Holger Petri, Bad Wildungen*
Stimulanzien
Methylphenidat
Methylphenidat ist Mittel der ersten Wahl in der medikamentösen Behandlung der Aufmerksamkeitsdefizits-/Hyperaktivitätsstörung (ADHS) [1]. Der Abbau erfolgt primär über die Carboxylesterase (CES) 1A1 vornehmlich zu pharmakologisch inaktiver Ritalinsäure [12, 20]. Zur großen interindividuellen Variabilität der Plasmaspiegel von Methylphenidat tragen Polymorphismen (SNP, single nucleotide polymorphisms) von CES1 bei [12, 19]. Es gibt Hinweise für arzneimittelbedingte Wechselwirkungen durch Hemmung des Enzyms CES1 [24]. Die klinische Bedeutung muss durch In-vivo-Studien jedoch erst noch evaluiert werden. Das Risiko für CYP-bedingte Wechselwirkungen ist vernachlässigbar.
Die D- und L-Enantiomere von Methylphenidat hemmen die Cytochrom-P450(CYP)-Enzyme 1A2, 2C8, 2C9, 2C19, 2D6, 2E1 oder 3A nicht in relevantem Ausmaß [6]. Dennoch wird aufgrund von Einzelfallberichten empfohlen, in Kombination mit Antikoagulanzien vom Cumarin-Typ den INR-Wert zu überwachen. Dosisanpassungen nach Bestimmung der Plasmakonzentrationen von Antikonvulsiva (z. B. Phenobarbital, Phenytoin, Primidon) und einigen Antidepressiva (trizyklische Antidepressiva und selektive Serotonin-Wiederaufnahmeinhibitoren) bei Beginn oder Absetzen einer Methylphenidat-Therapie können erforderlich sein [6, 15].
Modafinil
Modafinil ist zugelassen in der Therapie Erwachsener mit exzessiver Schläfrigkeit, die mit Narkolepsie einhergeht [8]. Es werden zwei pharmakologisch unwirksame Metaboliten gebildet. Über Esterasen entsteht der Hauptmetabolit Modafinilsäure (40–50 % der Dosis), über CYP3A4 in geringerem Ausmaß Modafinil-Sulfon [8, 16]. Somit könnten starke CYP3A4-Modulatoren zu veränderten Plasmaspiegeln führen. Ergebnisse aus pharmakokinetischen Interaktionsstudien liegen nicht vor.
Modafinil kann selbst den Metabolismus anderer Arzneimittel modulieren. Der AUC(Fläche unter der Konzentrations-Zeit-Kurve)-Wert des Testsubstrats Omeprazol (CYP2C19) erhöhte sich in einer Untersuchung durch Modafinil im Steady-State um 90 %, während die AUC des CYP3A4-Testsubstrats Midazolam um ein Drittel sank [18]. Der AUC-Wert von Triazolam, einem anderen CYP3A4-Substrat, verringerte sich in einer klinischen Studie um 57 % [17]. Somit ist Vorsicht geboten bei Kombination mit Arzneimitteln, deren Metabolismus klinisch relevant von CYP2C19 (z. B. Citalopram, Escitalopram) und CYP3A4 abhängig ist (z. B. Proteinkinase-Inhibitoren, selektive Immunsuppressiva).
Dexamfetamin und Lisdexamfetamin
Dexamfetamin ist das rechtsdrehende Enantiomer von Amphetamin, Lisdexamfetamin ist ein Prodrug von Dexamfetamin. Über CYP2D6 entsteht ein hydroxylierter Metabolit. Es ist nicht bekannt, wie sich CYP2D6-Inhibitoren auf die Dexamfetamin-Exposition auswirken. Nach Herstellerangaben gibt es keine Anhaltspunkte für einen Einfluss der Ethnie auf die Pharmakokinetik [4]. Da die Expression von CYP2D6 je nach Ethnie aber voneinander abweicht (Tab. 1), scheinen somit auch CYP-bedingte Interaktionen von untergeordneter Relevanz zu sein. Lisdexamfetamin verursacht in vitro keine relevanten Aktivitätsänderungen von Cytochrom-P450-Enzymen [10].
Nichtstimulanzien
Atomoxetin
Atomoxetin ist ein selektiver Noradrenalin-Wiederaufnahmehemmer. Der Metabolismus erfolgt primär über das polymorph exprimierte Enzym CYP2D6 zum äquipotenten Metaboliten 4-Hydroxyatomoxetin. Dieser wird aber sogleich in einer Phase-II-Reaktion glucuronidiert (Abb. 1) und trägt daher nur unwesentlich zur klinischen Wirksamkeit bei [23].
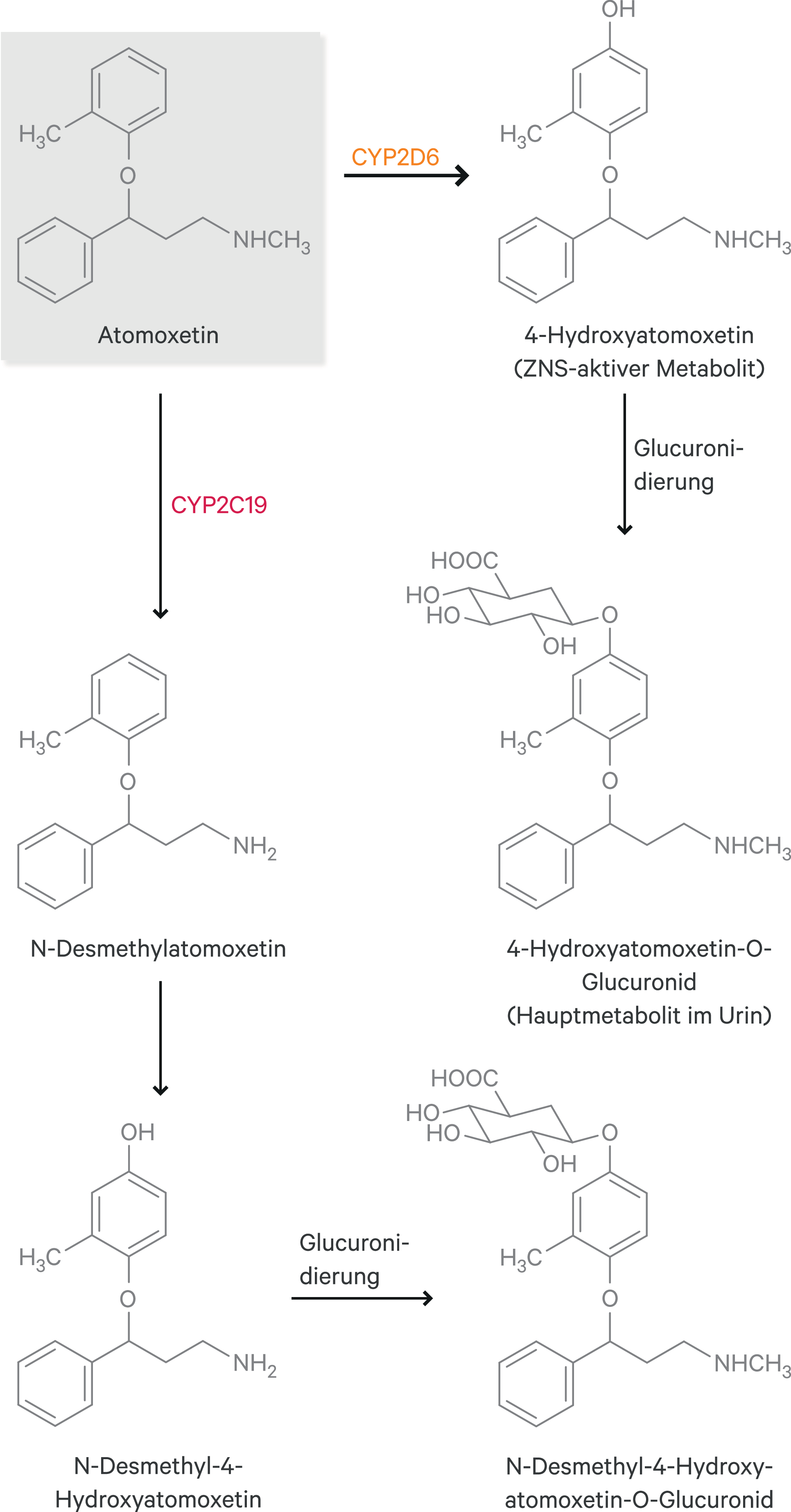
Abb. 1. Metabolismus von Atomoxetin (nach [23])
Homozygote Träger von Allelen, die für funktionsunfähige Enzyme kodieren (Poor Metabolizer, PM), haben bis zu 10-fach höhere AUC-Werte von Atomoxetin als Extensive Metabolizer (EM) [7]. Folglich erhöhten in klinischen Studien starke CYP2D6-Inhibitoren (Abb. 2) die Bioverfügbarkeit von Atomoxetin bei Probanden mit EM-Status in einem ähnlichen Ausmaß. Bupropion führte zu durchschnittlich 5,1-fach, Paroxetin zu 5,8- bis 6,5-fach höheren Plasmaspiegeln [2, 21, 22]. In einer Studie mit homozygoten CYP2D6*10-Trägern (Intermediate Metabolizer; IM) stiegen die AUC-Werte von Atomoxetin um das 3-Fache [9]. Vermutlich führen moderate CYP2D6-Inhibitoren zu ähnlich hohen AUC-Steigerungen.
In Kombination mit potenten CYP2D6-Hemmern können daher ein langsameres Auftitrieren und eine geringere Erhaltungsdosis notwendig werden [7]. In der US-amerikanischen Fachinformation wird konkret empfohlen, die initiale Dosis nur bei ausbleibendem klinischen Ansprechen und guter Verträglichkeit nach einem Zeitraum von vier Wochen zu erhöhen [14]. Patienten mit schnellem Metabolisierungsstatus (Ultrarapid Metabolizer) zeigen eine beschleunigte Clearance von CYP2D6-Substraten. Zur Dosisanpassung gibt es jedoch keine Daten [3]. Klinisch relevante CYP2D6-Induktoren sind nicht bekannt.
Über das ebenfalls polymorph exprimierte Enzym CYP2C19 wird im Nebenweg (3–6 % der Atomoxetin-Dosis) der pharmakologisch unwirksame Metabolit N-Desmethylatomoxetin gebildet (Abb. 1). In einer Studie hatte der CYP2C19-Polymorphismus einen Einfluss auf die Exposition von Atomoxetin [23]. Inwiefern hieraus klinische Konsequenzen besonders bei Kombination mit CYP2C19-Modulatoren erwachsen, ist nicht bekannt.
Guanfacin
Guanfacin ist ein selektiver Alpha2A-Rezeptoragonist [5]. Sein Abbau erfolgt primär über CYP3A4 zu unwirksamen Metaboliten. Der starke CYP3A4-Inhibitor Ketoconazol kann die AUC um das Dreifache erhöhen, der starke CYP3A4-Induktor Rifampicin um 70 % senken [5]. Die über ein physiologisch basiertes pharmakokinetisches Modell gewonnenen Daten sprechen dafür, dass es auch in Kombination mit moderaten Modulatoren zu klinisch signifikanten AUC-Veränderungen kommen kann [11]. Daher wird in Kombination mit starken und moderaten CYP3A4-Inhibitoren (Abb. 2) eine Dosisreduktion von Guanfacin um 50 % empfohlen. Mit starken CYP3A4-Induktoren kann eine neue Dosistitration erwogen werden. Gemäß US-Fachinformation soll bei Komedikation mit starken und moderaten Induktoren (Abb. 2) die Dosis verdoppelt werden [13].
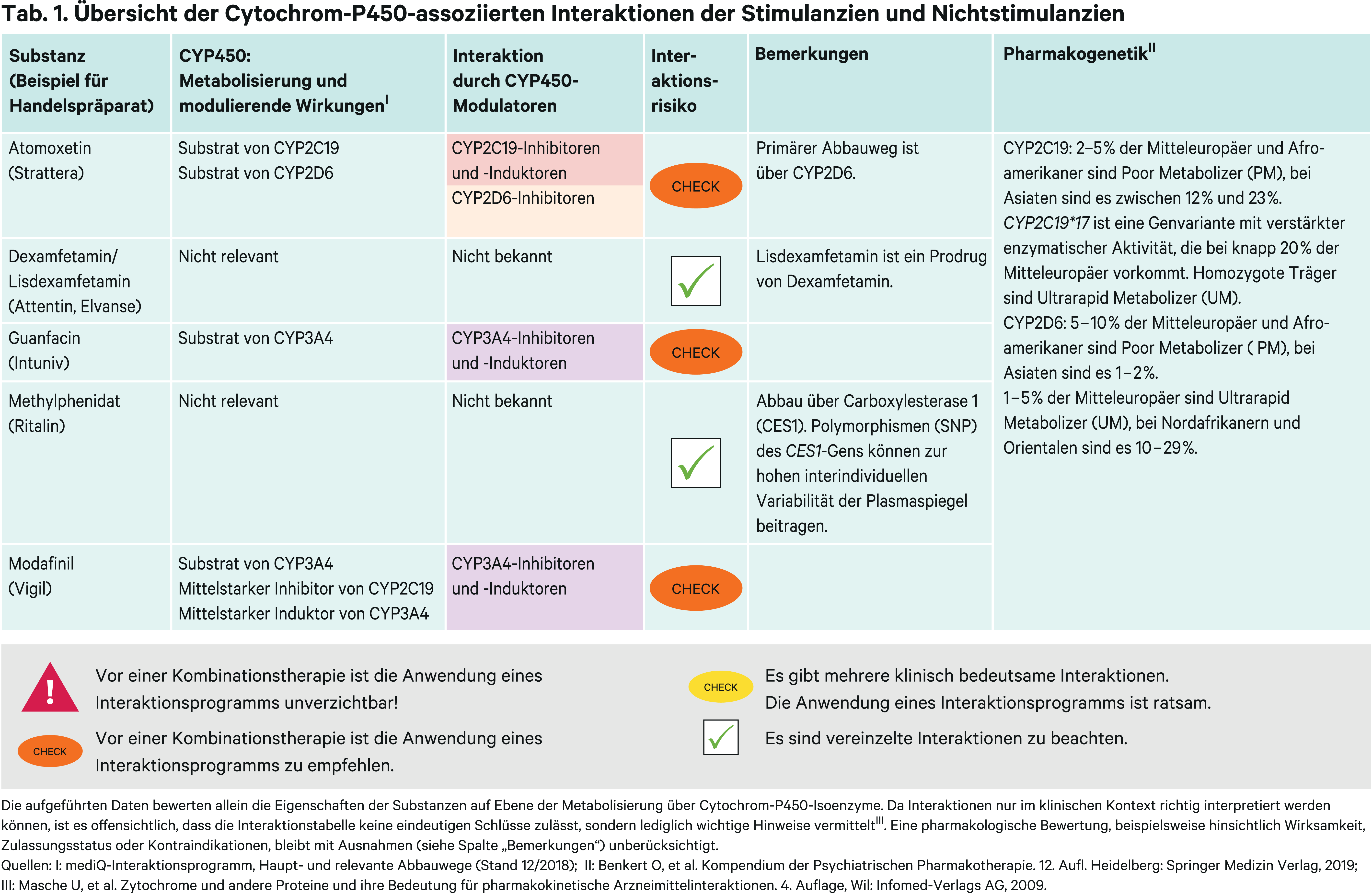
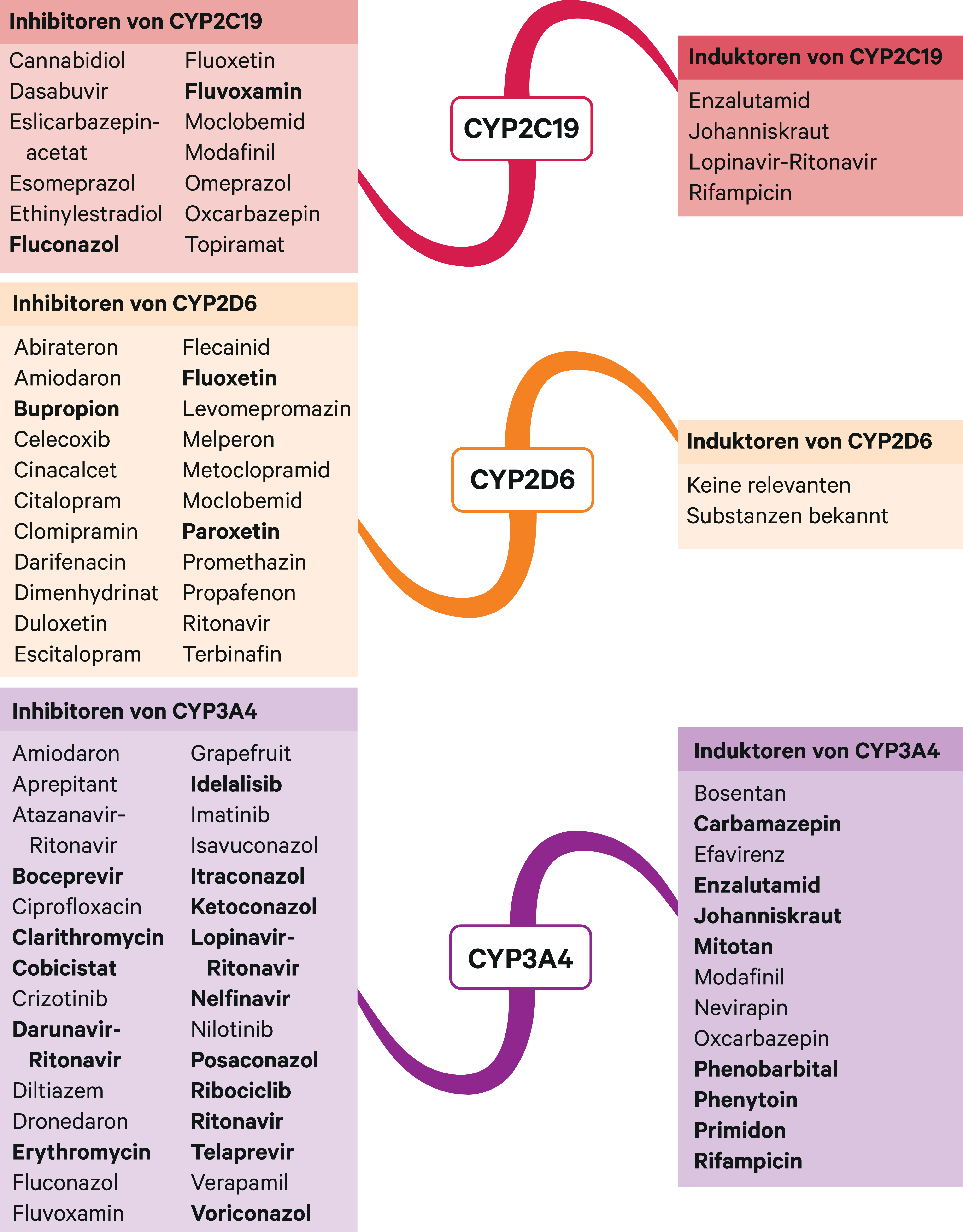
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C19, 2D6 und 3A4 (Stand 12/2018) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Banaschewski T, Hohmann S, Millenet S, et al. Leitlinie „ADHS bei Kindern, Jugendlichen und Erwachsenen“. Stand Mai 2017.
2. Belle DJ, Ernest CS, Sauer JM, et al. Effect of potent CYP2D6 inhibition by paroxetine on atomoxetine pharmacokinetics. J Clin Pharmacol 2002;11: 1219–27.
3. Dean L. Atomoxetine therapy and CYP2D6 genotype. Medical Genetics Summaries. September 2015.
4. Fachinformation Elvanse®. Stand: Oktober 2018.
5. Fachinformation Intuniv®. Stand: Oktober 2017.
6. Fachinformation Ritalin®. Stand: Mai 2018.
7. Fachinformation Strattera®. Stand: Juni 2015.
8. Fachinformation Vigil®. Stand: Dezember 2015.
9. Kim SH, Byeon JY, Kim YH, et al. Physiologically based pharmacokinetic modelling of atomoxetine with regard to CYP2D6 genotypes. Sci Rep 20188:12405. DOI:10.1038/s41598–018–308418.
10. Krishnan S, Moncrief S. An evaluation of the cytochrome p450 inhibition potential of lisdexamfetamine in human liver microsomes. Drug Metab Dispos 2007;35:180–4.
11. Li A, Yeo K, Welty D, et al. Development of guanfacine extended-release dosing strategies in children and adolescents with ADHD using a physiologically based pharmacokinetic model to predict drug-drug interactions with moderate CYP3A4 inhibitors or inducers. Paediatr Drugs 2018;20:181–94.
12. Lyauk YK, Stage C, Bergmann TK, et al. Population pharmacokinetics of methylphenidate in healthy adults emphasizing novel and known effects of several carboxylesterase 1 (CES1) variants. Clin Transl Sci 2016;9:337–45.
13. Prescribing information Intuniv®. Stand: Juli 2016.
14. Prescribing information Strattera®. Stand: Mai 2017
15. Nevels RM, Weiss, NH, Killebrew AE, et al. Methylphenidate and its under-recognized, underexplained, and serious drug interactions: A review of the literature with heightened concerns. German J Psychiatry 2013;16:29–42.
16. Robertson P Jr, Hellriegel ET. Clinical pharmacokinetic profile of modafinil. Clin Pharmacokinet 2003;42:123–37.
17. Robertson P Jr, Hellriegel ET, Arora S, et al. Effect of modafinil on the pharmacokinetics of ethinyl estradiol and triazolam in healthy volunteers. Clin Pharmacol Ther 2002;7:46–56.
18. Rowland A, van Dyk M, Warncken D, et al. Evaluation of modafinil as a perpetrator of metabolic drugdrug interactions using a model informed cocktail reaction phenotyping trial protocol. Br J Clin Pharmacol 2018;84:501–9.
19. Stage C, Jürgens G, Guski LS, et al. The impact of CES1 genotypes on the pharmacokinetics of methylphenidate in healthy Danish subjects. Br J Clin Pharmacol 2017;83:1506–14.
20. Sun Z, Murry DJ, Sanghani SP, et al. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J Pharmacol Exp Ther 2004;310:469–76.
21. Todor I, Popa A, Neag M, et al. Evaluation of a potential metabolism-mediated drug-drug interaction between atomoxetine and bupropion in healthy volunteers. J Pharm Pharm Sci 2016;2:198–207.
22. Todor I, Popa A, Neag M, et al. The influence of paroxetine on the pharmacokinetics of atomoxetine and its main metabolite. Clujul Med 2015;88:513–20.
23. Yu G, Li GF, Markowitz JS. Atomoxetine: A review of its pharmacokinetics and pharmacogenomics relative to drug disposition. J Child Adolesc Psychopharmacol 2016;26:34–26.
24. Zhu HJ, Appel DI, Peterson YK, et al. Identification of selected therapeutic agents as inhibitors of carboxylesterase 1: potential sources of metabolic drug interactions. Toxicology 2010;270:59–65.
*Nachdruck aus Krankenhauspharmazie 2019;40:53–7.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2019; 26(01):57-61