI), Los Angeles, CA, February 25–28, 2007.
33. Khalilieh S, Yee KL, Sanchez R, et al. Clinical pharmacokinetics of the novel HIV-1 non-nucleoside reverse transcriptase inhibitor doravirine: An assessment of the efect of patient characteristics and drug-drug interactions. Clin Drug Investig 2020;40:927–46.
34. Kharasch ED, Crafford A. Common Polymorphisms of CYP2B6 influence stereoselective bupropion disposition. Clin Pharmacol Ther 2019;105:142–52.
35. la Porte CJ, Colbers EP, Bertz R, et al. Pharmacokinetics of adjusted-dose lopinavir-ritonavir combined with rifampin in healthy volunteers. Antimicrob Agents Chemother 2004;48:1553–60.
36. la Porte CJ, Sabo JP, Béïque L, et al. Lack of effect of efavirenz on the pharmacokinetics of tipranavir-ritonavir in healthy volunteers. Antimicrob Agents Chemother 2009;53:4840–4.
37. Lamson M, Robinson P, Lamson M, et al. The pharmacokinetic interactions of nevirapine and ketoconazole. 12th World AIDS Conference, 1998, abstract 12218.
38. Lehmann C, Malin J, Suárez I, et al. Moderne HIV-Therapie. Internist 2019;60:411–19.
39. Marques SC, Ikediobi ON. The clinical application of UGT1A1 pharmacogenetic testing: gene-environment interactions. Hum Genomics 2010;4:238–49.
40. Ménard A, Solas C, Mokthari S, et al. Etravirine-raltegravir, a marked interaction in HIV-1-infected patients: about four cases. AIDS 2009;23:869–71.
41. Michaud V, Ogburn E, Thong N, et al. Induction of CYP2C19 and CYP3A activity following repeated administration of efavirenz in healthy volunteers. Clin Pharmacol Ther 2012;91:475–82.
42. Mukwaya G, MacGregor T, Hoelscher D, et al. Interaction of ritonavir-boosted tipranavir with loperamide does not result in loperamide-associated neurologic side effects in healthy volunteers. Antimicrob Agents Chemother 2005;49:4903–10.
43. Murphy RA, Marconi VC, Gandhi RT, et al. Coadministration of lopinavir/ritonavir and rifampicin in HIV and tuberculosis co-infected adults in South Africa. PLoS One 2012;7:e44793.
44. Nafziger AN, Bertino JS, Bertino JS. Probe Cocktail Studies. In: Piscitelli S., Rodvold K., Pai M. (eds) Drug Interactions in Infectious Diseases. Infectious Disease 2011. Humana Press.
45. Nijland HM, L’homme RF, Rongen GA, et al. High incidence of adverse events in healthy volunteers receiving rifampicin and adjusted doses of lopinavir/ritonavir tablets. AIDS 2008;22:931–5.
46. Petri H. Das Interaktionspotenzial der oralen Antikoagulanzien (OAK). Krankenhauspharmazie 2020;41:267–72.
47. Polk RE, Brophy DF, Israel DS, et al. Pharmacokinetic Interaction between amprenavir and rifabutin or rifampin in healthy males. Antimicrob Agents Chemother 2001;45:502–8.
48. Ramanathan S, Mathias AA, German P, et al. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin Pharmacokinet 2011;50:229–44.
49. Robertson SM, Maldarelli F, Natarajan V, et al. Efavirenz induces CYP2B6-mediated hydroxylation of bupropion in healthy subjects. J Acquir Immune Defic Syndr 2008;49:513–9.
50. Robinson P, Lamson P, Gigliotti M, et al. Pharmacokinetic interaction between nevirapine and rifampin. 12th World AIDS Conference, 1998, abstract 60623.
51. Sahai J, Gallicano K, Pakuts A. Effect of fluconazole on zidovudine pharmacokinetics in patients infected with human immunodeficiency virus. J Infect Dis 1994;169:1103–7.
52. Scholler M, Kraft M, Hoetelmans R, et al. Significant decrease in TMC125 exposures when co-administered with tipranavir boosted with ritonavir in healthy subjects. 13th Conference on Retroviruses and Opportunistic Infections, Denver, February, 2006, abstract 583.
53. Sekar V, Spinosa-Guzman S, Meyvisch P, et al. Cocktail study to investigate the in-vivo drug interaction potential of darunavir coadministered with low-dose ritonavir (DRV/r; RTV) on cytochrome P450 enzymes 2D6, 2C9 and 2C19. Presented at the 9th International Workshop on Clinical Pharmacology of HIV (IWCPHIV), New Orleans, USA, April 7–9, 2008.
54. Solas C, Poizot-Martin I, Drogoul MP, et al. Therapeutic drug monitoring of lopinavir/ritonavir given alone or with a non-nucleoside reverse transcriptase inhibitor. Br J Clin Pharmacol 2004;57:436–40.
55. Song I, Borland J, Chen S, et al. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Br J Clin Pharmacol. 2011;72:103–8.
56. Song I, Borland J, Chen S, et al. Effects of enzyme inducers efavirenz and tipranavir/ritonavir on the pharmacokinetics of the HIV integrase inhibitor dolutegravir. Eur J Clin Pharmacol 2014;70:1173–9.
57. Song I, Borland J, Min S, et al. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob Agents Chemother 2011;55:3517–21.
58. Song I, Weller S, Patel J, et al. Effect of carbamazepine on dolutegravir pharmacokinetics and dosing recommendation. Eur J Clin Pharmacol 2016;72:665–70.
59. Soon GH, Shen P, Yong EL, et al. Pharmacokinetics of darunavir at 900 milligrams and ritonavir at 100 milligrams once daily when coadministered with efavirenz at 600 milligrams once daily in healthy volunteers. Antimicrob Agents Chemother 2010;54:2775–80.
60. Stocker H, Kruse G, Kreckel P, et al. Nevirapine significantly reduces the levels of racemic methadone and (R)-methadone in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 2004;48:4148–53.
61. Tseng A, Hughes CA, Wu J, Seet J, Phillips EJ. Cobicistat versus ritonavir: similar pharmacokinetic enhancers but some important differences. Annals of Pharmacotherapy 2017;51:1008–22.
62. van der Lee MJ, Blenke AA, Rongen GA, et al. Interaction study of the combined use of paroxetine and fosamprenavir-ritonavir in healthy subjects. Antimicrob Agents Chemother 2007;51:4098–104.
63. van der Lee MJ, Dawood L, ter Hofstede HJ, et al. Lopinavir/ritonavir reduces lamotrigine plasma concentrations in healthy subjects. Clin Pharmacol Ther 2006;80:159–68.
64. Wenning LA, Hanley WD, Brainard DM, et al. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother 2009;53:28526.
65. Wenning LA, Petry AS, Kost JT, et al. Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin Pharmacol Ther 2009;85:623–7.
66. Wyen C, Fuhr U, Frank D, et al. Effect of an antiretroviral regimen containing ritonavir boosted lopinavir on intestinal and hepatic CYP3A, CYP2D6 and P-glycoprotein in HIV-infected patients. Clin Pharmacol Ther 2008;84:75–82.
67. www.fachinfo.de (letzter Zugriff am 08.03.2021).
68. Yeh RF, Gaver VE, Patterson KB, Rezk NL, et al. Lopinavir/ritonavir induces the hepatic activity of cytochrome P450 enzymes CYP2C9, CYP2C19, and CYP1A2 but inhibits the hepatic and intestinal activity of CYP3A as measured by a phenotyping drug cocktail in healthy volunteers. J Acquir Immune Defic Syndr 2006;42:52–60.
69. Zhang H, Custodio J, Wei X, et al. P176 Clinical pharmacology of the HIV integrase strand transfer inhibitor bictegravir. Sexually Transmitted Infektions 2017;93:A74.
70. Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet 2005;44:279–304.
71. Zhu L, Brüggemann RJ, Uy J, et al. CYP2C19 genotype-dependent pharmacokinetic drug interaction between voriconazole and ritonavir-boosted atazanavir in healthy subjects. J Clin Pharmacol 2017;57:235–46.
*Nachdruck aus Krankenhauspharmazie 2021;42:196–208.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, Dr. Jochen Weber, Bad Wildungen, und Prof. Dr. Jürgen Rockstroh, Bonn erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2021; 28(03):116-131