Holger Petri, Bad Wildungen*
Zur Behandlung der chronischen Hepatitis C werden in Deutschland koformulierte und fixdosierte Regime angewandt, die verschiedene Angriffspunkte über sogenannte nichtstrukturelle Proteine (NS) auf die Virusreplikation haben [6]. RNA-abhängige RNA-Polymeraseinhibitoren werden intrazellulär durch zelluläre Kinasen phosphoryliert, binden als Triphosphat im aktiven Zentrum der HCV-spezifischen NS5B-Polymerase und bewirken einen Kettenabbruch der wachsenden viralen RNA. Die generischen Namen der HCV-Polymeraseinhibitoren enden auf „-buvir“.
Proteaseinhibitoren sind gegen die HCV-NS3/4A-Serinprotease (Spaltung des HCV-Polyproteins) gerichtet, ihre generischen Namen enden auf „-previr“.
Das HCV-NS5A-Protein ist an der HCV-Replikation und an der Modulation von zellulären Funktionen beteiligt. Verschiedene NS5A-Inhibitoren wurden entwickelt, ihre generischen Namen enden auf „-asvir“. In Abbildung 1 sind der Replikationszyklus des Hepatitis-C-Virus und die Angriffspunkte der antiviralen Arzneimittel dargestellt.

Abb. 1. Replikationszyklus des Hepatitis-C-Virus und Angriffspunkte der antiviralen Arzneimittel
Die Erfolgsquoten für eine dauerhafte Viruseradikation („sustained virologic response“ [SVR]) liegen seit Einführung der direkt antiviral wirksamen Arzneistoffe („direct antiviral agents“ [DAA]) bei über 95 % und sind als Interferon-freies Therapiekonzept sehr gut verträglich [11].
Als einziger zurzeit verfügbarer nukleotidischer NS5B-Polymeraseinhibitor ist Sofosbuvir (SOF) in drei Kombinationsarzneimitteln enthalten. Sofosbuvir wird CYP-unabhängig metabolisiert. Seine pharmakokinetischen Wechselwirkungsrisiken beruhen darauf, dass durch Induktion des intestinalen Arzneimitteltransporters P-Glykoprotein (P-gp) seine systemische Bioverfügbarkeit deutlich reduziert wird. So sank durch Rifampicin als P-gp-Induktor die AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Sofosbuvir um 72 %. Als Substrat des organic anion-transporting polypeptide (OATP) 1B1 führt die Hemmung durch Ciclosporin, einem starken OATP1B1-Inhibitor, zu einer 4,5-fachen Erhöhung der AUC. Dies ist aufgrund der guten Verträglichkeit jedoch ohne klinische Relevanz [8].
Sofosbuvir(SOF)-haltige Fixkombinationen
Ledipasvir
Mit dem NS5A-Inhibitor Ledipasvir (LPV) wurde die erste Sofosbuvir-haltige Fixkombination zugelassen. Wie Sofosbuvir wird die systemische Aufnahme von Ledispavir durch intestinale P-gp-Induktion beeinträchtigt. Rifampicin verringerte die Exposition von Ledipasvir um die Hälfte. Starke P-gp-Induktoren sind unter SOF/LPV-Therapie kontraindiziert und mittelstarke werden nicht empfohlen. Als Inhibitor des Effluxtransporters BCRP (Breast cancer resistance protein) erhöhte Ledipasvir die systemische Verfügbarkeit des HMG-CoA-Reduktasehemmers Rosuvastatin, einem BCRP-Testsubstrat, um das 8-Fache. Rosuvastatin ist daher unter einer Ledipasvir-Therapie kontraindiziert. CYP-Wechselwirkungen sind nicht zu erwarten [2, 7].
Velpatasvir
Mit dem NS5A-Inhibitor Velpatasvir (VEL) kann Sofosbuvir bei allen bekannten HCV-Genotypen eingesetzt werden, was als pangenotypische Kombinationstherapie bezeichnet wird. Velpatasvir wird über CYP2B6, CYP2C8 und CYP3A4 metabolisiert. Rifampicin als Induktor dieser Enzyme führte zu einer Abnahme des AUC-Werts von Velpatasvir um 82 %. Starke CYP-Induktoren sind unter einer SOF/VEL-Therapie kontraindiziert. Das HIV-Mittel Efavirenz ist ein mittelstarker CYP3A4-Induktor. Unter Kombination mit Efavirenz wurde eine Reduktion der Velpatasvir-Exposition auf 47 % beobachtet. Mittelstarke CYP-Induktoren sollten folglich auch vermieden werden. Velpatasvir erhöhte die AUC von Rosuvastatin um das 2,7-Fache. Rosuvastatin kann in einer Dosis bis 10 mg am Tag mit Sofosbuvir/Velpatasvir komediziert werden. Starke CYP3A4-Hemmer wie Cobicistat, Ketoconazol und Ritonavir (Abb. 2) führen zu maximalen Steigerungen der Exposition von Velpatasvir um das 2,4-Fache. Dies erfordert keine Dosisanpassung [1].
Voxilaprevir
Der NS3/4-Protease-Inhibitor Voxilaprevir wird mit Sofosbuvir und Velpatasvir als Dreifachkombination vor allem nach Versagen einer vorhergehenden DAA-Therapie eingesetzt. Die SVR-Rate liegt auch bei diesen Patienten bei über 95 % [10].
Voxilaprevir ist Substrat von CYP3A4, P-gp und OATP1B1. Während der starke CYP3A4-Inhibitor Voriconazol die AUC von Voxilaprevir um 84 % erhöhte, führte die kombinierte Hemmung von CYP3A4, P-gp und OATP1B1 durch Atazanavir/Ritonavir zu einer 4,3-fachen Steigerung der AUC. Die Bedeutung von OATP1B1 für den Stoffwechsel von Velpatasvir zeigt sich an der 7,9-fachen AUC-Steigerung durch eine Einzeldosis Rifampicin, das auch ein kompetitiver Hemmstoff von OATP ist. Bei wiederholter Einnahme kommen die induktiven Effekte des Antibiotikums zu tragen und die Spiegel sinken um 73 % vom Ausgangswert. Ciclosporin führte zu einer 9,4-fachen AUC-Erhöhung von Voxilaprevir. Die Komedikation mit starken OATP1B1-Inhibitoren wird nicht empfohlen, da die Sicherheit bei erhöhten Voxilaprevir-Plasmaspiegeln nicht erwiesen ist. Voxilaprevir hat BCRP- und P-gp-hemmendes Potenzial und führt bei Komedikation mit Rosuvastatin (BCRP-Substrat) und Dabigatranetexilat (P-gp-Substrat) zu einer Steigerung der Plasmakonzentration um das 7,4- bzw. 2,6-Fache. Deshalb ist die Komedikation von Rosuvastatin und Dabigatranetexilat mit einer SOF/VEL/VOX-Therapie kontraindiziert [4].
Elbasvir/Grazoprevir
Der NS5A-Inhibitor Elbasvir (ELB) und der NS3/4-Protease-Inhibitor Grazoprevir (GRA) sind Substrate von CYP3A4 [6]. Efavirenz senkte die systemische Verfügbarkeit von Elbasvir um 54 % und von Grazoprevir um 83 %. Folglich sind nicht nur starke, sondern auch mittelstarke CYP3A4-Induktoren (Abb. 2) unter einer Elbasvir/Grazoprevir-Kombination kontraindiziert. Cobicistat- und Ritonavir-geboosterte HIV-Therapien können die AUC-Werte von Grazoprevir bis zum 12,9-Fachen anheben, wozu auch eine OATP1B1-Hemmung beiträgt. Ciclosporin erhöhte die Grazoprevir-Exposition um das 15,2-Fache. In der Konsequenz sind auch starke OATP1B1-Hemmer kontraindiziert und von einer Anwendung starker CYP3A4-Hemmer wird abgeraten. Dass sich die Effekte einer OATP1B1-Hemmung und der CYP3A4-Induktion aufheben können, zeigt sich am Verhalten von Grazoprevir zu Rifampicin. Bei Einmalgabe steigen die AUC-Werte durch OATP1B1-Hemmung von Grazoprevir um das 10,2-Fache. Bei Mehrfachgabe von Rifampicin und der daraus resultierenden CYP3A4-Induktion sinkt der Grazoprevir-Spiegel wieder auf das Niveau des Ausgangswerts [5].
Glecaprevir/Pibrentasvir
Die Fixkombination aus dem NS3/4-Protease-Inhibitor Glecaprevir (GLE) und dem NS5A-Inhibitor Pibrentasvir (PIB) wirkt pangenotypisch [11]. Glecaprevir ist Substrat von CYP3A4. Die starke Hemmung dieses Enzyms durch Darunavir-Ritonavir erhöhte die Glecaprevir-Exposition um das 5-Fache. Rifampicin in einer Mehrfahrdosis reduzierte die Exposition von Glecaprevir auf 12 %.
Als Substrate bzw. Inhibitoren der Arzneimitteltransporter BCRP, P-gp und OATP1B1/3 resultieren weitere Interaktionsrisiken.
Über eine OATP1B1-Hemmung wird die Bioverfügbarkeit von Glecaprevir durch 400 mg Ciclosporin um das 5-Fache und durch eine Einzeldosis Rifampicin um das 8,4-Fache angehoben.
Andererseits erhöhte Glecaprevir/Pibrentasvir die systemische Verfügbarkeit von Rosuvastatin (BCRP-Testsubstrat) um das 2,2-Fache, Dabigatranetexilat (P-gp-Substrat) um das 2,4-Fache sowie die OATP1B1-Substrate Atorvastatin und Simvastatinsäure (Metabolit von Simvastatin) um das 8,3-Fache bzw. 4,5-Fache. Atorvastatin, Dabigatranetexilat und Simvastatin sind daher kontraindiziert [3].
Fazit
Die Behandlung der Hepatitis C hat durch Einführung der direkt antiviralen Agenzien die bisherige Interferon-basierte Therapie in Kombination mit Ribavirin abgelöst. Eine sehr hohe Eradikationsrate, die bessere Verträglichkeit und eine in der Regel kürzerer Therapiedauer führten zu diesem Therapieswitch. Jedoch zeigen DAA-basierte Kombinationstherapien ein hohes Potenzial für pharmakokinetische Wechselwirkungen, die 30 bis 44 % der Patienten betreffen können [9]. Die Anwendung einer Interaktionsdatenbank und/oder eine Prüfung über die Internetseite der Universität Liverpool https://www.hep-druginteractions.org/checker ist bei Kombinationstherapie daher dringend zu empfehlen, zumal die hochpreisigen Fixkombinationen nur in einer Stärke verfügbar sind und Dosisanpassungen nicht vorgenommen werden können.
Literatur
1. Fachinformation Epclusa®. Stand: Dezember 2018.
2. Fachinformation Harvoni®. Stand: Februar 2019.
3. Fachinformation Maviret®. Stand: April 2019.
4. Fachinformation Vosevi®. Stand: Januar 2019.
5. Fachinformation Zepatier®. Stand: Dezember 2018.
6. Geddawy A, Ibrahim YF, Elbahie NM, et al. Direct acting anti-hepatitis C virus drugs: clilnical pharmacology and future direction. J Transl Int Med 2017;5:8–17.
7. German P, Mathias A, Brainard DM, et al. Drug-drug interaction profile of the fixed-dose combination tablet regimen ledipasivr/sofosbuvir. Clin Pharmacokinet 2018;57:1369–83.
8. Kirby BJ, Symonds WT, Kearney BP, et al. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor Sofosbuvir. Clin Pharmacokinet 2015;54:677–90.
9. Kondili LA, Gaeta GB, Ieluzzi D, et al. Real-life data on potential drug-drug interactions in patients with chronic hepatitis C viral infection undergoing antiviral therapy with interferon-free DAAs in the PITER Cohort Study. PLoS One. 2017;12(2):e0172159.
10. Sarrazin C, Zimmermann T, Berg T, et al. S3-Leitlinie „Prophylaxe, Diagnostik und Therapie der Hepatitis-C-Virus (HCV) -Infektion“. Z Gastroenterol 2018; 56:756–838.
11. Zeuzem S. Chronische Hepatitis C. Internist 2018;59:528–35.
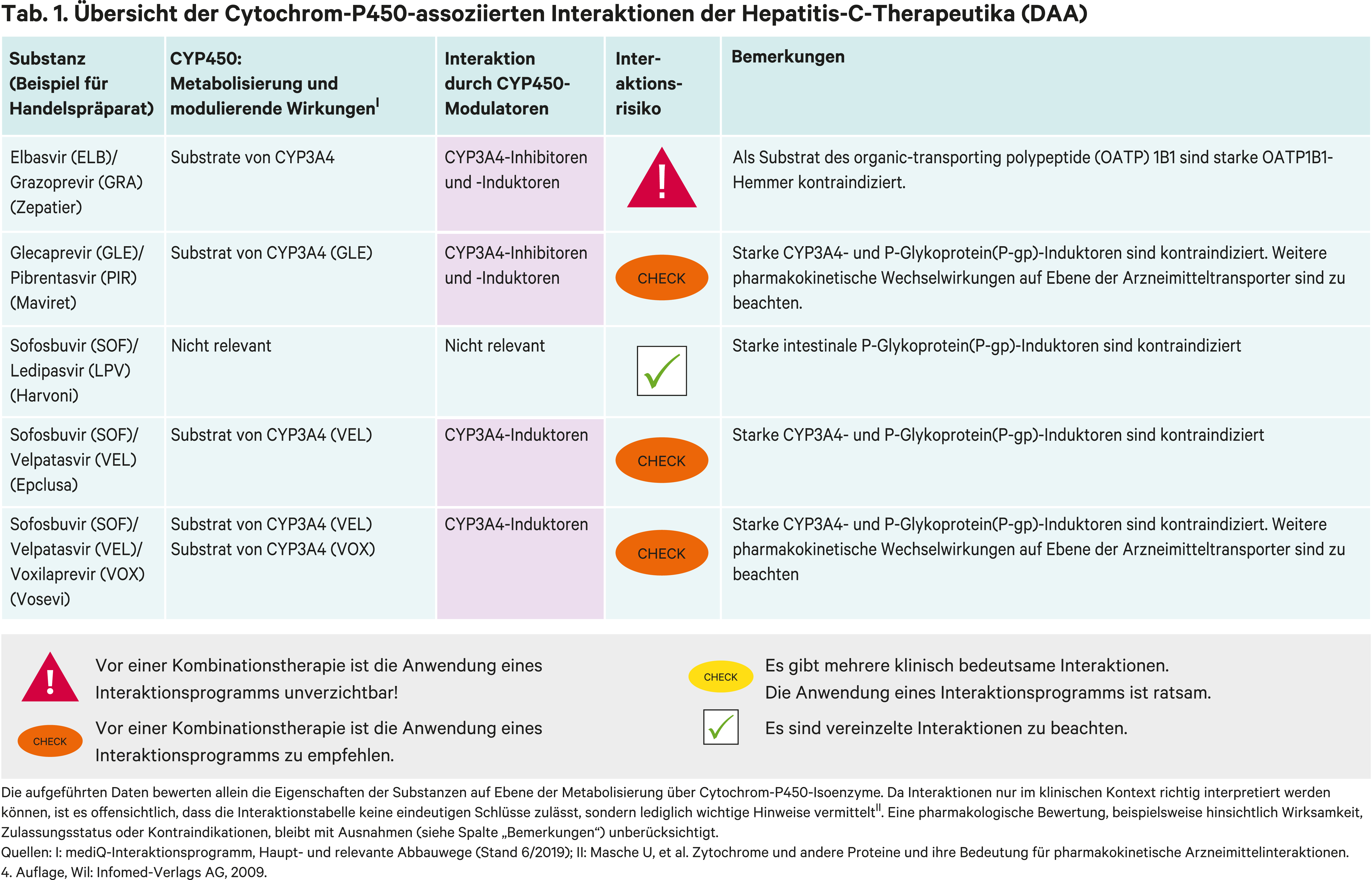
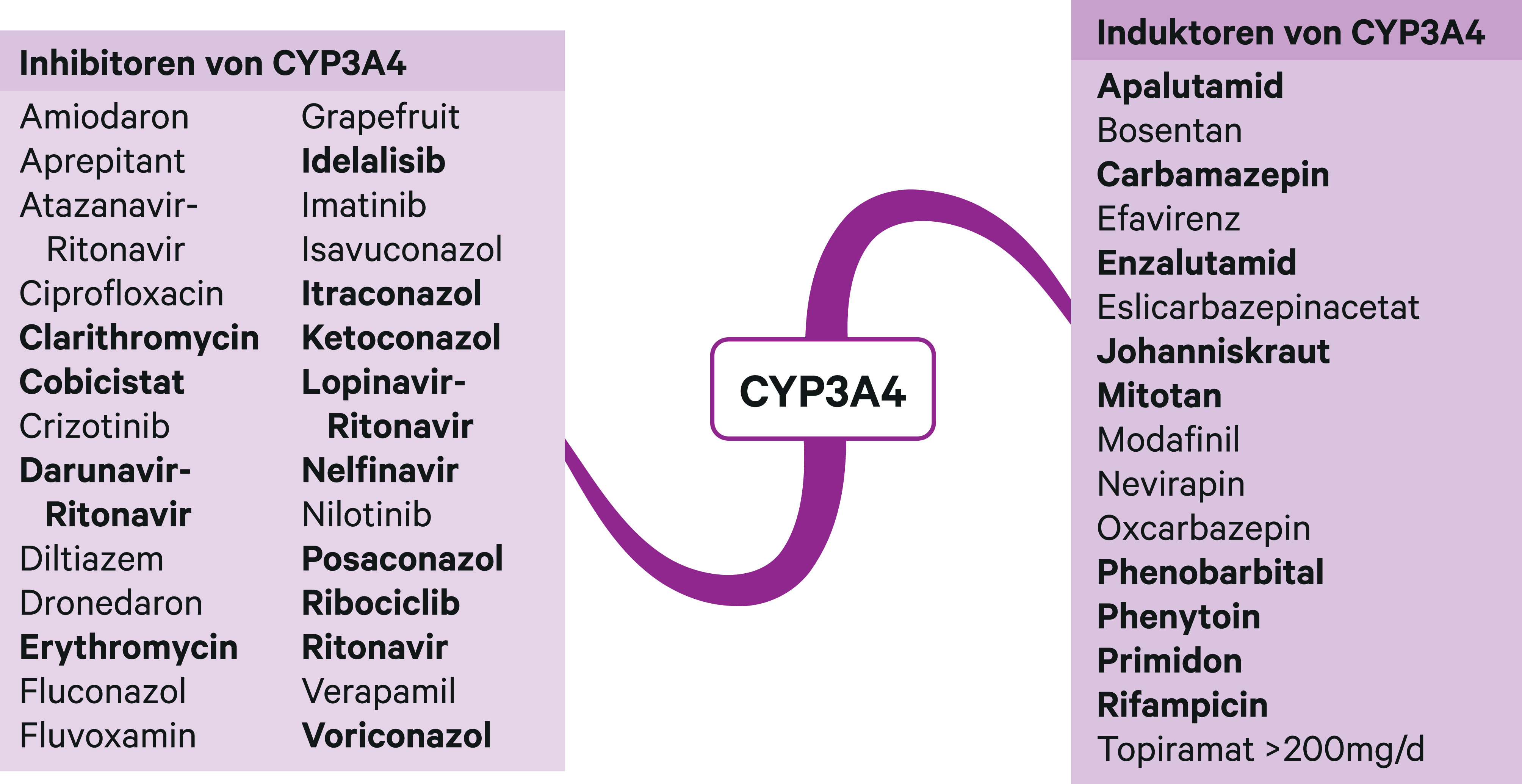
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 3A4 (Stand 8/2019) [Quelle: mediQ-Interaktionsprogramm]
*Nachdruck aus Krankenhauspharmazie 2019;40:441–3.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2019; 26(05)