Holger Petri, Bad Wildungen*
Orale Antikoagulanzien (OAK) werden eingeteilt in Vitamin-K-Antagonisten (VKA) und direkte orale Antikoagulanzien (DOAK), die auch als Nicht-Vitamin-K-abhängige orale Antikoagulanzien (NOAK) bezeichnet werden. Während Vitamin-K-Antagonisten die Bildung von Gerinnungsfaktoren hemmen, wirken die direkten oralen Antikoagulanzien auf die Gerinnungsfaktoren Thrombin (Dabigatran) und Xa (Apixaban, Edoxaban und Rivaroxaban) (Abb. 1) [2].
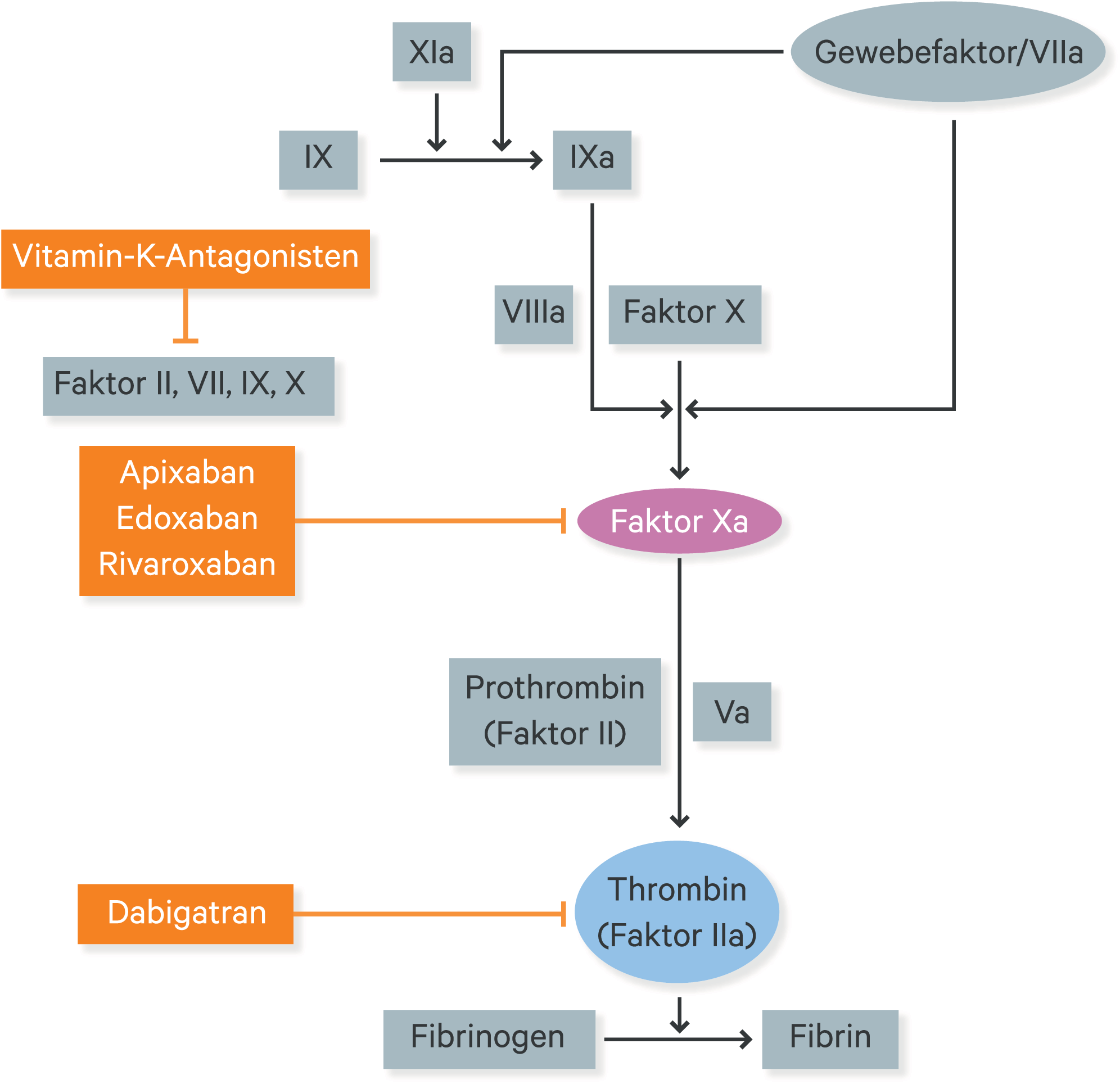
Abb. 1. Angriffspunkte der verschiedenen oralen Antikoagulanzien (mod. nach [2])
Vitamin-K-Antagonisten (VKA)
Phenprocoumon
Phenprocoumon ist der in Deutschland primär verordnete Vitamin-K-Antagonist [43]. Sein Metabolismus erfolgt über die Cytochrom-P450(CYP)-Isoenzyme 2C9 und 3A4 [44]. Daten zu Wechselwirkungen mit Modulatoren dieser beiden Enzyme (Abb. 2) sind von begrenzter Aussagekraft [19, 44].
In einer Untersuchung wurde der Einfluss verschiedener Antiinfektiva auf das Blutungsrisiko unter bestehender Phenprocoumon-Therapie analysiert. Es zeigten sich keine Signale, dass das Blutungsrisiko durch eine pharmakokinetische Interaktion im Sinne eines Plasmaspiegelanstiegs durch Abbauhemmung erhöht sein könnte. Eine Komedikation mit Azolantimykotika, die ein hohes pharmakokinetisches Interaktionspotenzial besitzen [40], bewirkte überhaupt kein erhöhtes Blutungsrisiko [1]. Dies bestätigten Ergebnisse einer älteren Studie, bei der die vermehrten Blutungsraten durch pharmakodynamische Wechselwirkungen begründet wurden [29].
In einer Kasuistik wird über eine 78-jährige Patientin berichtet, deren INR-Wert von 2,2 nach Beginn der Einnahme von Tilidin/Naloxon über mehrere Wochen trotz Anpassung der Phenprocoumon-Dosierung auf einen INR-Wert von 5 stieg und dieser nach wenigen Tagen der Beendigung der Tilidin/Naloxon-Einnahme wieder auf 1,3 fiel, wodurch ein kausaler Zusammenhang als wahrscheinlich angenommen wird. Eine CYP3A4-bedingte Interaktion wird vermutet, obwohl Tilidin und seine Metaboliten keine relevanten CYP3A4-hemmenden Eigenschaften besitzen [5].
Fallberichte über erniedrigte INR-Werte durch Induktoren wie Carbamazepin und Rifampicin weisen auf einen beschleunigten Metabolismus von Phenprocoumon hin, der bis zu 14 Tage nach Absetzen des Induktors anhalten kann [35, 42].
Warfarin
Warfarin besteht als racemisches Gemisch zu gleichen Teilen aus S- und R-Warfarin. S-Warfarin besitzt eine 2- bis 5-fach höhere blutgerinnungshemmende Aktivität als R-Warfarin [15]. S-Warfarin wird primär über das polymorph exprimierte Enzym CYP2C9 abgebaut [44]. Auf Basis des CYP2C9- und des Vitamin-K-Epoxid-Reduktase(VKORC1)-Genotyps gibt es angepasste Dosierungsleitlinien [13].
Auch für Warfarin wurde das Blutungsrisiko durch eine antiinfektiöse Therapie untersucht [7, 26, 31, 41]. Im Gegensatz zu Phenprocoumon deuten die Daten darauf hin, dass pharmakokinetische Wechselwirkungen zu einer vermehrten Blutungsrate beitragen. Der moderate CYP2C9 Hemmer Cotrimoxazol und besonders Fluconazol, ein starker CYP2C9-Inhibitor, führten zu einem erhöhten Blutungsrisiko. So hatten beispielsweise in einer Studie 9,7 % der Patienten unter Fluconazol-Komedikation einen INR-Wert von über 6 [31]. Bei Anwendung des starken CYP2C9-Hemmers Miconazol als Mundgel können die Plasmaspiegel von Warfarin steigen [34]. Nach der gleichzeitigen Anwendung von Miconazol-Mundgel wurde über Blutungen, teils mit tödlichem Ausgang, berichtet [16]. Wie bei Phenprocoumon sind auch für Warfarin durch zahlreiche Untersuchungen und Fallberichte INR-Absenkungen durch Induktoren in den subtherapeutischen Bereich belegt [11, 23, 32].
Direkte orale Antikoagulanzien (DOAK)
Apixaban
Etwa 15 % einer oralen Apixaban-Dosis wird über CYP3A4 abgebaut. Es entstehen inaktive Metaboliten. Apixaban ist zudem ein Substrat der Effluxtransporter P-Glykoprotein (P-gp) und Breast cancer resistance protein (BRCP) [10, 24]. Ketoconazol ist ein starker CYP3A4- und P-gp-Hemmer. In einer klinischen Studie erhöhte das Azolantimykotikum die Exposition des Antikoagulans um durchschnittlich das 2-Fache. Ketoconazol und andere starke CYP3A4- und P-gp-Hemmer wie die Azolantimykotika Itraconazol, Posaconazol und Voriconazol sowie HIV-Proteasehemmer wie Ritonavir sollen nicht zusammen mit Apixaban angewendet werden [17].
Clarithromycin ist ein starker CYP3A4- und nur schwacher P-gp-Hemmer. Das Antibiotikum erhöhte die mittlere AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Apixaban um das 1,6-Fache. Der moderate CYP3A4- und schwache P-gp-Hemmer Diltiazem führte zu einer Steigerung der AUC um das 1,4-Fache. Naproxen als schwacher P-gp-Hemmer erhöhte die Exposition um das 1,5-Fache [24]. Diese Plasmaspiegelerhöhungen erfordern keine Dosisanpassung [17].
Rifampicin als starker CYP3A4- und P-gp-Induktor reduzierte die Apixaban-Exposition mit 54 % um mehr als die Hälfte [24]. Apixaban sollte daher in der prophylaktischen Indikation mit Rifampicin und anderen kombinierten starken CYP- und P-gp-Induktoren wie Carbamazepin, Phenytoin und Phenobarbital nur mit Vorsicht eingesetzt und in der Behandlung von tiefen Venenthrombosen (TVT) und Lungenembolien (LE) vermieden werden [17].
Dabigatranetexilat
Dabigatranetexilat wird CYP-unabhängig verstoffwechselt. Der größte Anteil wird als Dabigatran renal eliminiert [24]. Dabigatranetexilat ist ein Prodrug und im Gegensatz zu Dabigatran ein Substrat von P-gp. Die intestinale Aufnahme kann durch P-gp-Hemmung oder -Induktion beeinflusst werden. Ketoconazol und Dronedaron erhöhten die AUC von Dabigatran um das 2,5- bzw. 2,1-Fache [24]. Die gleichzeitige Behandlung mit Ketoconazol und Dronedaron ist kontraindiziert. Das gilt auch für die Komedikation mit anderen potenten P-gp-Inhibitoren, zum Beispiel Ciclosporin, Itraconazol und der Fixkombination aus Glecaprevir/Pibrentasvir [20]. Schwächere P-gp-Inhibitoren wie Amiodaron, Chinidin oder Verapamil erhöhten die Dabigatran-Exposition in einen Bereich von 53 bis 70 % und können unter Dosisreduktion verordnet werden [20, 24].
Die gemeinsame Verordnung von Dabigatranetexilat mit Simvastatin und Lovastatin war in einer bevölkerungsbasierten Fall-Kontroll-Studie mit einer erhöhten Rate an schweren Blutungen assoziiert. Der Grund könnte eine Wechselwirkung sein. Die beiden Statine werden als Lacton-Prodrugs eingenommen, die in vitro P-gp hemmen [4].
Es sollte bedacht werden, dass weitere P-gp-Hemmer mit Dabigatranetexilat kontraindiziert sein können, obwohl diese nicht in den Fachinformationen des DOAK aufgeführt sind. Beispielsweise ist Dabigatranetexilat unter Einnahme der Hepatitis-C-Fixkombination Sofosbuvir/Velpatasvir/Voxilaprevir kontraindiziert. In einer klinischen Studie erhöhte die Dreifachkombination die Dabigatran-Exposition um das 2,87-Fache [22]. Das Beispiel zeigt zum einen, dass auch die Fachinformationen der Kombinationspartner bei der Prüfung auf Wechselwirkungen des DOAK zu Rate gezogen werden sollten. Zum anderen können die P-gp-hemmenden Effekte summarisch mit der Anzahl der gleichzeitig verordneten Arzneimittel und Naturstoffe (z. B. Grapefruit, Roter Reis) steigen.
Nach Herstellerangaben sollten Proteasehemmer wie Ritonavir aufgrund fehlender Daten nicht gleichzeitig mit Dabigatranetexilat angewendet werden. In einer Studie mit gesunden Probanden wurde der Einfluss von Ritonavir und Cobicistat auf die Exposition des DOAK geprüft. Bei simultaner Einnahme führte Ritonavir zu keiner signifikanten Änderung des AUC-Werts, während bei zeitversetzter Einnahme von zwei Stunden nach DOAK-Einnahme die Exposition um 27 % sank. Cobicistat erhöhte in beiden Konstellationen den AUC-Wert um mehr als Doppelte [30].
Rifampicin als P-gp-Induktor verminderte über sieben Tage eingenommen die Dabigatran-Exposition um zwei Drittel [24]. Die gleichzeitige Anwendung von P-gp-Induktoren mit Dabigatranetexilat sollte vermieden werden [20]. In einem Fallbericht konnten bei Komedikation mit dem Induktor Phenytoin keine Dabigatran-Spiegel gemessen werden [25].
Edoxaban
Edoxaban wird nur zu einem geringen Anteil verstoffwechselt. Weniger als 10 % einer oralen Dosis werden über die Carboxylesterase (CES) 1 und CYP3A4 metabolisiert [24, 37]. Es entstehen die aktiven Metaboliten M4 und M6, die aufgrund ihrer geringen Plasmaspiegel nicht wesentlich zur antikoagulatorischen Wirksamkeit beitragen. Als Substrat von P-gp sind Wechselwirkungen mit P-gp-Modulatoren zu beachten. Eine Dosisreduktion muss bei gleichzeitiger Therapie mit den potenten P-gp-Hemmern Ciclosporin, Dronedaron, Erythromycin und Ketoconazol vorgenommen werden, da sich die Edoxaban-Exposition um 73 bis 87 % erhöhen kann [18]. Weniger starke P-gp-Inhibitoren steigerten die mittleren AUC Werte in geringem Ausmaß (Chinidin: 63 %, Verapamil: 53 %, Amiodaron: 40 %), was keine Dosisanpassung erfordert [18].
Verglichen mit anderen DOAK fiel die Abnahme der mittleren AUC durch den Induktor Rifampicin mit 34 % in einer klinischen Studie deutlich geringer aus. Die äquipotenten Metaboliten wurden vermehrt gebildet und kompensierten den Plasmaspiegelrückgang der Muttersubstanz [33]. Der Summenspiegel der aktiven Fraktion wäre mit unter 10 % kaum geringer als ohne Rifampicin-Komedikation. Die Induktion von CYP3A4 und P-gp erfolgt über dieselben Mechanismen, sodass CYP3A4-Induktoren immer auch P-gp-induktive Effekte besitzen, auch wenn in schwächerer Ausprägung [14]. In einem Fallbericht erlitt ein Patient unter Apixaban-Einnahme eine transitorische ischämische Attacke. Der Patient nahm wegen einer Trigeminusneuralgie Carbamazepin ein. Die Spitzenplasmaspiegel von Apixaban lagen deutlich unter den zu erwartenden Mindestwerten. Nach Umstellung auf Edoxaban und unter Kontrolle der Plasmaspiegel zeigten sich in 18 Monaten Nachbeobachtung keine Auffälligkeiten [12]. Somit stellt Edoxaban aus der Stoffgruppe der zurzeit verfügbaren DOAK im Hinblick auf Wechselwirkungen mit starken CYP3A4-Induktoren und damit auch potenten P-gp-Induktoren die vorteilhafteste Substanz dar.
Rivaroxaban
Rivaroxaban wird zu etwa 18 % über CYP3A4 metabolisiert [24, 36]. Es entstehen keine pharmakologisch aktiven Metaboliten. Das Antikoagulans ist Substrat von P-gp und BCRP. In pharmakokinetischen Studien war die Rivaroxaban-Exposition durch Inhibitoren nur dann in einem relevanten Ausmaß erhöht, wenn diese stark hemmende Wirkungen sowohl auf CYP3A4 als auch auf P-gp hatten. So bewirkte Ketoconazol einen Anstieg der mittleren AUC-Werte um das 2,6-Fache, Ritonavir um das 2,5-Fache. Dies führte zu einer verstärkten antikoagulatorischen Wirkung. Wie bei Apixaban sollten dieselben Azolantimykotika und HIV-Proteasehemmer gemieden werden [21].
Inhibitoren wie Ciclosporin, Clarithromycin, Erythromycin und Fluconazol, die nur einen Eliminationsweg von Rivaroxaban blockieren, erhöhten die AUC bei normaler Nierenfunktion nur um 30 bis 50 %, was beim Großteil der Patienten als nicht klinisch relevant angesehen wird [21, 24]. Jedoch erhöhte Erythromycin die Rivaroxaban-Exposition bei Patienten mit leichter und mittelschwerer Nierenfunktionsstörung um das 1,8- bzw. 2-Fache [24]. Gemäß deutscher Herstellerangaben soll bei Patienten mit einer Creatinin-Clearance von 30 bis 80 ml/min Rivaroxaban nur mit Vorsicht eingesetzt werden, wenn sie gleichzeitig andere Arzneimittel erhalten, die zu erhöhten Rivaroxaban-Plasmaspiegeln führen können [21].
In einer klinischen Studie wurde untersucht, wie sich die Kombination eines starken P-gp-Inhibitors mit einem moderaten CYP3A4-Hemmer auf die Exposition von Rivaroxaban auswirkt. Ciclosporin als starker P-gp-Inhibitor und der moderate CYP3A4-Inhibitor Fluconazol ohne P-gp-hemmende Eigenschaften erhöhten gemeinsam die mittlere AUC von Rivaroxaban bei gesunden Probanden um 86 % [9].
Rifampicin reduzierte die Rivaroxaban-Exposition um die Hälfte [24]. Von der gleichzeitigen Anwendung von Rivaroxaban und Rifampicin sowie anderen CYP3A4-Induktoren wird abgeraten, es sei denn, die Patienten werden engmaschig auf Zeichen und Symptome einer Thrombose überwacht [21].
In zwei Fallberichten wurde der beschleunigte Metabolismus von Rivaroxaban durch Phenytoin und Rifampicin mit den daraus folgenden subtherapeutischen Plasmaspiegeln dokumentiert. Im Rifampicin-Fallbericht machen die Autoren die Wechselwirkung für die Entwicklung einer tödlich verlaufenden Lungenembolie mitverantwortlich [3, 8].
Fazit
Orale Antikoagulanzien bieten ein breites Spektrum an möglichen Interaktionsrisiken. Die Vitamin-K-Antagonisten sind Arzneimittel mit enger therapeutischer Breite. Sie haben jedoch den Vorteil, dass durch Messen der INR-Werte die antikoagulatorische Therapie engmaschig überwacht werden kann. Im Gegensatz dazu fehlen diese einfachen Gerinnungstests bei den direkten oralen Antikoagulanzien [6], Patienten mit problematischer Begleitmedikation waren in den zulassungsrelevanten Studien unterrepräsentiert. Im klinischen Alltag stellt sich die Frage nach einer validen Therapiekontrolle, wenn zum Risikofaktor Polymedikation noch eine eingeschränkte Nierenfunktion, ein geringes Körpergewicht oder ein hohes Alter hinzukommen [28]. Dosisreduktionen können nach Herstellerempfehlungen bei Apixbaban und Edoxaban nur um die Hälfte vorgenommen werden [2]. Bei Dabigatranetexilat zeigte sich in der RE-LY-Studie, dass die 110-mg- im Vergleich zur 150-mg-Stärke das Risiko für starke Blutungen verminderte, verbunden mit einem erhöhten Schlaganfallrisiko [28]. Die reduzierte Dosis von 30 mg Edoxaban führte bei Patienten unter Kombination mit dem moderaten P-gp-Inhibitor Amiodaron in der ENGAGE-AF-TIMI-48-Studie nicht zu einem schlechteren klinischen Outcome im Vergleich zu der vom Hersteller empfohlenen 60-mg-Dosis [24]. Es gibt jedoch noch keine pharmakokinetischen Zielparameter, nach denen eine Dosisanpassung eines DOAK vorgenommen werden kann [24]. Daten aus einer retrospektiven Studie zeigen, dass auch kombinierte P-gp- und moderate CYP3A4-Hemmer das Blutungsrisiko von Apixaban und Rivaroxaban erhöhen können [27].
Bei gleichzeitiger Anwendung von Induktoren können den Herstellerangaben keine praktischen Empfehlungen zur Dosisanpassung entnommen werden. In einer retrospektiven Studie nahmen von 1596 mit einem DOAK behandelten Patienten 22 (1,4%) gleichzeitig einen Enzyminduktor ein. Von elf Patienten (10 mit Apixaban, 1 mit Rivaroxaban) wurden die Plasmaspiegel bestimmt. Bei sechs Personen lagen die DOAK-Plasmaspiegel unterhalb der aus Studien erwarteten 5. Perzentile [38]. Hierdurch ist zu befürchten, dass subtherapeutische Plasmaspiegel bis zum Eintritt ischämischer Komplikationen unbemerkt bleiben [39]. Es bleibt in solchen Situationen entweder der Wechsel des Induktors auf weniger interaktionsträchtige Arzneimittel oder die Verwendung von Vitamin-K-Antagonisten unter Kontrolle der INR-Werte [25]. Edoxaban stellt aus der Gruppe der DOAK eine Alternative dar, dessen positives-Nutzen-Risiko-Verhältnis aber noch durch weitere Studien mit Induktoren untermauert werden sollte.
Durch Bestimmung der Anti-Xa-Aktivität bzw. einer verdünnten Thrombinzeit oder Ecarinzeit (für Dabigatran) können die Plasmaspiegel der einzelnen DOAK quantitativ gemessen werden. Im Einzelfall ist besonders bei niedrigen Spiegeln eine Bestimmung über LC-MS/MS im Speziallabor notwendig [45]. Für eine Dosierung im Sinne einer personalisierten Medizin vergleichbar der INR-Wertbestimmung unter einer VKA-Therapie sind weitere Studien notwendig [45].
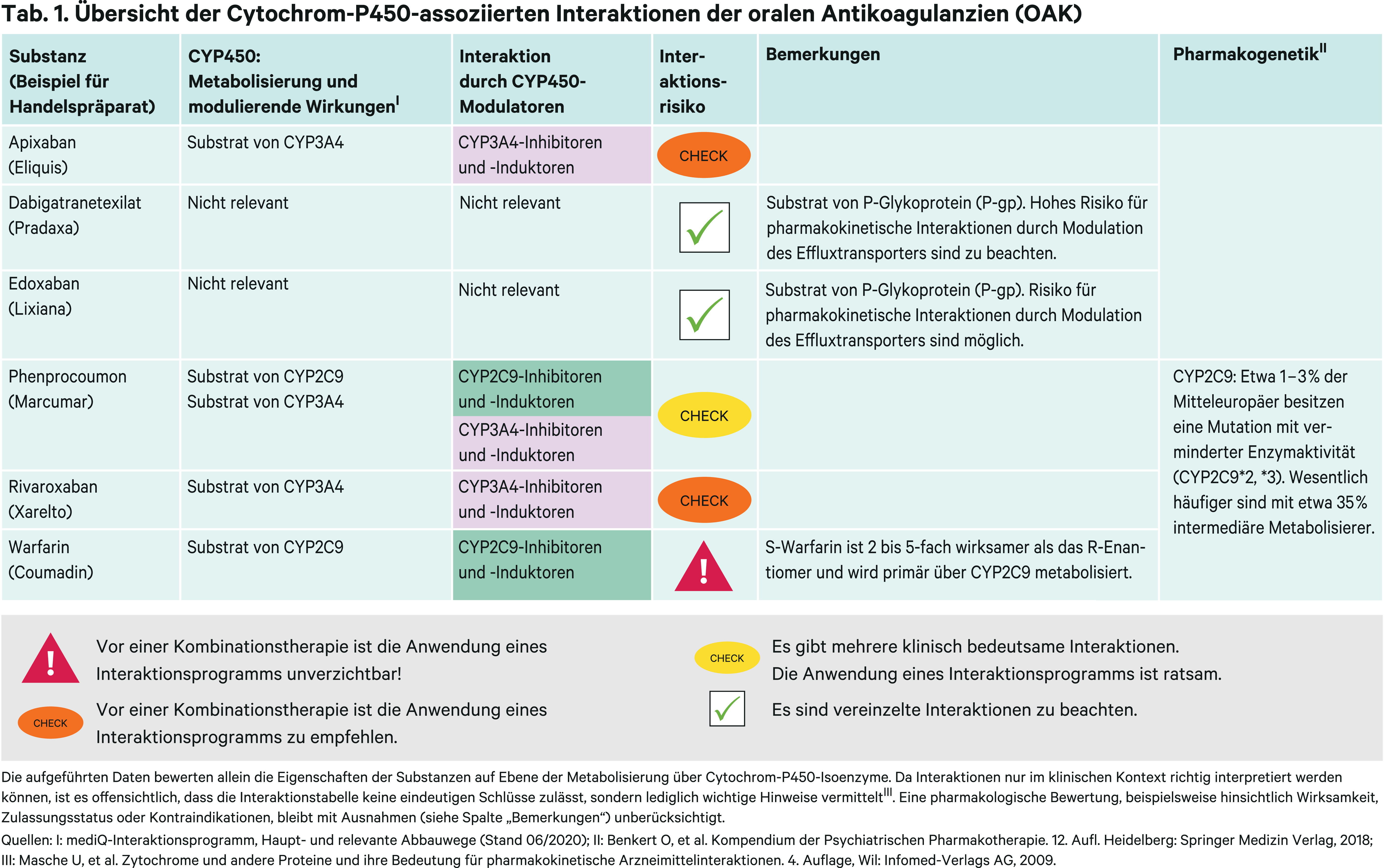
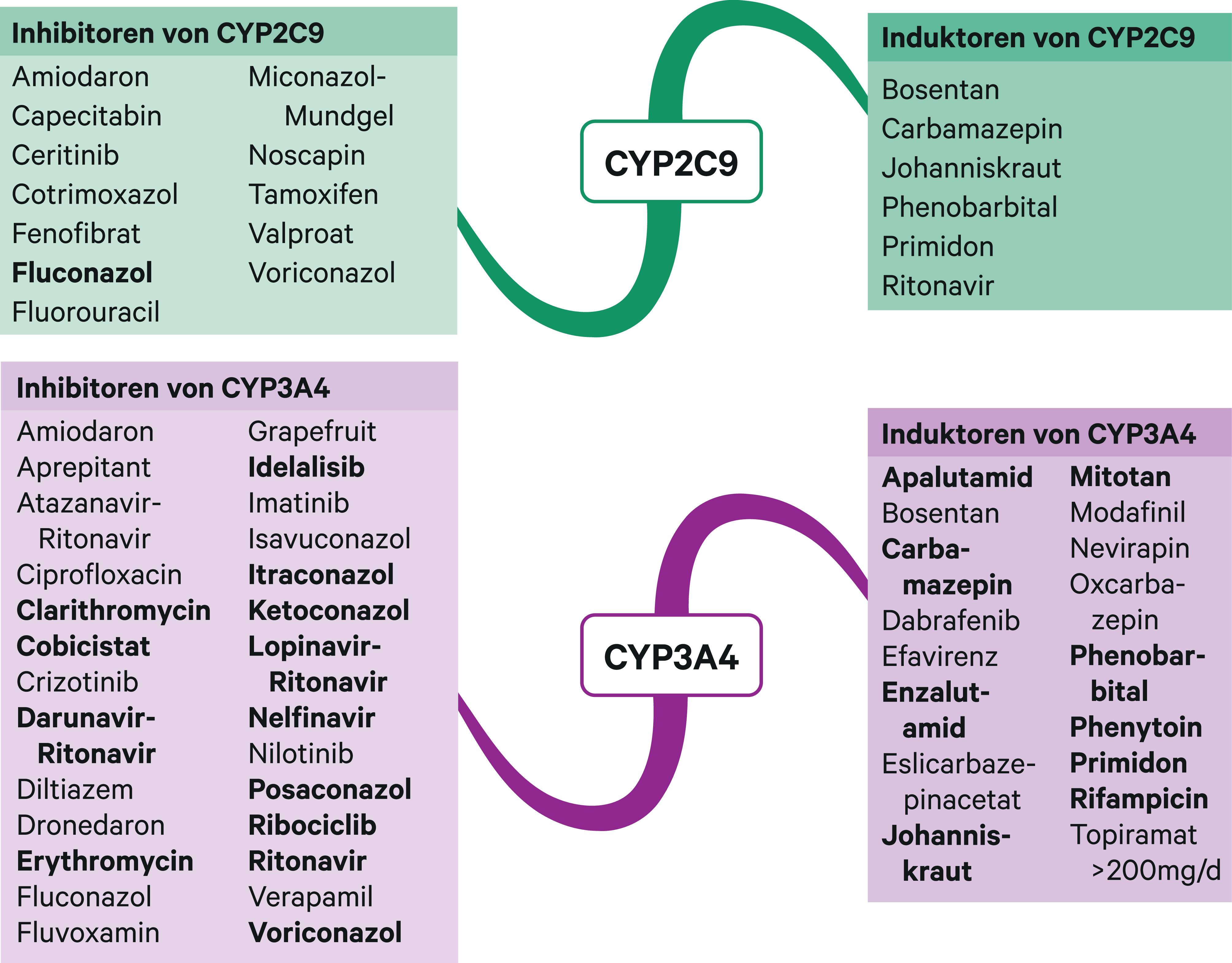
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C9 und 3A4 (Stand 06/2020) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Abbas S, Ihle P, Harder S, et al. Risk of bleeding and antibiotic use in patients receiving continuous phenprocoumon therapy. A case-control study nested in a large insurance- and population-based German hohort. Thromb Haemost 2014;111:912–22.
2. Alban S. Antikoagulation mit DOAK-ein Update. Med Monatsschr Pharm 2019;8:284–92.
3. Altena R, van Roon E, Folkeringa R, et al. Clinical challenges related to novel oral anticoagulants: drug-drug interactions and monitoring. Haematologica 2014;99:e26‐e27.
4. Antoniou T, Macdonald EM, Yao Z, et al. Association between statin use and ischemic stroke or major hemorrhage in patients taking dabigatran for atrial fibrillation. CMAJ 2017;189:E4–E10.
5. Arzneimittelkommission der deutschen Ärzteschaft. Interaktion zwischen Phenprocoumon und Tilidin. Drug Safety Mail 2017–16.
6. Arzneimittelkommission der deutschen Ärzteschaft. Orale Antikoagulation bei nicht valvulärem Vorhofflimmern. 3., überarbeitete Auflage, November 2019.
7. Baillargeon J, Holmes HM, Lin YL, et al. Concurrent use of warfarin and antibiotics and the risk of bleeding in older adults. Am J Med 2012;125:183–9.
8. Becerra AF, Amuchastegui T, Tabares AH. Decreased rivaroxaban levels in a patient with cerebral vein thrombosis receiving phenytoin. Case Rep Hematol 2017;2017: 4760612
9. Brings A, Lehmann ML, Foerster KI, et al. Perpetrator effects of ciclosporin (P-glycoprotein inhibitor) and its combination with fluconazole (CYP3A inhibitor) on the pharmacokinetics of rivaroxaban in healthy volunteers. Br J Clin Pharmacol 2019;85:1528–37.
10. Byon W, Garonzik S, Boyd RA, et al. Apixaban: A clinical pharmacokinetic and pharmacodynamic review. Clin Pharmacokinet 2019;58:1265–79.
11. Clark NP, Hoang K, Delate T, et al. Warfarin interaction with hepatic cytochrome P-450-enzyme-inducing anticonvulsants. Clin Appl Thromb Hemost 2018;24:172–8.
12. Di Gennaro L, Lancellotti S, De Cristofaro R, et al. Carbamazepine interaction with direct oral anticoagulants: help from the laboratory for the personalized management of oral anticoagulant therapy. J Thromb Thrombolysis 2019;48: 528–31.
13. Dingermann T, Zündorf I. Stratifizierte Pharmakotherapie. Eschborn: Govi Verlag, 2017.
14. Elmeliegy M, Vourvahis M, Guo C, et al. Effect of p-glycoprotein (P-gp) inducers on exposure of -gp Substrates: Review of clinical drug-drug interaction studies [published online ahead of print, 2020 Feb 13]. Clin Pharmacokinet 2020;10.1007/s40262-020-00867-1.
15. Fachinformation Coumadin®. Stand: Juni 2017.
16. Fachinformation Daktar® 2 % Mundgel. Stand: März 2017.
17. Fachinformation Eliquis®. Stand: Februar 2020.
18. Fachinformation Lixiana®. Stand: Februar 2020.
19. Fachinformation Marcumar®. Stand: Juni 2018.
20. Fachinformation Pradaxa®. Stand: Dezember 2019.
21. Fachinformation Xarelto®. Stand: Oktober 2019.
22. Fachinformation Vosevi® 400 mg/100 mg/100 mg. Stand: November 2019.
23. Fahmi AM, Abdelsamad O, Elewa H. Rifampin-warfarin interaction in a mitral valve replacement patient receiving rifampin for infective endocarditis: a case report. Springerplus 2016;5:8.
24. Foerster KI, Hermann S, Mikus G, et al. Drug-drug interactions with direct oral anticoagulants. Clin Pharmacokinet 2020 /doi.org/10.1007/s40262-020-00879-x.
25. Galgani A, Palleria C, Iannone LF, et al. Pharmacokinetic interactions of clinical interest between direct oral anticoagulants and antiepileptic drugs. Front Neurol 2018;9:1067.
26. Glasheen JJ, Fugit RV, Prochazka AV. The risk of overanticoagulation with antibiotic use in outpatients on stable warfarin regimens. J Gen Intern Med 2005;20:653–6.
27. Hanigan S, Das J, Pogue K, et al. The real world use of combined P-glycoprotein and moderate CYP3A4 inhibitors with rivaroxaban or apixaban increases bleeding. J Thromb Thrombolysis 2020;49:636–43.
28. Herink MC, Zhuo YF, Williams CD, DeLoughery TG. Clinical management of pharmacokinetic drug interactions with direct oral anticoagulants (DOACs). Drugs 2019;79:1625–34.
29. Jobski K, Behr S, Garbe E. Drug interactions with phenprocoumon and the risk of serious haemorrhage: a nested case-control study in a large population-based German database. Eur J Clin Pharmacol 2011;67(9):941–51.
30. Kumar P, Gordon LA, Brooks KM, et al. Differential influence of the antiretroviral pharmacokinetic enhancers ritonavir and cobicistat on intestinal p-glycoprotein transport and the pharmacokinetic/pharmacodynamic disposition of dabigatran. Antimicrob Agents Chemother 2017;61:e01201–17.
31. Lane MA, Zeringue A, McDonald JR. Serious bleeding events due to warfarin and antibiotic co-prescription in a cohort of veterans. Am J Med 2014;127: 657–63.
32. Mannheimer B, Andersson ML, Järnbert-Pettersson H, Lindh JD, et al. The effect of carbamazepine on warfarin anticoagulation: a register-based nationwide cohort study involving the Swedish population. J Thromb Haemost 2016;14:765–71.
33. Mendell J, Chen S, He L, Desai M, Parasramupria DA. The effect of rifampin on the pharmacokinetics of edoxaban in healthy adults. Clin Drug Investig 2015;35:447–53.
34. Miki A, Ohtani H, Sawada Y. Warfarin and miconazole oral gel interactions: analysis and therapy recommendations based on clinical data and a pharmacokinetic model. J Clin Pharm Ther 2011;36:642–50.
35. Mizera L, Geisler, T, Mörike K, et al. Problems in anticoagulation of a patient with antibiotic treatment for endocarditis: Interaction of rifampicin and vitamin K antagonists. BMJ Case Reports 2018 doi: 10.1136/bcr-2016-215155.
36. Mueck W, Stampfuss J, Kubitza D, et al. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet 2014;53:1–16.
37. Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non-vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet 2016;55:641–55.
38. Perlman A, Hochberg-Klein S, et al. Management strategies of the interaction between direct oral anticoagulant and drug-metabolizing enzyme inducers. J Thromb Thrombolysis 2019;47:590–5.
39. Perlman A, Wanounou M, Goldstein R, et al. Ischemic and thrombotic events associated with concomitant Xa-inhibiting direct oral anticoagulants and antiepileptic drugs: Analysis of the FDA adverse event reporting system (FAERS). CNS Drugs 2019;33:1223–8.
40. Petri H. Das Interaktionspotenzial der Azol-Antimykotika. Krankenhauspharmazie 2016;37:506–10.
41. Schelleman H, Bilker WB, Brensinger CM, et al. Warfarin with fluoroquinolones, sulfonamides, or azole antifungals: interactions and the risk of hospitalization for gastrointestinal bleeding. Clin Pharmacol Ther 2008;84:581–8.
42. Schlienger R, Kurmann M, Drewe J, et al. Inhibition of Phenprocoumon Anticoagulation by Carbamazepine. Eur Neuropsychopharmacol 2000;10:219–21.
43. Schwabe U, Paffrath D, Ludwig WD, et al. Arzneiverordnungs-Report 2019. Berlin, Heidelberg: Springer, 2019.
44. Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet 2005;44:1227–46.
45. Wieland E, Shipkova M. Pharmacokinetic and pharmacodynamic drug monitoring of direct-acting oral anticoagulants: Where do we stand? Ther Drug Monit 2019;41:180–91.
*Nachdruck aus Krankenhauspharmazie 2020;41:267–72.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2020; 27(04):210-215