Hans-Jürgen Möller, München, und Karl Broich, Bonn
Allgemeine Grundlagen der Therapieforschung
Man kann die Therapieforschung unterteilen in retrospektive und prospektive, nichtexperimentelle, quasi-experimentelle und experimentelle Verfahren [5, 10]. Ohne weiter auf Methodik sowie Vor- und Nachteile dieser unterschiedlichen prinzipiellen Forschungsansätze einzugehen, soll zusammenfassend ihre Wertigkeit hinsichtlich des Erkenntnisgewinns klassifiziert werden. Als hypothesengenerierende Verfahren sind nichtexperimentelle Studien, in denen korrelative Zusammenhänge beschrieben werden, fruchtbar. Dabei haben prospektive Studien Vorrang gegenüber retrospektiven Studien (Abb. 5–1), die wegen der größeren Gefahr der Beobachtungsverfälschung und der höheren Wahrscheinlichkeit zufälliger Korrelationen problematisch sind.
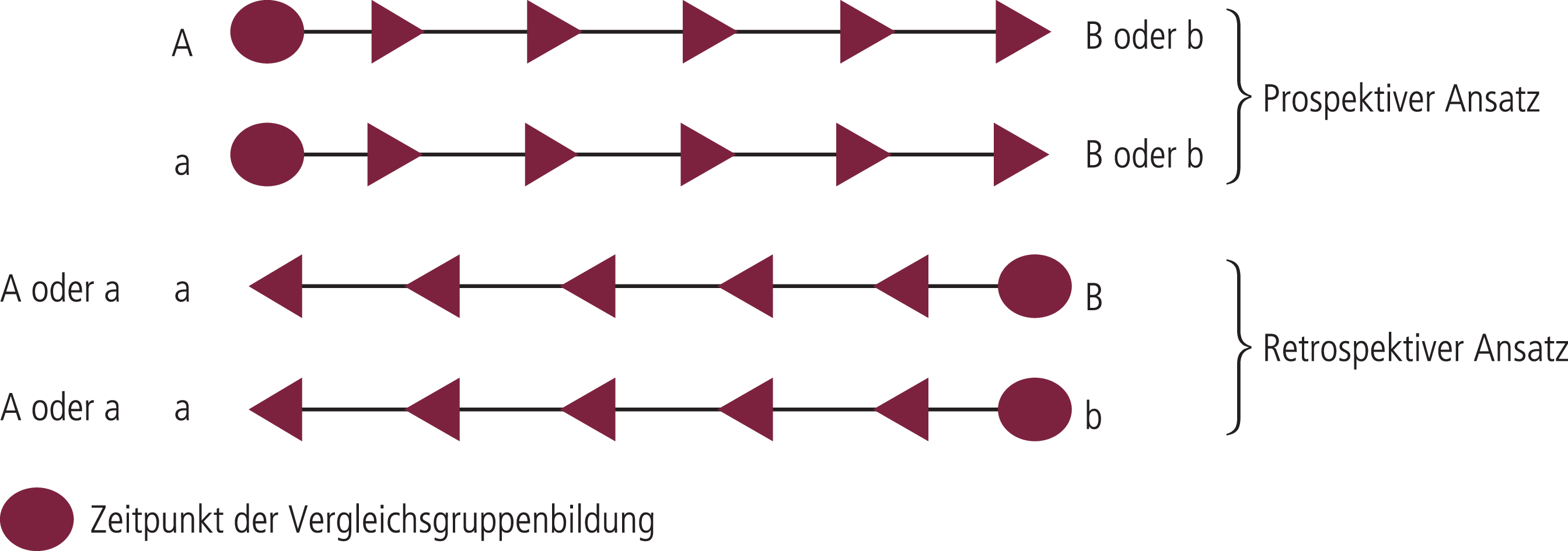
Abb. 5–1. Schematische Darstellung von prospektiven und retrospektiven Ansätzen (mod. nach [5]). Große Buchstaben symbolisieren das Vorhandensein, kleine das Fehlen eines Merkmals. Die beiden Linien bedeuten jeweils die Vergleichsgruppen
Die so gefundenen Zusammenhänge können nur in prospektiven experimentellen Untersuchungen auf das Vorhandensein kausaler Beziehungen überprüft werden. Je mehr ein quasi-experimenteller Ansatz sich hinsichtlich des Ausmaßes der Variablenkontrolle dem experimentellen Ansatz nähert, desto stringenter ist die Beweisführung (Kriterium der internen Validität). Allerdings wird die Generalisierbarkeit der Ergebnisse (Kriterium der externen Validität) zunehmend erschwert, je reduktionistischer ein experimentelles Vorgehen ist. Ist die experimentelle Überprüfung eines im Rahmen einer nichtexperimentellen Untersuchung gefundenen korrelativen Zusammenhangs zwischen zwei Variablen aus ethischen oder forschungspraktischen Gründen nicht möglich, kann ein kausaler Zusammenhang nur postuliert werden, sofern dieser im Theoriekontext als plausibel hingestellt werden kann; eine empirische Beweisführung im engeren Sinne ist aber nicht möglich.
Das experimentelle Vorgehen wird für psychopharmakologische Therapiestudien am Menschen allgemein akzeptiert und praktiziert und als methodischer Standard für den Nachweis der Wirksamkeit eines Medikaments angesehen (Tab. 5–1). Aber auch nichtexperimentelle Verfahren spielen eine Rolle; dazu gehört beispielsweise die Erfassung von möglichen Nebenwirkungen während routinemäßig durchgeführter medikamentöser Therapien.
Tab. 5–1. Mögliche Studiendesigns zur Therapie-Evaluation
|
Einzelfallstudie |
|
Einzelgruppenstudie |
|
Kontrollgruppenstudie
|
|
u. a. |
Für die Entwicklung neuer Psychopharmaka sind Tierversuche unabdingbar. Durch sie werden wichtige pharmakologische, toxikologische, biochemische, neurophysiologische und neuroendokrinologische Grunddaten zur Charakterisierung eines potenziellen Psychopharmakons gewonnen. Die Kenntnisse über diese Wirkungseigenschaften der Substanz werden dann durch weitere präklinische Untersuchungen an gesunden Probanden ergänzt. Erst in dieser Weise gut untersuchte Substanzen werden in Therapiestudien an psychiatrischen Patienten auf ihre Wirksamkeit und Verträglichkeit geprüft werden.
Therapiebezogene Studien am Tier
Tierexperimentelle Studien sind weiterhin in der Entwicklung und Weiterentwicklung von Psychopharmaka von Wichtigkeit, wenn auch versucht wird, ihr Ausmaß einzuschränken und durch sinnvolle Alternativen zu ersetzen. In der tierexperimentellen Forschung wird unter anderem in translationalen Ansätzen versucht, Bedingungsgefüge psychischer Erkrankungen an geeigneten Tiermodellen zu untersuchen und Möglichkeiten für psychopharmakologische Interventionen zu identifizieren. Für die Entwicklung und Weiterentwicklung von Psychopharmaka werden unter anderem systematische tierpharmakologische Suchprogramme („screenings“) durchgeführt, über die bestimmte pharmakologische Wirkungen von Arzneimitteln erkannt werden können. Dabei werden heute die aus vergleichenden tierpharmakologischen Untersuchungen abgeleiteten Indikatoren (z. B. für Neuroleptika: kataleptische Wirkung, Apomorphin-Antagonismus, Hemmung bzw. Aufhebung bedingter Fluchtreaktionen) nicht mehr als absolut sichere Prädiktoren für die spezielle therapeutische Wirksamkeit eines potenziellen Psychopharmakons beim Menschen angesehen. Die Grenzen des tierexperimentellen Screenings wurden schon bei der Entdeckung der antipsychotischen Wirksamkeit des Clozapins, das bekanntlich im Tierversuch einige der für Neuroleptika bisher als unabdingbar angesehenen Effekte nicht hat, aufgezeigt. Gerade das Fehlen von im Tiermodell darstellbaren Risiken für extrapyramidale Nebenwirkungen bei gleichzeitig guter antipsychotischer Wirksamkeit war das Innovative in dieser Geburtsstunde der atypischen Neuroleptika.
Wenn auch die klassischen tierpharmakologischen Untersuchungsprogramme in ihrer Aussagefähigkeit bezüglich möglicher therapeutischer Wirkungen begrenzter sind, als längere Zeit angenommen wurde, so liefern sie doch eine Reihe wichtiger Daten, die für die Erprobung eines Psychopharmakons am Menschen erforderlich sind: pharmakokinetische Daten, toxikologische Daten und andere. Sie werden ergänzt durch biochemische, neuroendokrinologische, molekularbiologische, genetische Methoden und Bildgebungsverfahren, die auch im Rahmen der präklinischen und klinischen Forschung am Menschen eingesetzt werden können. Dieser Zweig der Psychopharmakologie hat seine Bedeutung vor allem darin, dass die aus ihm erwachsenden Perspektiven in enger Beziehung zur Erforschung der Wirkungsmechanismen (z. B. biochemische Änderungen im Bereich der Synapsen) bekannter und neuer Psychopharmaka und vor allem auch zur Erforschung der neurobiologischen Grundlagen psychischer Störungen beim Menschen stehen.
Das 4-Phasen-Modell der Prüfung eines Psychopharmakons am Menschen
Experimentelle Therapiestudien am Menschen sind in der Psychopharmakologie unverzichtbar, da die Ergebnisse tierexperimenteller Studien nur begrenzt auf den Menschen übertragbar sind und es obendrein für die psychischen Erkrankungen keine zufriedenstellenden Tiermodelle gibt, an denen die spezifische Wirksamkeit eines Pharmakons überprüft werden könnte. Die Untersuchung von Psychopharmaka am Menschen wird – wie die Prüfung anderer Pharmaka – konventionsgemäß in vier Phasen eingeteilt (Abb. 5–2).

Abb. 5–2. Das 4-Phasen-Modell der Medikamentenprüfung in der klinischen Psychopharmakologie (mod. nach [5])
Untersuchungen in Phase I werden in der Regel nicht bei Patienten, sondern bei Probanden durchgeführt; sie gehören damit zur präklinischen Forschung. Wenn Untersuchungen der Phase I in Ausnahmefällen bei psychiatrischen Patienten erfolgen, sollten Psychologen und Psychiater mitwirken, damit schon während der Verträglichkeitsuntersuchung auch pharmakopsychologische und eventuell biochemische Befunde erhoben werden, die für die Planung und Indikationsfestlegung der nachfolgenden Phase-II-Untersuchungen Hinweise liefern können.
Im Allgemeinen beginnt die klinische Prüfung eines potenziellen Pharmakons mit der Phase II. Erste Erfahrungen mit einer neuen Substanz werden unter der Mitwirkung von Psychiatern in offenen Prüfungen an einem heterogenen Patientengut gewonnen, um den möglichen Indikationsbereich abzugrenzen. Dieses an sich sinnvolle heuristische Vorgehen wird aber aus verschiedenen pragmatischen Gründen seit vielen Jahren immer seltener praktiziert. Wenn ausreichende Hypothesen zur Wirksamkeit der Substanz in einer bestimmten Indikation vorliegen, werden heute in der Regel schon in Phase II randomisierte, Placebo-kontrollierte Kontrollgruppenuntersuchungen, in der Regel an relativ kleinen Stichproben, durchgeführt, um Hinweise für Wirksamkeit und Verträglichkeit an Patienten des Indikationsbereichs zu bekommen und die diesbezüglichen optimalen Dosierungen einzugrenzen. Dieses methodische, im Vergleich zu früher anspruchsvollere Vorgehen soll die Validität der Ergebnisse sichern.
Die Ergebnisse von Phase II bilden die Planungsgrundlagen für die Untersuchungen der Phase III, in der an großen Stichproben die Wirksamkeit und Verträglichkeit gegen Placebo oder ein Standardpräparat im doppelblinden randomisierten Kontrollgruppenvergleich (RCT = „randomised controlled trial“) getestet werden. Sind die Ergebnisse positiv kann damit die Zulassung des Medikaments in der geprüften Indikation bei der zuständigen Zulassungsbehörde – BfArM in Deutschland, EMA in Europa, FDA in den USA – beantragt werden. Allerdings werden diese Phasen der klinischen Prüfung nun häufig komprimiert, so dass manche Entwicklungsprogramme auch scheitern, weil zum Beispiel die Dosisfindung in Phase II nicht adäquat durchgeführt wurde und Phase III dann mit nicht optimaler Dosis im Wirksamkeitsnachweis scheitert.
Ist ein Arzneimittel auf dem Markt eingeführt, so dient seine weitere Erforschung in Phase IV durch prospektive oder retrospektive Beobachtungsstudien (NIS = nichtinterventionelle Studien) unter den Bedingungen der alltäglichen Anwendung der Verbesserung der Kenntnisse über Wirksamkeit und Nebenwirkungen (insbesondere der Erkenntnis seltener Nebenwirkungen, die bei der begrenzten Fallzahl in den RCTs nicht erkannt werden) und gegebenenfalls der Erweiterung des Indikationsbereichs. In Phase IV werden in neuerer Zeit zunehmend auch sogenannte „Real-World“-Studien durchgeführt, nicht mit speziell selektierten Patienten, wie in Phase-III-Studien, sondern mit Patienten, die der üblichen klinischen Klientel (z. B. einschließlich der Problematik von Komorbiditäten, Therapierefraktärität) entsprechen. Diese Studien folgen im Gegensatz zu den in der Regel naturalistischen Studienansätzen in Phase IV – Beobachtungsstudien, nichtinterventionelle Studien – methodisch stringenteren Studiendesigns, beispielsweise im Sinne von offenen oder verblindeten Kontrollgruppenstudien, die in Ausnahmefällen sogar annähernd (aber nicht vollständig! siehe [6]) den methodischen Standard von RCTs erreichen. Sie werden auch „Effectiveness“-Studien (im Gegensatz zu den „Efficacy“-Studien der Phase III) genannt. Mit „Effectiveness“ ist gemeint: die Effektivität unter den Bedingungen der alltäglichen Behandlung, die nicht nur durch die in Phase III belegte Wirksamkeit, sondern auch durch Nebenwirkungen, Praktikabilität, Einnahmeverhalten und anderes bedingt wird. Ein sehr gut wirksames Medikament kann im Alltag der klinischen Behandlung versagen, wenn es beispielsweise schwere oder unangenehme Nebenwirkungen hat und/oder unpraktisch hinsichtlich der Einnahmemodalitäten (z. B. Einnahme eine Stunde nach Nahrungsaufnahme) ist.
Methodisches Spektrum bei der Evaluation eines Psychopharmakons
Bei den folgenden Ausführungen wird der Aspekt des Wirksamkeitsnachweises in den Vordergrund gestellt. Selbstverständlich gelten die dargestellten Methoden und Probleme in analoger Weise auch für andere psychopharmakologische Fragestellungen, beispielsweise Verträglichkeit (Tab. 5–2).
Tab. 5–2. Spektrum von Kontrollgruppenstudien
|
Prospektive Parallelgruppenstudie |
Nicht-prospektive Parallelgruppenstudie |
|
Ohne randomisierte Zuordnung |
|
|
Mit randomisierter Zuordnung
|
|
|
2-Arm-Studie: Verum (experimentelle Substanz)-Gruppe vs. Placebo-Gruppe |
|
|
2-Arm Studie: experimentelle Stubstanz-Gruppe vs. Standardpräparat-Gruppe |
|
|
3-Arm Studie: experimentelle Substanz- vs. Placebo- vs. Standardpräparat-Gruppe |
|
Die wichtigste Methode zum Nachweis der Wirksamkeit eines Pharmakons ist die prospektiv geplante randomisierte, doppelblinde Kontrollgruppen-Studie [9]. Dabei werden die Effekte einer zu prüfenden Substanz auf die randomisiert zugeteilten Patienten der Experimentalgruppe mit den Effekten eines Placebos oder eines bereits eingeführten Pharmakons gleicher Indikation (Standardpräparat) auf die Patienten der Kontrollgruppe verglichen. Die Patienten der beiden Gruppen werden zeitgleich randomisiert. Wartegruppen, wie sie lange Zeit in der Psychotherapieforschung als Kontrollgruppe verwendet wurden, sind nicht zulässig.
Beim gekreuzten Kontrollgruppenvergleich (Cross-over-Verfahren) wird nach dem Schema: Gruppe 1: Substanz A → Substanz B; Gruppe 2: Substanz B → Substanz A verfahren. Durch dieses Verfahren lässt sich die Aussagefähigkeit bei bestimmten Fragestellungen erhöhen, beispielsweise individuelles Ansprechen auf bestimmte Medikamente. Allerdings sind „Carry-over-Effekte“ (Überhangsphänomene) zu berücksichtigen, weshalb dieses Verfahren von Zulassungsbehörden nicht als ausreichend aussagekräftig für die Zulassung eines Pharmakons angesehen wird.
Bei der Prüfung gegen Placebo wird auf Überlegenheit des Psychopharmakons gegen Placebo getestet. Die Einbeziehung von Placebo in die Wirksamkeitsprüfung wird in und außerhalb der Medizin immer wieder aus ethischen Gründen kritisch diskutiert [2]. Die Zulassungsbehörden, unter anderem die europäische Zulassungsbehörde EMA, haben aber immer wieder die Notwendigkeit der Placebo-Kontrolle bei bestimmten psychiatrischen Indikationen wie Depression, Angststörung oder schizophrener Psychose betont. Nur durch die Prüfung auf Überlegenheit gegen Placebo ist ein ausreichend sicherer Beweis der Wirksamkeit möglich [3, 9]. Bei Einhaltung verschiedener Vorsichtsmaßnahmen ist die Prüfung gegen Placebo auch ethisch zu rechtfertigen [1].
Immer wieder in die Diskussion gebrachte alternative Verfahren, wie insbesondere die Prüfung auf Gleichwirksamkeit in einer Äqivalenzprüfung („equivalence study“, „non-inferiority study“) gegen ein Standardpräparat, haben methodische Probleme, die schwer zu lösen sind. Wenn eine solche Äquivalenzprüfung durchgeführt wird, muss vorher, basierend auf Vorerfahrungen in dem jeweiligen Indikationsgebiet, eine Unterschiedsschranke (kritisches Delta, „margin“, z. B. eine bestimmte Score-Differenz) definiert werden, die bei Gleichwirksamkeit nicht überschritten werden darf (Abb. 5–3). Die Festlegung dieses kritischen Delta gibt Möglichkeiten, das Ergebnis von vornherein im erwünschten Sinne zu beeinflussen, beispielsweise durch relativ großzügige Definition der Unterschiedsschranke. Man kann je nach Definition diese Schranke noch unterscheiden zwischen Gleichwertigkeit im strengen Sinn oder aus klinischer Sicht „nur unwesentlich schwächerer Wirksamkeit“. Bei der Auswertung von Äquivalenzstudien muss eine andere statistische Auswertungsmethodik angewandt werden als bei Prüfung auf Überlegenheit, eine speziell auf die Gleichwertigkeitsprüfung zugeschnittene Statistik, die unter anderem höhere Fallzahlen erforderlich macht. Eine abstrakte formale Analyse der Gegebenheiten bei Prüfung auf Gleichwertigkeit gibt Einsicht in die Problematik.
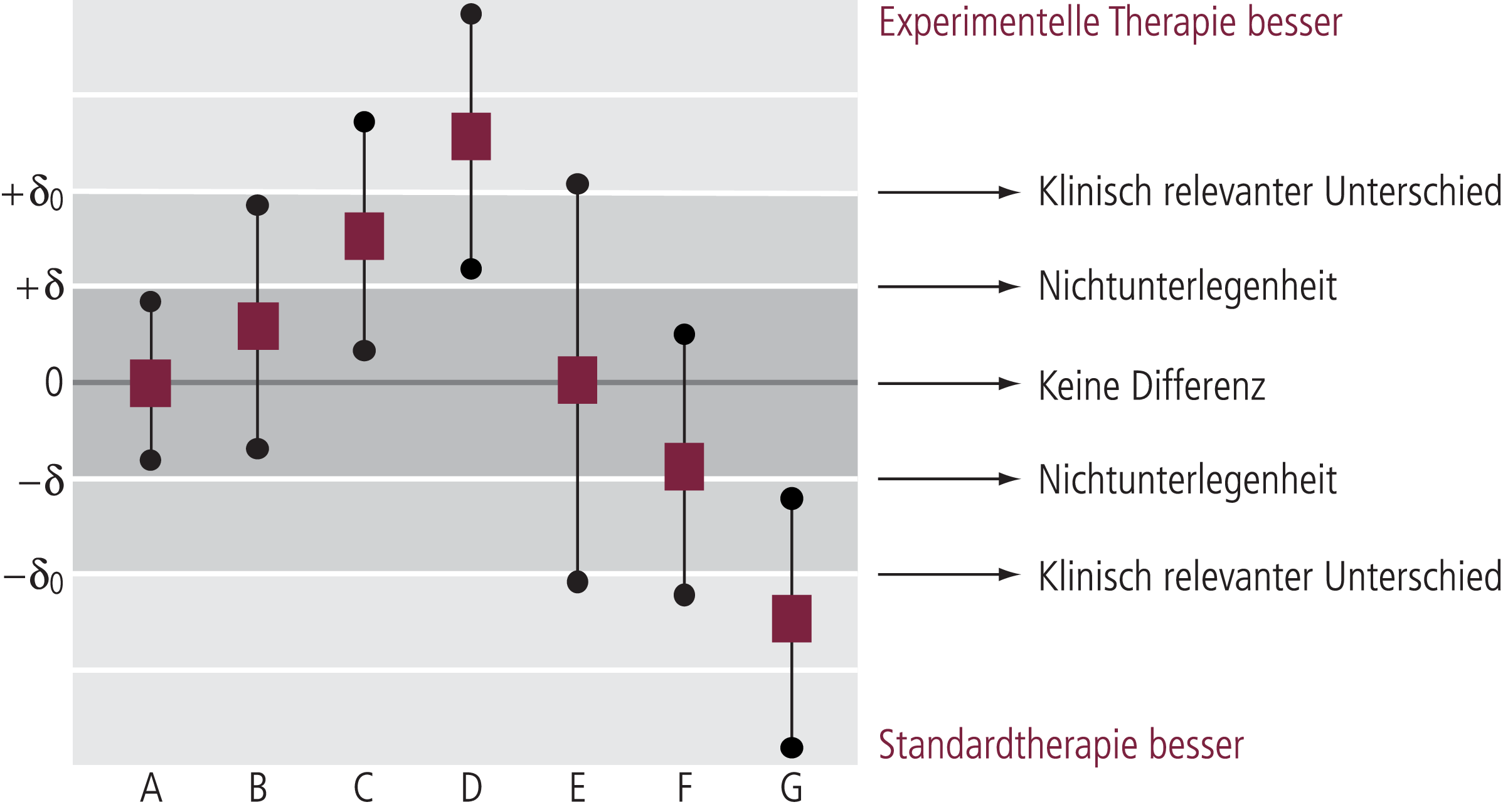
Abb. 5–3. Überlegenheit, Nichtunterlegenheit und die Rolle aktiver Komparatoren: Dargestellt sind die Punktschätzer und zweiseitigen 95%-Konfidenzintervalle für die Differenz der Effekte zwischen experimenteller und bewährter Standardtherapie (mod. nach [5])
Wenn in Abbildung 5–3 laut Protokoll auf Nichtunterlegenheit geprüft werden sollte, entsprechen A, B und C einer Nichtunterlegenheit. Im Falle D besteht sogar Überlegenheit der experimentellen Therapie gegenüber der Standardmedikation. Bei E und F ragen die Konfidenzintervalle über die Nichtunterlegenheitsschranke hinaus, damit sind die Ergebnisse nicht beweiskräftig und Nichtunterlegenheit kann nicht attestiert werden. Ergebnis G würde einer Unterlegenheit der experimentellen Therapie gegenüber der Standardmedikation entsprechen.
Das größte Problem bei Prüfung gegen Standardpräparat in einer Äquivalenzstudie ist die sogenannte „Assay-Sensitivität“. Man kann sich beispielsweise nicht sicher sein, dass das gewählte Standard-Antidepressivum auch in der Untersuchungsstichprobe wirksam ist. Bestimmte Konditionen wie ein zu niedriger Depressions-Schweregrad der einbezogenen Patienten, ein zu hoher Anteil an therapierefraktären Patienten oder zu viel Komedikation mit Hypnotika/Anxiolytika und andere können die Wirksamkeit des Standardpräparats reduzieren und damit gegebenenfalls einem nur schwach wirksamen zu prüfenden neuen Antidepressivum die Chance geben, als gleichwirksam zu erscheinen.
Als weitere Möglichkeit, die ethische Problematik der Prüfung gegen Placebo zu umgehen, wurde die Prüfung auf Überlegenheit (Superioritätsstudie, „superiority study“) des neuen zu prüfenden Psychopharmakons gegen ein Standardpräparat vorgeschlagen. Damit würde gleichzeitig ein Problem der Äquivalenzprüfung umgangen: die Definition des kritischen Delta (s. o.). So verlockend diese Alternative scheint, sie ist aber wenig realistisch, da die meisten neu einzuführenden Psychopharmaka erfahrungsgemäß diesen Test nicht bestehen würden. Falls bei Prüfung auf Superiorität diese statistisch nicht bewiesen werden kann, darf daraus nicht geschlossen werden, dass die untersuchten Medikamente in ihrer Wirksamkeit gleichwertig sind!
Je nach Fragestellung (z. B. Wirksamkeit, Wirksamkeitsvergleich, Nebenwirkungen) stehen diesen aufwendigen Verfahren ökonomischere und praktikablere gegenüber, die insbesondere im Rahmen von Erkundungsstudien über neue Psychopharmaka angewandt werden, beispielsweise Verfahren ohne Kontrollgruppe, einfachblinde oder nichtblinde Verfahren. Hier sind auch nichtexperimentelle Untersuchungen in Form retrospektiver oder prospektiver Verlaufsbeobachtungen zu nennen. Diese haben vor allem als heuristische Methoden ihren Stellenwert zur Überprüfung von Langzeiteffekten und Langzeitnebeneffekten von bereits eingeführten Präparaten.
Die gesamte Palette dieser letztgenannten Verfahren hat in der klinischen Erprobung von Psychopharmaka ihre Berechtigung, sofern die für die einzelnen Verfahren geltenden Einschränkungen hinsichtlich des Erkenntniswerts beachtet werden. Diese methodischen Limitierungen werden aber nicht immer ausreichend berücksichtigt, wie man beispielsweise bei der oft unkritischen Interpretation der Ergebnisse der im letzten Jahrzehnt so populär gewordenen „Effectiveness“-Studien beobachten konnte [6].
Die randomisierte doppelblinde Kontrollgruppenstudie und ihre Charakteristika
Die prospektive randomisierte Kontrollguppenstudie (RCT = randomised controlled trial) wird als methodische Basis der klinischen psychopharmakologischen Forschung zur Wirksamkeit und Verträglichkeit eines Psychopharmakons (Abb. 5–4a und 5–4b) angesehen [5, 9]. Verglichen werden zwei parallele Gruppen (Parallelgruppenvergleich!, nicht zeitversetze Stichproben wie z. B. Wartegruppen). Obendrein sollten möglichst Doppelblind-Bedingungen vorliegen, um eine faire Beurteilung ohne Beeinflussung durch Einstellungen oder vorgefasste Meinungen unter anderem der Untersucher zu garantieren. Von den Zulassungsbehörden wird für die Zulassung eines neuen Psychopharmakons bzw. für eine zusätzliche neue Indikation eines bereits zugelassenen Psychopharmakons in den meisten Indikationen die Untersuchung im Vergleich zu Placebo verlangt. Die Prüfung gegen Standardpräparate wird nur in Ausnahmefällen (s. u.) als ausreichend für den Beweis der Wirksamkeit angesehen. Der Königsweg der Psychopharmakaprüfung ist also der prospektive, randomisierte, Placebo-kontrollierte, doppelblinde Parallelgruppenvergleich. Bei Prüfung der Akutbehandlung wird in der Regel eine 6- bis 8-wöchige Therapie durchgeführt, bei Langzeitbehandlung (Erhaltungstherapie, Rezidivprophylaxe) ein bis zwei Jahre.
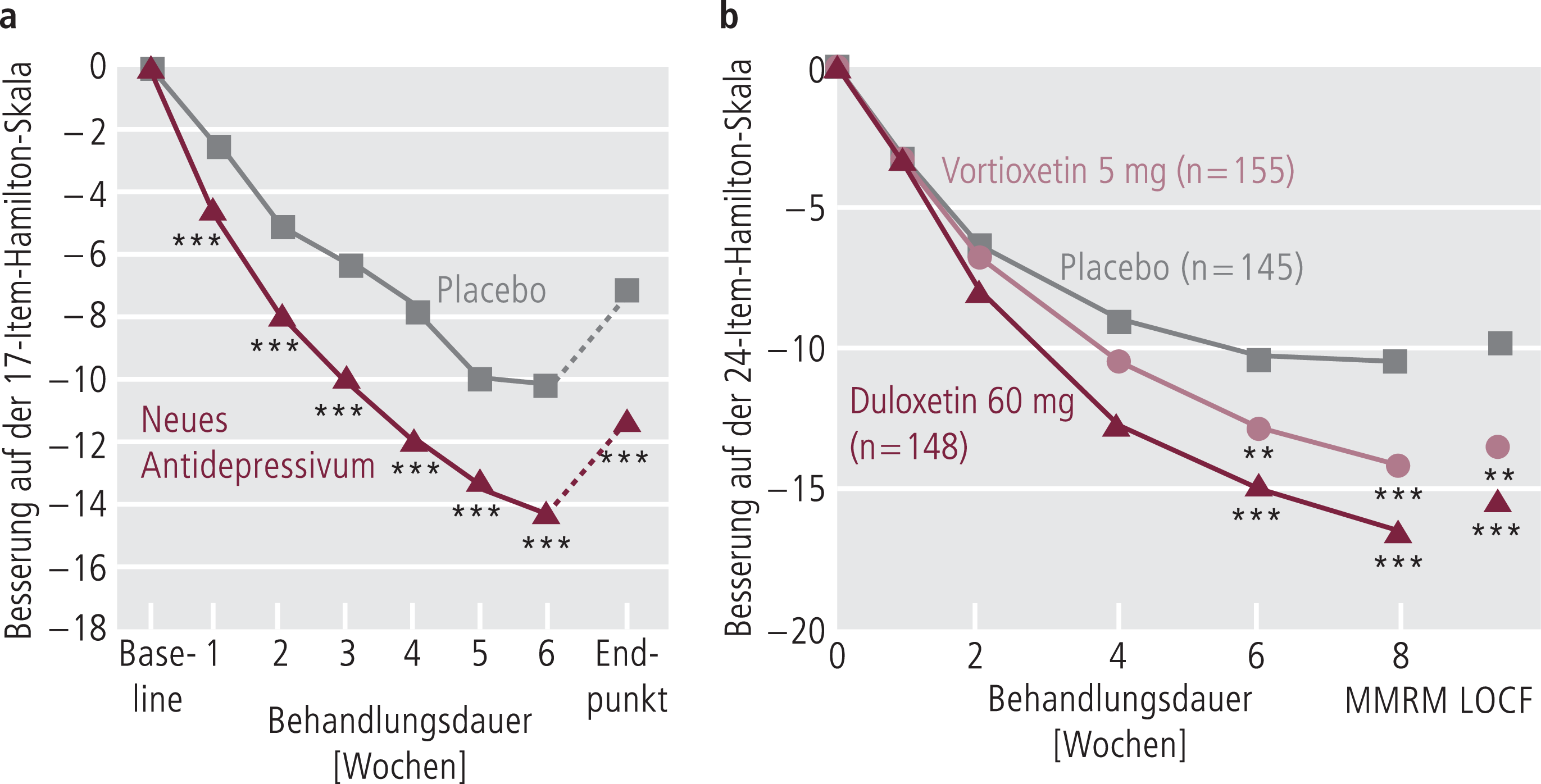
Abb. 5–4a. Mögliches Ergebnis einer prospektiven, randomisierten, Placebo-kontrollierten Antidepressiva-Studie: HAMD-Mittelwertsvergleich. Hier zeigt das neue Antidepressivum eine statistisch signifikant stärkerer Abnahme (***) der depressiven Symptomatik im Vergleich mit Placebo (mod. nach [3]) Abb. 5–4b. Dreiarmige Antidepressiva-Studie zu Vortioxetin (mod. nach [4]); ** p < 0,01; *** p < 0,001; MMRM: mixed model repeat measurement; LOCF: last observation carried forward
Durch Randomisierung wird eine streng zufällige Zuteilung (Münzwurfprinzip, Zufallszahlentabelle u. a.) der Patienten zur Experimentalgruppe und zur Kontrollgruppe und so die Strukturgleichheit beider Gruppen angestrebt. Jeder Patient hat absolut die gleiche Chance, der einen oder anderen Gruppe zugeteilt zu werden. Eine zufällige Zuteilung lässt erwarten, dass störende Einflussgrößen die Ergebnisse nicht verfälschen, da sie im Gruppenvergleich gleichermaßen zu Buche schlagen. Das gilt aber nur für große Stichproben. Gerade bei kleinen Fallzahlen besteht die Gefahr, dass sich die beiden Gruppen trotz Zufallszuteilung hinsichtlich verschiedener Variablen wie psychopathologischer Befund, Erkrankungsdauer usw. unterscheiden. Dieser mangelnden Ausbalancierung muss bei der Auswertung Rechnung getragen werden, um zu vermeiden, dass dadurch bedingte unterschiedliche Resultate fälschlicherweise der therapeutischen Intervention zugeschrieben werden.
Durch Parallelisierung bzw. Stratifizierung (Schichtung) kann man auch bei kleinen Stichproben erreichen, dass sich die relevanten Einflussgrößen auf die beiden Gruppen gleich verteilen. Bei der Parallelisierung werden die Patienten, die sich in bestimmten Variablen ähneln, zu verschiedenen Paaren oder Blöcken zusammengefasst, sodass die Unterschiede zwischen den Beobachtungseinheiten innerhalb eines Blocks gering, aber zwischen den Blöcken relativ groß sind. Die Patienten der beiden Blöcke der Experimentalgruppe und der Kontrollgruppe werden dann nach Zufallsprinzip den Untersuchungsgruppen zugeteilt. Mit diesem Verfahren kann man die Strukturgleichheit beider Gruppen hinsichtlich bestimmter, als relevant angesehener Variablen als gegeben ansehen. Dieses Verfahren ist zwar bei zwei oder drei bekannten und als relevant angesehenen Einflussgrößen noch praktikabel, erreicht aber seine Grenze, wenn hinsichtlich einer größeren Zahl von Einflussgrößen parallelisiert werden soll. In diesen Fällen können komplizierte Verfahren weiterführen.
Neben der so geschaffenen Strukturgleichheit der untersuchten Patientengruppen ist die Beobachtungsgleichheit wesentlich, das heißt, alle Patienten sollen von den gleichen Untersuchern mit den gleichen Verfahren zu gleichen Zeitpunkten beobachtet und beurteilt werden. Zentrales Kriterium in der psychopharmakologischen Wirksamkeitsprüfung ist der psychopathologische Befund. Neben Veränderungen des psychopathologischen Befunds müssen Veränderungen des körperlich-neurologischen Befunds sowie von klinisch oder theoretisch relevanten biochemischen Parametern registriert werden, insbesondere um Nebenwirkungen zu erfassen.
Da sich die einfache klinische Befunderhebung für die psychopharmakologische Forschung als zu undifferenziert erwies, wurden standardisierte Beurteilungsskalen zur quantifizierten Befunddokumentation entwickelt [8]. Geläufig sind beispielsweise die Hamilton Depressionsskala (HAMD), die Montgomery-Åsberg Depression Rating Scale (MADRS), die Brief Psychiatric Rating Scale (BPRS), die Positive and Negative Syndrome Scale (PANSS) u. a. Diese Fremdbeurteilungsskalen werden zur Evaluation von Antidepressiva oder Antipsychotika eingesetzt. Selbstbeurteilungsskalen wie das Beck Depression Inventory (BDI) können ergänzend eingesetzt werden, sind aber als alleinige Dokumentation der Therapieeffekte problematisch und werden von den Zulassungsbehörden als alleinige Erfassung der Wirksamkeit nicht akzeptiert. Wichtig ist zu wissen, dass die verschiedenen Depressionsskalen unterschiedliche Charakteristika haben, die sich unter anderem auch in der Verlaufsdokumentation zeigen (Abb. 5–5).
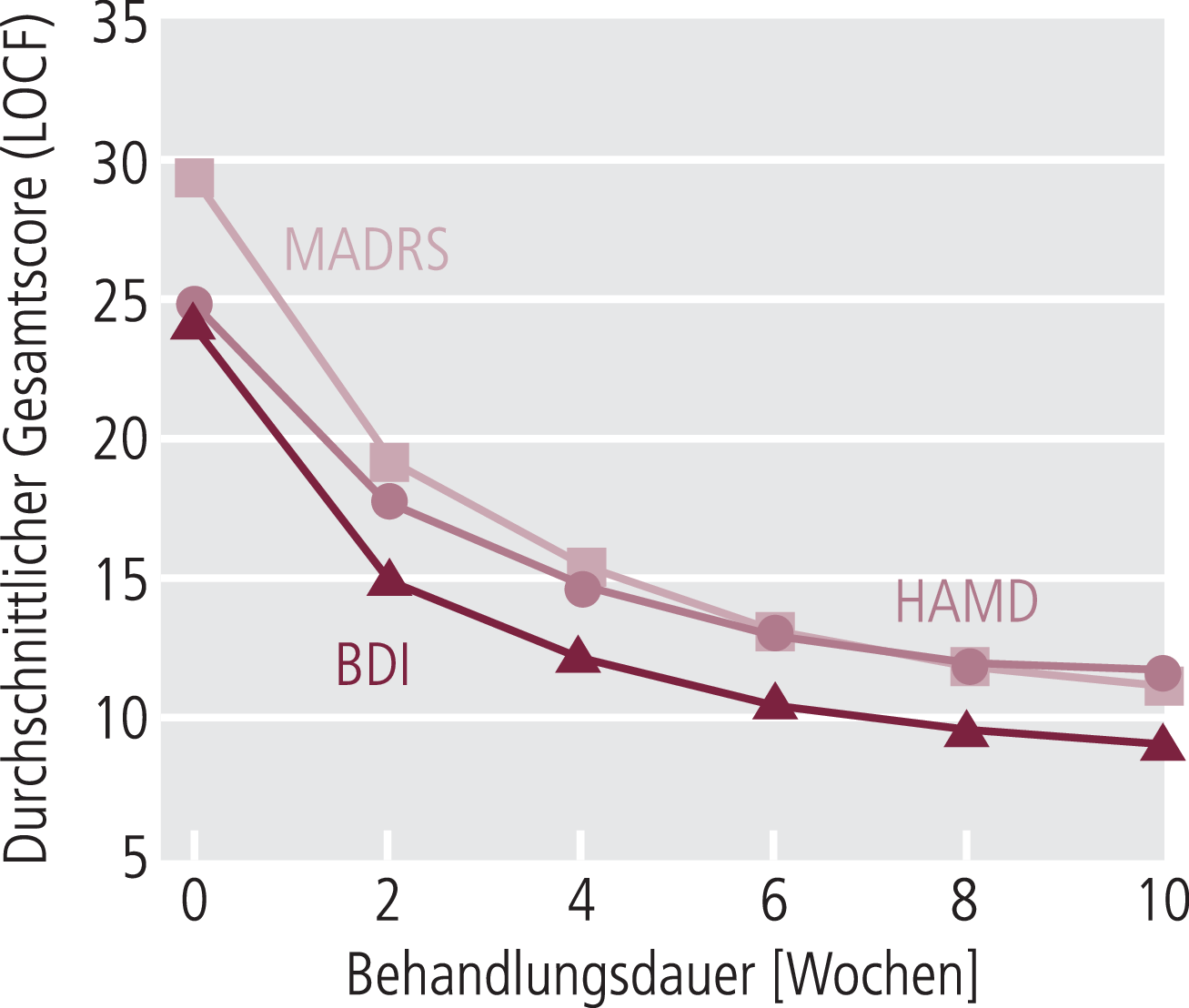
Abb. 5–5. Depressionsverlauf gemessen mit den Depressionsskalen MADRS, HAMD und BDI. Daten aus einer naturalistischen Depressionsstudie an 1000 Patienten. BDI: Beck Depression Inventory; HAMD: Hamilton-Depressionsskala; LOCF: last observation carried forward; MADRS: Montgomery-Åsberg Depression Rating Scale [Möller, bisher unpublizierte Ergebnisse aus dem Forschungsnetzwerk Depression/Suizidalität]
Durch Verblindung – Untersucher und/oder Untersuchte sind nicht informiert, welches Medikament appliziert wird – sollen Erwartungshaltungen des Patienten und des Untersuchers und dadurch ausgelöste Autosuggestions- und Heterosuggestionseffekte, die das Resultat der Studie verfälschen könnten, ausgeschlossen werden. Als ideal und von den Zulassungsbehörden gefordert wird das doppelblinde Vorgehen (weder Untersucher noch Patient sind informiert, welches Medikament gegeben wird) angesehen. Aus pragmatischen Gründen werden in weniger relevanten Studien auch einfach-blinde Verfahren angewendet. Die methodische Forderung der Verblindung wird allgemein akzeptiert, ihre Realisierbarkeit bereitet aber Probleme. Immer wieder gelingt es Untersuchern oder Patienten, an bestimmten Phänomenen (Äußerlichkeiten des Medikaments, physikochemische Eigenschaften, Nebenwirkungen) das Placebo vom Verum zu unterscheiden. In solchen Fällen kann das Versuchsresultat maßgeblich durch Erwartungshaltungen der Untersucher/Patienten beeinflusst werden, die in ihren Auswirkungen sehr schwer abzuschätzen sind.
Grundsätzlich ist es wünschenswert, dass der Wirksamkeitsnachweis in kontrollierten Parallelgruppenvergleichen gegen Placebo erfolgt (Tab. 5–3). Dies wird von der europäischen und der US-amerikanischen Zulassungsbehörde für die meisten Indikationsbereiche gefordert, ist aber aus ethischen Gründen nicht immer möglich [1, 2]. Dann kann die Wirksamkeit eines neuen Psychopharmakons durch Doppelblindvergleich gegen ein Standardpräparat analysiert werden. Das führt aber zu einer Reihe methodischer Probleme, unter anderem dem des Beta-Fehlers bei zu geringer statistischer „Power“. In einem solchen Fall wird fälschlicherweise aus der Tatsache, dass ein Unterschied zwischen zwei Beobachtungsgruppen nicht gefunden werden konnte, auf die Gleichheit der Gruppen geschlossen. Diese Problematik ist bei Prüfungen gegen Standardpräparate nicht mit ausreichender Sicherheit zu umgehen.
Tab. 5–3. Vor- und Nachteile Placebo-kontrollierter und aktiv kontrollierter Studien (mod. nach [5])
|
Studientyp |
Vorteile |
Nachteile |
|
Placebo-kontrollierte Studien |
|
|
|
Rein aktiv kontrollierte Studien |
|
|
Am aussagekräftigsten sind 3-Arm-Studien, in denen die neue Substanz mit Placebo und einem Standardpräparat verglichen wird. Dieser Ansatz wird von der EMA als idealer Forschungsansatz zur Wirksamkeits- und Verträglichkeitsprüfung nahegelegt. Dieser hat unter anderem den Vorteil, dass man die Wirksamkeit nicht nur gegen Placebo, sondern auch gegen ein Standardpräparat vergleichen kann. Auch dient dieser Ansatz dazu, die Assay-Sensitivität einer Stichprobe zu prüfen. Wenn beispielsweise ein experimentell zu testendes neues Antidepressivum in einer Stichprobe depressiver Patienten keine Überlegenheit gegen Placebo zeigt, kann das bedeuten, dass es keine/keine ausreichende antidepressive Wirksamkeit besitzt. Das Ergebnis kann aber auch bedeuten, dass die Stichprobe so geartet ist (zu geringer Ausprägungsgrad der Depressivität, zu hoher Anteil therapierefraktärer Patienten), dass ein ausreichendes Ansprechen auf Antidepressiva nicht zu erwarten ist.
Wenn gleichzeitig neben der Placebo-Gruppe eine Standardpräparat-Gruppe untersucht wird, kann zwischen diesen beiden Möglichkeiten differenziert werden. Wenn auch das Standardpräparat keine Überlegenheit gegen Placebo zeigt („failed study“), liegt offensichtlich eine für Antidepressiva nicht-sensitive Stichprobe vor. Wenn das Standardpräparat Überlegenheit gegenüber Placebo zeigt, nicht aber die zu prüfende neue Substanz („negative study“), dann muss von einer unzureichenden Wirksamkeit der neuen Substanz ausgegangen werden. Sowohl „negative studies“ wie auch „failed studies“ sind relativ häufig, nicht nur in der Prüfung von Antidepressiva, sondern auch in der Prüfung von Antipsychotika und Antimanika. Dies hängt damit zusammen, dass die Unterschiede gegen Placebo in den meisten Studien nicht sehr groß sind, beispielsweise bei Antidepressiva-Studien im Mittelwertsvergleich eine Differenz von 2 bis 3 HAMD-Scorewerten oder im Vergleich der Responder-Raten eine Differenz von etwa 15 % [7]. Während die Placebo-Response unter anderem in den Antidepressiva-Studien in den letzten Jahrzehnten generell zugenommen hat, scheint die Placebo-Verum-Differenz abgenommen zu haben.
Interessenkonflikterklärung
Keine Interessenkonflikte.
Literatur
1. Adam D, Kasper S, Moller HJ, Singer EA, et al. Placebo-controlled trials in major depression are necessary and ethically justifiable: how to improve the communication between researchers and ethical committees. Eur Arch Psychiatry Clin Neurosci 2005;255:258–60.
2. Baldwin D, Broich K, Fritze J, Kasper S, et al. Placebo-controlled studies in depression: necessary, ethical and feasible. Eur Arch Psychiatry Clin Neurosci 2003;253:22–8.
3. Broich K. Klinische Prüfungen mit Antidepressiva und Antipsychotika. Bundesgesundheitsblatt 2005;48:541–7.
4. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol 2012;27:215–23.
5. Möller H-J, Broich K. Prinzipien der Methodik empirischer Forschung in der Psychiatrie. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017:463–90.
6. Möller HJ. Do effectiveness („real world“) studies on antipsychotics tell us the real truth? Eur Arch Psychiatry Clin Neurosci 2008;258:257–70.
7. Möller HJ. Isn’t the efficacy of antidepressants clinically relevant? A critical comment on the results of the metaanalysis by Kirsch et al. 2008. Eur Arch Psychiatry Clin Neurosci 2008;258:451–5.
8. Möller HJ. Standardisierte Befunddiagnostik in der Psychiatrie. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017:577–600.
9. Möller HJ, Broich K. Principle standards and problems regarding proof of efficacy in clinical psychopharmacology. Eur Arch Psychiatry Clin Neurosci 2010;260:3–16.
10. Volz H-P, Kasper S, Möller H-J. Psychopharmakotherapie – klinisch-empirische Grundlagen. In: Möller H-J, Laux G, Kapfhammer H-P (Hrsg.). Psychiatrie, Psychosomatik, Psychotherapie. Berlin, Heidelberg: Springer-Verlag, 2017:795–842.
Herrn Prof. Dr. med. Dipl.-Psych. Gerd Laux zum 70. Geburtstag in großer Dankbarkeit für eine fast lebenslange erfolgreiche Zusammenarbeit vom Erstautor gewidmet.
Prof. Dr. med. Dr. h.c. mult. Hans-Jürgen Möller, Ehemaliger Direktor der Klinik für Psychiatrie und Psychotherapie, Nußbaumstraße 7, 80336 München, E-Mail: hans-juergen.moeller@med.uni-muenchen.de
Prof. Dr. Karl Broich, Präsident des Bundesinstituts für Arzneimittel und Medizinprodukte (BfArM), Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn
Methodology of clinical treatment research on psychopharmaceuticals (Part I): General principles, 4-phase model, design of clinical studies
The paper presents the range of methodological options for clinical treatment research on psychopharmaceuticals. The prospective, randomised, double-blind, placebo-controlled trial, performed as a parallel group comparison, is thereby considered to be the most informative method overall. All other methods have various disadvantages.
Key words: Psychopharmaceutical study, randomised control group study, RCT, placebo-controlled study, proof of efficacy
Psychopharmakotherapie 2018; 25(05):251-258