Holger Petri, Bad Wildungen*
Zu den Basistherapeutika (Disease modifying antirheumatic drugs, DMARDs) gehören die klassischen, synthetischen Basismedikamente (conventional synthetic DMARDs, csDMARDs), Biologika (biological DMARDs, bDMARDs) und die Januskinase(JAK)-Inhibitoren (targeted synthetic DMARDs, tsDMARDs). Januskinasen sind intrazelluläre Tyrosinkinasen, die sogenannte STAT-Proteine (STAT: signal transducers and activators of transcription) aktivieren. Januskinasen sind an der Bildung von Interleukinen und Interferonen beteiligt. Es sind vier Januskinasen bekannt. Diese werden als JAK-1, JAK-2, JAK-3 und TYK-2 abgekürzt. Die erwünschten und unerwünschten Arzneimittelwirkungen hängen von der spezifischen Blockade der einzelnen Enzyme durch einen JAK-Inhibitor ab [2].
Im Sinne einer Drug-Disease-Wechselwirkung führen Inflammationen über die vermehrte Bildung von Immunmediatoren zu einer reduzierten Expression von Cytochrom-P450(CYP)-Enzymen und hierdurch sinkt die Metabolisierungskapazität [1, 3, 5, 13]. Für Psychopharmaka wie Clozapin und Risperidon sowie dem HMG-CoA-Reduktasehemmer Simvastatin wurden hierdurch klinisch relevante Plasmaspiegelerhöhungen gezeigt [11, 17]. Somit kann sich durch eine suffiziente Senkung der entzündlichen Parameter der Arzneistoffmetabolismus normalisieren. Dies sollte bei der Interpretation von Daten aus pharmakokinetischen Interaktionsstudien bedacht werden, da sie üblicherweise mit gesunden Probanden durchgeführt werden.
Januskinasen werden auch in der Behandlung chronisch entzündlicher Darmerkrankungen (CED) sowie dermatologischer und hämatologischer Erkrankungen eingesetzt.
Tofacitinib
Der metabolische Abbau von Tofacitinib erfolgt primär über das CYP-Isoenzym 3A4 (CYP3A4) und nachgeordnet über CYP2C19. Die Metaboliten tragen weniger als 10 % zur Wirkung bei [4]. Ketoconazol als starker CYP3A4-Inhibitor erhöhte in einer pharmakokinetischen Studie mit gesunden Probanden die AUC(Fläche unter der Konzentrations-Zeit-Kurve) um das Doppelte. Fluconazol als moderater CYP3A4- und starker CYP2C19-Inhibitor konnte die Tofacitinib-Exposition um 79 % erhöhen [10]. Folglich wird empfohlen, in Kombination mit starken CYP3A4-Hemmern (Abb. 1) oder Substanzen, die gleichzeitig moderate CYP3A4- und starke CYP2C19-Hemmer sind, die Dosis zu halbieren [9]. Ebenso soll Tofacitinib bei einer Creatinin-Clearance von unter 30 ml/min in der empfohlenen Dosis halbiert werden [9]. Bei einer empfohlenen Dosis von zweimal täglich 5 mg und Vorliegen einer stark eingeschränkten Nierenfunktion (< 30 ml/min) kann ein starker oder moderater CYP3A4-Hemmer und gleichzeitig starker CYP2C19-Inhibitor nicht mit Tofacitinib kombiniert werden. Wenn aus diesem Grund die Tagesdosis auf 5 mg reduziert werden müsste, kann eine Therapie mit der 11-mg-Retard-Formulierung nicht fortgeführt werden.
Bei Poor-Metabolizern (PM) von CYP2C19-Substraten zeigte sich kein klinisch relevanter Effekt auf die Pharmakokinetik von Tofacitinib. In einer Studie hatten Poor-Metabolizer nur eine 17 % höhere Exposition [4]. Jedoch sollte berücksichtigt werden, dass der Geno- bzw. Phänotyp des Patienten in der Regel nicht bekannt ist. Analog zu Fluconazol können mittelstarke CYP3A4-Inhibitoren wie Diltiazem bei CYP2C19-PM-Patienten durch eine erhöhte Tofacitinib-Exposition vermehrt Nebenwirkungen verursachen, die eine Dosisreduktion notwendig machen. Genauso ist Vorsicht geboten bei Kombination mit potenten CYP2C19-Inhibitoren (Abb. 1).
Ciclosporin ist weder ein starker CYP3A4- noch starker CYP2C19-Inhibitor. Pharmakokinetische Interaktionen sind vielmehr möglich über Hemmung von P-Glykoprotein (P-gp) und OATP1B1 (organic anion-transporting polypeptide 1B1) [15]. Diese Arzneimitteltransporter beeinflussen die Pharmakokinetik von Tofacitinib wahrscheinlich jedoch nur unwesentlich. Dennoch kann der Calcineurin-Inhibitor die AUC von Tofacitinib um 73 % erhöhen [9]. Aufgrund additiver immunsuppressiver Effekte sind Calcineurin-Inhibitoren wie Ciclosporin und Tacrolismus aber unabhängig hiervon zusammen mit JAK-Inhibitoren kontraindiziert [9].
Der starke CYP3A4-Induktor Rifampicin (Abb. 1) senkte die AUC von Tofacitinib in einer Studie um 84 % [4]. Es droht ein Therapieversagen. Starke CYP3A4-Induktoren sollten daher nicht mit Tofacitinib kombiniert werden [9].
Upadacitinib
Upadacitinib ist primär ein Substrat von CYP3A4. Durch den starken CYP3A4-Hemmer Ketoconazol stieg die AUC bei gesunden Probanden um 75 %. Ein Abfallen der Exposition um 60 % erfolgte nach achttägiger Einnahme des starken Induktors Rifampicin [12]. Upadacitinib ist nur in einer Stärke verfügbar [5]. Eine langfristige Einnahme starker CYP3A4-Inhibitoren sollte gemieden und Alternativen in Betracht gezogen werden. Veränderungen der Krankheitsaktivität sind bei Kombination mit CYP3A4-Induktoren zu überwachen [8]. Weitere pharmakokinetische Interaktionen von Upadacitinib sind nicht zu erwarten [12].
Baricitinib und Filgotinib
Baricitinib wird zu über 80 % unverändert größtenteils über die Nieren ausgeschieden, ein kleiner Rest wird über CYP3A4 metabolisiert [16]. Pharmakokinetische Interaktionen sind über renale Arzneimitteltransporter denkbar. So verdoppelte sich die Exposition von Baricitinib bei Kombination mit Probenecid durch Blockade des Organic anion transporters 3 (OAT3) [16]. Die Baricitinib-Dosis ist bei gleichzeitiger Anwendung mit starken OAT3-Hemmern zu halbieren [7]. Wenn die niedrigere Dosis schon aufgrund des höheren Alters oder einer eingeschränkten Nierenfunktion gewählt wurde, kann ein potenter OAT3-Hemmer nicht mit Baricitinib kombiniert werden.
Filgotinib wird umfangreich metabolisiert, wobei der Hauptabbau über die Carboxylesterase 2 (CES2) erfolgt. Einer der aktiven Metaboliten ist zwar zehnfach geringer potent als die Muttersubstanz, wird jedoch etwa zehnmal mehr gebildet. Somit wird die Exposition der aktiven Fraktion unter Berücksichtigung von Aktivität und Molekulargewicht bestimmt [6]. Diese erhöhte sich durch eine P-gp-Hemmung mit Itraconazol um 21 % und sank durch Rifampicin um 33 %. Beides erfordert keine Dosisanpassung [6].
Das Potenzial pharmakokinetischer Interaktionen von Filgotinib ist insgesamt gering [14].
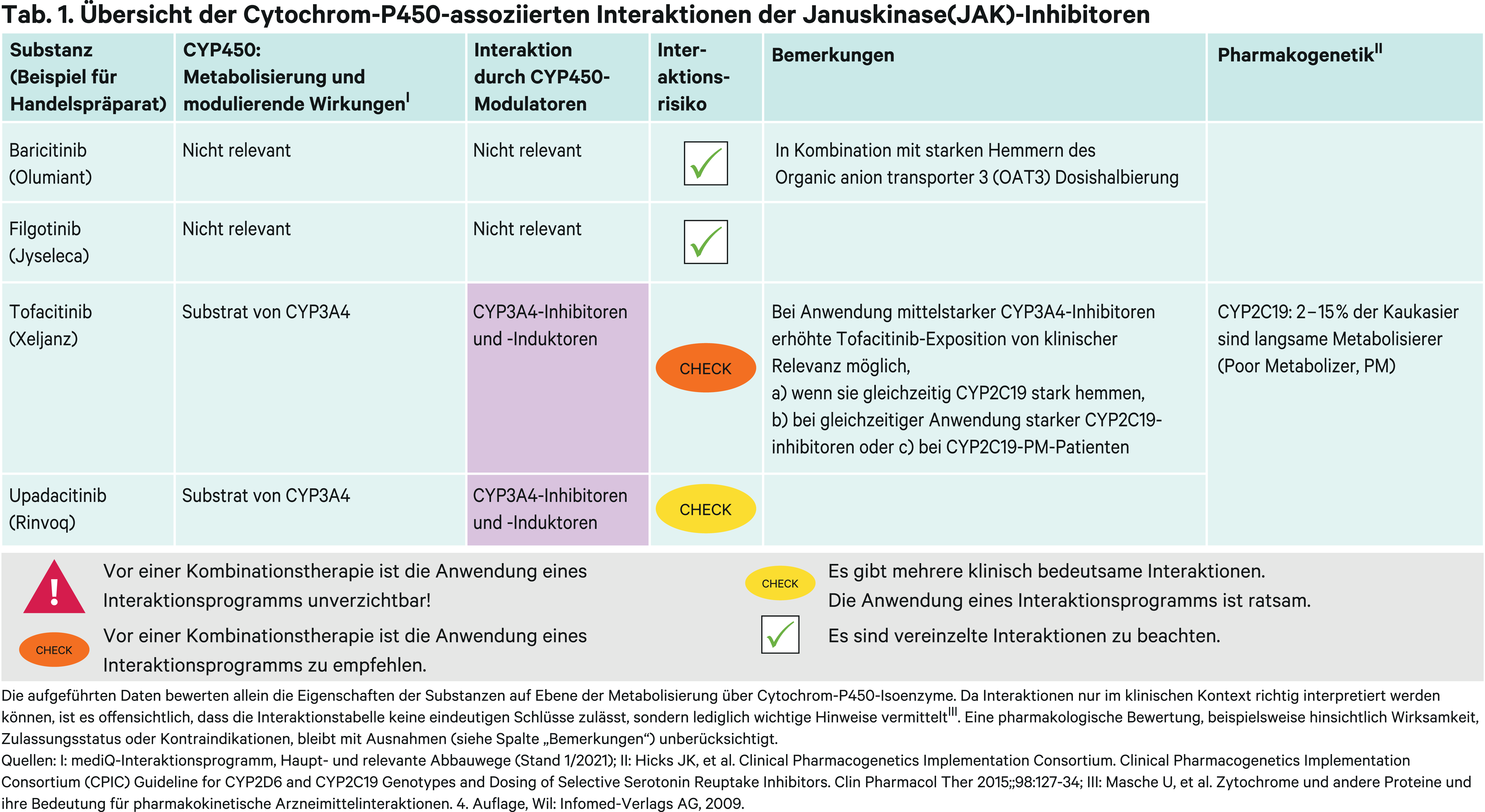
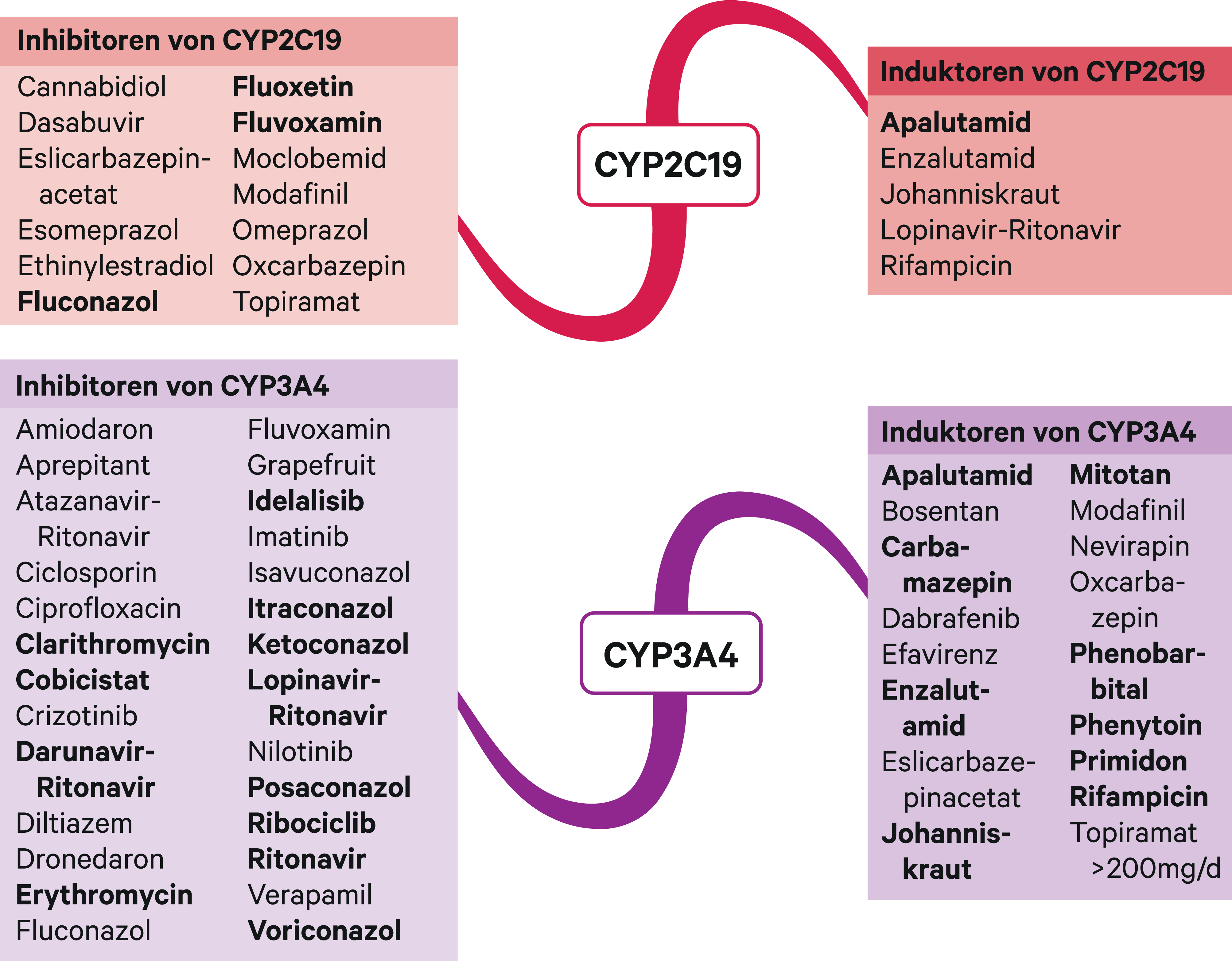
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C19 und 3A4 (Stand 02/2021) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–49
2. Alten R, Mischkewitz M, Stefanski AL, et al. Januskinase-Inhibitoren. Z Rheumatol 2020;79,241–54.
3. Christensen H, Hermann M. Immunological response as a source to variability in drug metabolism and transport. Front Pharmacol 2012;10;3:8.
4. Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab Dispos 2014;42:759–73.
5. Elkahwaji J, Robin MA, Berson A, et al. Decrease in hepatic cytochrome P450 after interleukin-2 immunotherapy. Biochem Pharmacol 1999;57:951–4.
6. Fachinformation Jyseleca®. Stand: September 2020.
7. Fachinformation Olumiant®. Stand: Dezember 2020.
8. Fachinformation Rinvoq®. Stand: November 2020.
9. Fachinformation Xeljanz®. Stand: November 2020.
10. Gupta P, Chow V, Wang R, et al. Evaluation of the effect of fluconazole and ketoconazole on the pharmacokinetics of tofacitinib in healthy adult subjects. Clin Pharmacol Drug Dev 2014;3:72–7.
11. Hefner G, Shams ME, Unterecker S, et al. Inflammation and psychotropic drugs: the relationship between C-reactive protein and antipsychotic drug levels. Psychopharmacology (Berl). 2016;233:1695–705.
12. Mohamed MF, Klünder B, Othman AA. Clinical pharmacokinetics of upadacitinib: Review of data relevant to the rheumatoid arthritis indication. Clin Pharmacokinet 2020;59:531–44.
13. Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther 2009;85:434–8.
14. Namour F, Desrivot J, Van der Aa A, et al. Clinical confirmation that the selective JAK1 inhibitor filgotinib (GLPG0634) has a low liability for drug-drug interactions. Drug Metab Lett 2016;10:38–48.
15. Petri H. Das Interaktionspotenzial der selektiven Immunsuppressiva. Krankenhauspharmazie 2018;39:496–9.
16. Richez C, Truchetet ME, Kostine M, et al. Efficacy of baricitinib in the treatment of rheumatoid arthritis. Expert Opin Pharmacother 2017;18:1399–1407.
17. Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther 2011;89:735–40.
*Nachdruck aus Krankenhauspharmazie 2021;42:112–5.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2021; 28(02):82-85