Jörn Peter Sieb, Stralsund
Die Myasthenia gravis (MG) umfasst eine Gruppe unterschiedlicher Autoimmunerkrankungen, deren gemeinsames Merkmal eine postsynaptische Störung der synaptischen Signalübertragung zwischen Nerv und Skelettmuskel an der motorischen Endplatte ist (Abb. 1) [19, 28]. Klinisches Charakteristikum dieser Erkrankungen ist eine unter Belastung zunehmende oder erst unter Belastung auftretende Muskelschwäche. Es kommt zu einer mehr oder weniger stark fluktuierenden, unterschiedlich rasch (über Tage, Wochen oder Monate) zunehmenden, schmerzlosen, bei Belastung sich akzentuierenden Muskelschwäche. Die Muskelschwäche ist zumeist generalisiert, wobei die stammnahen Muskeln deutlicher betroffen sind als die distalen, oder sie tritt mit einem oropharyngealen oder okulären Schwerpunkt auf. Häufig beginnt die Erkrankung mit Doppelbildern und einer gegebenenfalls auch asymmetrisch ausgeprägten Ptose in den Abendstunden. Manchmal zeigt sich als Initialsymptom eine Beeinträchtigung des Kauens, Schluckens und Sprechens; gerade im Senium entwickelt sich dies manchmal innerhalb von Stunden, sodass dann sogar die Verkennung als Schlaganfall möglich ist. Die Muskelschwäche kann als myasthene Krise mit Ausbildung einer respiratorischen Insuffizienz akut lebensbedrohlich werden [19].
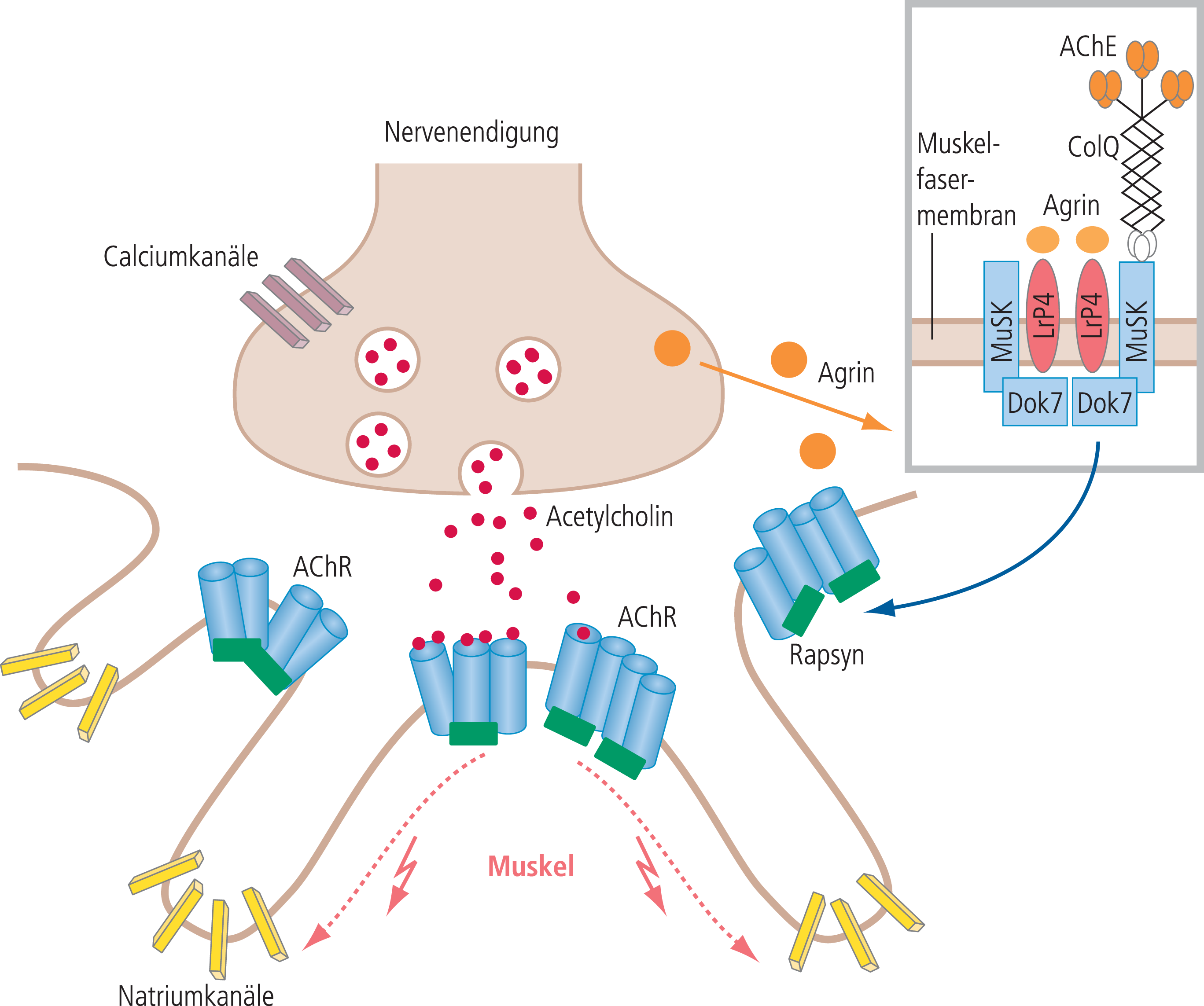
Abb. 1. Schematische Darstellung der neuromuskulären Signalübertragung an der motorischen Endplatte. Präsynaptisch öffnen sich mit der Depolarisation während eines nervalen Aktionspotenzials spannungsgesteuerte Calciumkanäle, durch die Calciumionen in die Nervenendigung einströmen. Calciumionen sind erforderlich für die Ausschüttung der Acetylcholinvesikel. Acetylcholin öffnet postsynaptisch einen Kationen-selektiven Acetylcholinrezeptor-Ionenkanal (AChR). Dies führt zur Depolarisation der Postsynapse und damit gegebenenfalls zur Öffnung von spannungsgesteuerten Natriumkanälen, was ein muskuläres Aktionspotenzial und konsekutiv die Muskelfaserkontraktion auslöst. Der AChR ist ein Pentamer, wobei die beiden Alpha-Untereinheiten über je eine Acetylcholin-Bindungsstelle verfügen. Sind beide Bindungsstellen mit Acetylcholin besetzt, dann öffnet sich der Kationen-selektive Ionenkanal im Millisekundenbereich [19]. Der postsynaptische Faltenapparat ist eine Besonderheit der motorischen Endplatte. Mit Ausbildung des Faltenapparats erhöht sich die postsynaptische Membranoberfläche. Er wirkt elektrophysiologisch als Verstärker. Die über das Protein Rapsyn vernetzten AChR befinden sich in Clustern an der Aufwölbung der synaptischen Falten. Die Ausbildung der AChR-Cluster wird von der Nervenendigung durch den Faktor Agrin induziert. Agrin bindet an LRP4 (Low-density lipoprotein receptor-related protein 4), und über die Aktivierung der muskelspezifischen Tyrosinkinase MuSK, woran Dok7 (Docking protein 7) beteiligt ist, kommt es zur Ausbildung der AChR-Cluster. Die Acetylcholinesterase (AChE) sitzt mit einer Kollagen-ähnlichen Verankerung (ColQ) im synaptischen Spalt [19].
Die Einteilung der Myasthenia gravis erfolgt nach den folgenden Kriterien [28]:
- Verlaufstyp:
- Okulär (bei etwa 20 % aller MG-Patienten)
- Oropharyngeal bzw. generalisiert
- Erkrankungsbeginn:
- Neonatal (passager, durch den transplazentaren Transfer mütterlicher Antikörper intrauterin)
- Vor Beginn der Pubertät
- Bei jüngeren Erwachsenen (in der Literatur uneinheitlich Altersgrenze: meist Erkrankungsbeginn vor dem 50. Geburtstag = „early onset“)
- Bei älteren Erwachsenen (nach dem 50. Geburtstag = „late onset“)
- Antikörperspezifität:
- Anti-AChR (Acetylcholinrezeptor)
- Anti-MuSK (Muskel-spezifische Rezeptor-Tyrosinkinase)
- Anti-LRP4 (Low-density lipoprotein receptor-related protein 4)
- Seronegativ: kein Nachweis krankheitsspezifischer Autoantikörper
- Assoziierte Antikörper Anti-Agrin, Anti-ColQ u. a.
- Thymushistologie
- Normal/atroph
- Thymitis
- Bei 10 bis 20 % Thymom (paraneoplastische Myasthenia gravis)
Nach wie vor ist unklar, was diese Antikörper-vermittelte, T-Zell-abhängige Autoimmunerkrankung auslöst [30]. Bei etwa 80 % aller Fälle von generalisierter Myasthenie finden sich Autoantikörper gegen den Acetylcholinrezeptor (AChR). Bei anderen Myasthenie-Patienten ohne Nachweis von Anti-AChR-Antikörpern beruht die myasthene Schwäche auf einem Autoimmunprozess, der nicht gegen den AChR, sondern gegen andere postsynaptische Zielproteine gerichtet ist. Bei etwa 40 % der Fälle von generalisierter Myasthenie ohne Anti-AChR-Antikörper-Nachweis sind dies Antikörper gegen eine muskelspezifische Tyrosinkinase (MuSK) [23]. An der Anti-MuSK-positiven Myasthenia gravis erkranken zumeist Frauen im mittleren Erwachsenenalter. Typischerweise sind die oropharyngealen Muskeln und der Kopfhalteapparat besonders betroffen. Im Vergleich zur anti-AChR-positiven Myasthenie ist das Risiko myasthener Krisen hoch und therapeutisch wird weniger häufig eine anhaltende Remission erreicht. Auch immunologisch unterscheiden sich beide Myasthenie-Formen. Bei der Anti-AChR-Myasthenie werden Autoantikörper vom IgG1- und IgG3-Typ gebildet, die an der motorischen Endplatte zu einer Komplementaktivierung führen (Abb. 2) [14]. Bei den Anti-MuSK-Antikörpern handelt es sich dagegen um IgG4-Antikörper, die das Komplement nicht aktivieren. Auch gibt es derzeit keinen Anhalt dafür, dass der Thymus an der Entstehung der Anti-MuSK-positiven Myasthenie beteiligt ist [23].
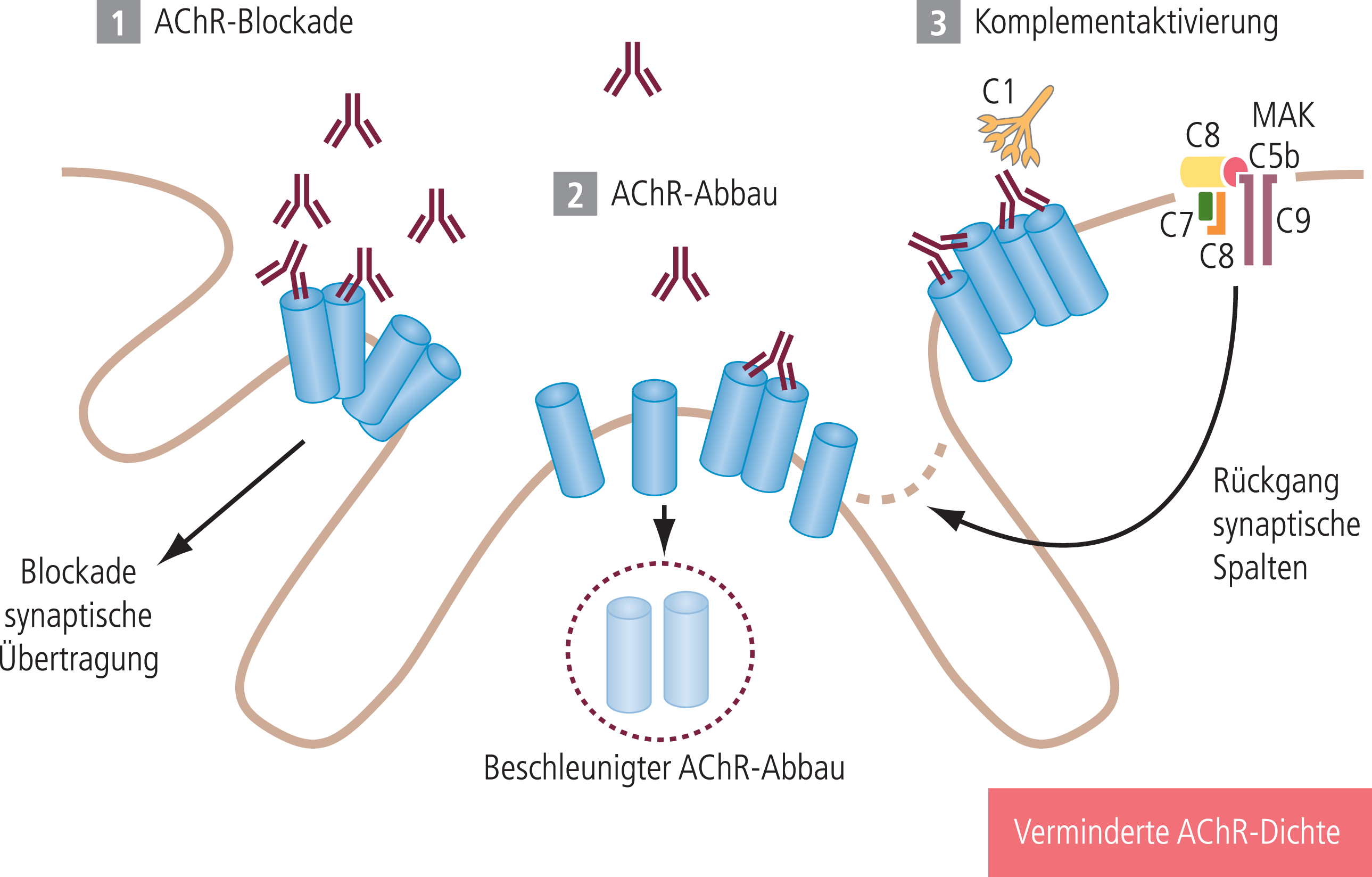
Abb. 2. Zur Störung der neuromuskulären Signalübertragung an der motorischen Endplatte kommt es bei der Myasthenia gravis durch drei unterschiedliche Effekte der Anti-AChR-Antikörper. (1) Ein Teil der Antikörper blockiert direkt die Acetylcholinrezeptor-Ionenkanäle und hemmt somit die synaptische Übertragung. (2) Weitere Antikörper vernetzen die AChR und beschleunigen den AChR-Abbau mit der Folge einer verminderten AChR-Dichte. (3) Die Komplementaktivierung als dritter Antikörpereffekt mit Ausbildung des Membranangriffskomplexes (MAK) führt über eine Destruktion des postsynaptischen Faltenapparats ebenfalls zu einer Reduktion der AChR-Dichte. Die Verminderung der AChR-Dichte beeinträchtigt die neuromuskuläre Signalübertragung. Klinisches Korrelat ist die myasthene Schwäche [30].
Darüber hinaus gibt es immunologisch weitere Myasthenie-Subtypen. Im Jahr 2011 wurde eine Myasthenie mit Antikörpern gegen den Agrin-Rezeptor LRP4 beschrieben [13, 27]. Während der Endplattenreifung führt die Bindung von Agrin, das von der Nervenendigung gebildet wird, an LRP4 als Agrinrezeptor über die Aktivierung der MuSK zur Ausbildung von vernetzten Gruppen der vorab auf der Myotubusoberfläche verstreut liegenden AChR (Abb. 1). Insgesamt wurde inzwischen ein ganzes Spektrum von bei Myasthenie auftretender Autoantikörper beschrieben, wie beispielsweise gegen Agrin [9] oder Cortactin [4], deren Bedeutung jedoch bislang unklar ist, und nach wie vor gibt es Myasthenie-Fälle ohne Nachweis eines spezifischen Autoantikörpers.
Behandlung
Folgende Behandlungsoptionen stehen bei der Myasthenie zur Verfügung [6]:
- Symptomatische Therapie (Verbesserung der neuromuskulären Signalübertragung durch Acetylcholinesterase-Inhibitoren): Pyridostigmin
- Akutbehandlung krisenhafter Verschlechterungen (myasthene Krise): Plasmapherese, Immunadsorption bzw. intravenös Immunglobuline. Die subkutane Gabe von Immunglobulinen ist in Deutschland nicht für die Myasthenie-Behandlung zugelassen [2].
- Immunsuppression: Mittel der 1. Wahl: Azathioprin. Gemäß Leitlinien der Deutschen Gesellschaft für Neurologie sollte bei Behandlungsresistenz insbesondere eine B-Zell-depletierende Behandlung mit Rituximab erwogen werden (s. u.) [6].
- Operativ: Thymektomie zur Verlaufsbeeinflussung mit individueller Indikationsstellung [34] bzw. bei paraneoplastischer Myasthenie zur Thymombehandlung.
- Therapierefraktäre Myasthenia gravis: Eculizumab
Immer ist ein individueller Therapieplan vonnöten, der im Krankheitsverlauf weiter angepasst werden muss. Durch eine konsequente Therapie wird das Risiko myasthener Krisen reduziert und vielfach sind die Erkrankten im Alltag kaum oder gar nicht mehr eingeschränkt. Leider zeigt sich jedoch bei mutmaßlich 10 bis 15 % der Patienten keine ausreichende Wirkung der hergebrachten Therapieoptionen [21].
Zu wichtigen therapeutischen Fragen der Myasthenie-Therapie fehlen nach wie vor Evidenzen aus kontrollierten Studien. Es ist unklar, ob eine frühzeitig im Krankheitsverlauf begonnene und intensive Immunsuppression entsprechend der rheumatologischen „hit hard and early“-Therapiestrategie den Krankheitsverlauf abmildert und das Risiko einer Generalisation reduziert [29]. Erst aktuell wurde durch eine erste prospektive, kontrollierte Studie bestätigt, dass die seit Jahrzehnten praktizierte Thymektomie den Verlauf der nicht-paraneoplastischen, anti-AChR-positiven Myasthenie bei jüngeren Erwachsenen tatsächlich günstig beeinflusst [34]. Dies gilt jedoch nicht für die anti-MuSK-Antikörper-positive Myasthenia gravis, bei der eine Thymektomie zur Beeinflussung des Krankheitsverlaufs derzeit nicht empfohlen wird [23]. Nach wie vor werden in der Myasthenie-Behandlung die hergebrachten, unselektiven Immunsuppressiva, wie Azathioprin, eingesetzt, während bei anderen immunologischen Erkrankungen, wie der multiplen Sklerose und der rheumatoiden Arthritis, die Forschungsaktivität aufgrund deren Häufigkeit deutlich intensiver ist und der Wechsel zu immer selektiver in den Krankheitsprozess eingreifenden Therapieoptionen bereits gelungen ist.
Symptomatische Therapie: Acetylcholinesterase-Inhibitoren
Zumeist wird das seit mehr als 60 Jahren verfügbare Pyridostigminbromid zur Hemmung der Acetylcholinesterase (AChE) eingesetzt. AChE-Inhibitoren wirken symptomatisch. Bei sehr hohen Tagesdosen besteht das Risiko einer Intoxikation („cholinerge Krise“), sodass nur ausnahmsweise mit mehr als 600 mg Pyridostigmin täglich therapiert werden sollte. Zumeist werden AChE-Inhibitoren zusätzlich zu einer Immunsuppression eingesetzt. Eine Monotherapie empfiehlt sich nur bei Patienten mit einem langfristig überaus gutartigen Krankheitsverlauf. Nicht wenige Patienten klagen unter Pyridostigmin über ein störend vermehrtes Schwitzen, Muskelkrämpfe, einen vermehrten Harndrang oder eine Diarrhö. Es kann zu einer relevanten Bradykardie führen und die Bronchialsekretion steigern [19].
Immunsuppression
Ziel der Immunsuppression ist es, das Risiko myasthener Krisen als lebensbedrohliche Akzentuierung der Erkrankung zu mindern und den Krankheitsverlauf günstig zu beeinflussen. Das bereits 1967 in die Myasthenie-Therapie eingeführte Azathioprin ist weiterhin das einzige in Deutschland ausdrücklich für die Myasthenie-Behandlung zugelassene Immunsuppressivum [5]. Es wird in einer Tagesdosis von 1,5 bis 3 mg/kg Körpergewicht eingesetzt. Ein direkter Zusammenhang der durch Azathioprin induzierten Blutbildveränderungen mit dem Therapieeffekt ist nicht gesichert, trotzdem wird zumeist eine Leukozytenzahl um 4000/mm3 mit einer Lymphozytenzahl von 800 bis 1000/mm3 angestrebt. Der Effekt von Azathioprin setzt erst verzögert nach bis zu zwölf Monaten allmählich ein. Vielfach muss die Zeit bis dahin mit Prednisolon überbrückt werden [19].
Glucocorticoide wirken rasch und zuverlässig. Eine Dauertherapie verbietet sich jedoch aufgrund der massiven Begleitwirkungen. Wird direkt mit einer hohen Steroid-Tagesdosis behandelt, beispielsweise mit 60 bis 100 mg Prednisolon täglich, kann dies in den ersten Tagen der Therapie eine myasthene Schwäche erheblich akzentuieren. Eine allmähliche Eindosierung ist deshalb häufig individuell günstiger trotz des dann verzögerten Wirkbeginns. Bei der okulären Myasthenie führt Prednisolon häufig bereits in niedriger Dosierung für einen begrenzten Zeitraum, beispielsweise 20 mg für zwei bis vier Wochen, zu einer signifikanten Verbesserung.
Die Azathioprin-Therapie ist gelegentlich nicht ausreichend wirksam oder verträglich, sodass auf ein anderes Immunsuppressivum ausgewichen werden muss:
Mycophenolat mofetil
Die Therapie ist einfach durchführbar und in Tagesdosen von zumeist 1 bis 2,5 g allgemein gut verträglich. Mycophenolat mofetil hat vom Gemeinsamen Bundesausschuss im Rahmen eines Off-Label-Verfahrens 2012 ein positives Votum für die Behandlung der generalisierten Myasthenia gravis mit unzureichender Wirksamkeit bzw. Verträglichkeit von Azathioprin erhalten [10].
Ciclosporin, Tacrolimus
Beide Immunsuppressiva finden in der Transplantationsmedizin und in der Behandlung von Autoimmunerkrankungen Anwendung. Es besteht keine Strukturähnlichkeit: Ciclosporin ist ein elf Aminosäuren umfassendes Peptid mit einer D-Aminosäure, während Tacrolimus eine Makrolid-Lacton-Struktur aufweist. Beide Substanzen greifen über die Inhibition von Calcineurin in die Calcium-abhängige Signaltransduktion bei der T-Zellaktivierung ein. Zum Nebenwirkungsspektrum beider Immunsuppressiva gehören die Nephrotoxizität, das Risiko einer Enzephalopathie, eine Hypertonie und eine Tremorinduktion. In einer randomisierten Studie mit 80 unter einer Glucocorticoid-Therapie stabilen Patienten mit generalisierter Myasthenie konnte ein zusätzlicher steroidsparender Effekt durch Tacrolimus nach 28 Wochen nicht erreicht werden, wobei dies möglicherweise auf die zu kurze Dauer dieser Studie zurückzuführen ist [36].
Methotrexat, Cyclophosphamid
Methotrexat wird gelegentlich als Ausweichpräparat bei Azathioprin-Unverträglichkeit empfohlen. In einer aktuell publizierten, kontrolliert durchgeführten Studie hat es sich aber als nicht wirksam erwiesen [25]. Wegen der erheblichen Toxizität sollte Cyclophosphamid nur eingesetzt werden, wenn ansonsten keine ausreichende Stabilisierung erreicht werden konnte. Hochdosiertes Cyclophosphamid ist eine Option bei therapierefraktärer Myasthenie [8].
Rituximab
Gemäß Leitlinien der Deutschen Gesellschaft für Neurologie kann bei Therapieresistenz insbesondere eine B-Zell-depletierende Behandlung mit Rituximab als Therapieeskalation erfolgreich sein [6]. Dies ist ein chimärer Antikörper, der spezifisch an das Transmembran-Antigen CD20 bindet. CD20 ist auf Prä-B- und reifen B-Lymphozyten, aber nicht auf Plasmazellen lokalisiert. Die Gabe von Rituximab führt zu einer über Monate anhaltenden B-Zell-Depletion. Es ist für die Therapie bestimmter Lymphome, der chronischen, lymphatischen Leukämie (CLL), der schweren rheumatoiden Arthritis in Kombination mit Methotrexat und der Wegenerschen Granulomatose zugelassen. Rituximab hat sich insbesondere bei der anti-MuSK-positiven Myasthenie als sehr wirksam erwiesen [12, 32]. Eine Rituximab-Therapie geht mit dem Risiko einer progressiven, multifokalen Leukenzephalopathie als lebensbedrohliche Komplikation einher. Dies ist die opportunistische Aktivierung einer JC-Virus-Infektion im Zentralnervensystem, was inzwischen auch im Rahmen der Myasthenie-Therapie beobachtet wurde [17].
Mit dem aktuell für die Multiple-Sklerose-Therapie zugelassenen humanisierten Anti-CD20-Antikörper Ocrelizumab gibt es zumindest keine bislang publizierten Erfahrungen bei der Myasthenie.
Eculizumab
Im Herbst 2017 wurde Eculizumab (Soliris®) zur Behandlung der therapierefraktären generalisierten, anti-AChR-positiven Myasthenia gravis im Erwachsenenalter in der Europäischen Union zugelassen. Als therapierefraktär gilt gemäß der Fachinformation eine Myasthenie, wenn Folgendes erfüllt ist [1]:
- Seit mindestens einem Jahr erfolglose Behandlung mit zwei oder mehr immunsuppressiven Therapien (entweder in Kombination oder als Monotherapie), das heißt Patienten, deren Alltagsaktivitäten trotz immunsuppressiver Therapien weiterhin eingeschränkt waren.
Oder
- Erfolglose Behandlung mit mindestens einer immunsuppressiven Therapie und Notwendigkeit eines dauerhaften Plasmaaustauschs oder von intravenösen Immunglobulinen zur Kontrolle der Symptome, das heißt, die Patienten benötigten mindestens alle drei Monate über die letzten zwölf Monate regelmäßig Plasmaaustausch oder intravenöse Gabe von Immunglobulinen zur Behandlung von Muskelschwäche.
Parallel erfolgten die Zulassungen in den USA und in Japan für die Myasthenie-Behandlung, jedoch mit etwas von der europäischen Zulassung abweichenden Definitionen des Indikationsbereichs. In den USA wurde Eculizumab für die Behandlung der generalisierten, anti-AChR-positiven Myasthenie zugelassen und in Japan für Patienten mit generalisierter, anti-AChR-positiver Myasthenie, deren Symptome nicht ausreichend durch die Gabe von hochdosierten Immunglobulinen oder Apheresen beeinflusst werden kann [7]. Bereits seit einigen Jahren ist Eculizumab für die Behandlung zweier anderer Komplement-vermittelter Erkrankungen zugelassen, und zwar für die Therapie der paroxysmalen, nächtlichen Hämoglobinurie und des atypischen hämolytisch-urämischen Syndroms. Die Ergebnisse der PREVENT-Studie zum Einsatz von Eculizumab bei den Neuromyelitis-optica-Spektrumerkrankungen liegen derzeit noch nicht vor [26].
Substanz und Wirkungsmechanismus
Eculizumab ist ein rekombinanter, humanisierter, monoklonaler IgG2/4k-Antikörper, der die Aktivierung des terminalen Komplements verhindert [7]. Er bindet mit hoher Affinität und Spezifität an das humane Komplementprotein C5 und verhindert dadurch die Spaltung von C5 in die Komplementkomponenten C5a und C5b. C5a wirkt chemotaktisch auf proinflammatorische Zellen und C5b ist an der Ausbildung des Membranangriffkomplexes beteiligt. Insgesamt wird die Endplattendestruktion durch den myasthenen Immunprozesses verhindert (Abb. 2). Die dauerhafte Verabreichung von Eculizumab führt zu einer sofortigen, vollständigen und nachhaltigen Hemmung der terminalen Komplementaktivität. Erhalten bleiben aber die frühen Komponenten der Komplementaktivierung, die wichtig bei der Elimination von Immunkomplexen und der Opsonierung von Fremdzellen sind. Opsonierung ist der Mechanismus, durch den die Oberfläche von Fremdzellen mit Antikörpern und Faktoren des Komplementsystems bedeckt wird. Nach der Opsonierung können die Fremdzellen von phagozytierenden Zellen des angeborenen Immunsystems eliminiert werden. Das wichtigste Opsonin ist C3b.
Studiendaten
Nach einer Phase-II-Studie wurde die Therapiesicherheit und Wirksamkeit von Eculizumab bei der anti-AChR-positiven, therapierefraktären und generalisierten Myasthenie in der REGAIN-Studie untersucht [15, 16, 33]. In dieser multizentrischen, randomisierten, doppelblinden und Placebo-kontrollierten Phase-III-Studie wurden 62 Myasthenie-Patienten mit Eculizumab und 63 Patienten mit Placebo behandelt, und zwar als Zusatztherapie zur laufenden Basistherapie. Zu den Ausschlusskriterien gehörten
- Thymusneoplasma in der Vorgeschichte
- Thymektomie in den letzten zwölf Monaten
- Okuläre Myasthenie (Stadium I nach den Kriterien der Myasthenia Gravis Foundation of America [MGFA]) bzw. myasthene Krise (MGFA-Stadium V)
- i. v. Gabe von Immunglobulinen bzw. Apherese-Therapie in den letzten vier Wochen
- Rituximab-Gabe während der letzten sechs Monate
Die 26-wöchige Behandlungsphase umfasste die wöchentliche i. v. Gabe von 900 mg Eculizumab während der ersten vier Wochen. Ab der fünften Woche wurden dann in zweiwöchentlichen Abständen 1200 mg Eculizumab i. v. verabreicht. Die Placebo-Gabe erfolgte entsprechend diesem Schema. Der Therapieeffekt wurde wiederholt mit den folgenden Skalen erfasst:
- Myasthenia Gravis-Activities of Daily Living (MG-ADL): ein acht Punkte umfassender Fragenkatalog, mit dem die Beeinträchtigung im täglichen Leben durch den Patienten erfasst wird
- Quantitative Myasthenia Gravis (QMG) Score: standardisierte Erfassung der Muskelkraft durch den Arzt
- Myasthenia Gravis Composite (MGC) Score: Patient und Arzt erfassen hiermit die Beeinträchtigung durch die Myasthenie
- 15-Item Myasthenia Gravis Quality of Life Questionnaire (MG-QoL15): Erfassung der Lebensqualität durch den Patienten mit 15 Kategorien
Auf der Website der MGFA findet sich eine umfassende Darstellung dieser Skalen mit Hinweisen auf die erfolgten Validierungsstudien [24]. Von den initial 125 Studienteilnehmern schlossen 118 (94 %) die 26-wöchige Behandlungsphase ab. Zu schwerwiegenden Therapiekomplikationen, zum Beispiel einer Meningokokken-Infektion, kam es während der REGAIN-Studie nicht. 117 (94 %) Patienten wurden anschließend in die offene, multizentrische Verlängerungsstudie zur Untersuchung der langfristigen Wirksamkeit und Sicherheit aufgenommen, bei der alle Teilnehmer Eculizumab erhalten.
Der primäre Studienendpunkt – die mit dem MG-ADL erfasste Verbesserung der Aktivitäten des täglichen Lebens nach 26 Behandlungswochen – erreichte keine Signifikanz (p = 0,0698). Die Herstellerfirma Alexion war mit der amerikanischen Zulassungsbehörde FDA bei der Studienplanung übereingekommen, dass der Worst-Rank ANCOVA (Analysis of covariance, Kovarianzanalyse) als statischer Test zur Datenanalyse eingesetzt werden sollte. Dabei erwies es sich als Problem, dass Studienabbrecher, beispielsweise durch das Auftreten eines Prostatakarzinoms, das statistisch schlechteste Ranking erhielten, und zwar gegebenenfalls trotz einer im Studienverlauf bereits eingetretenen Verbesserung im MG-ADL. Im QMG und im MG-QoL15 zeigte sich auch im Worst-Rank ANCOVA eine signifikante Verbesserung, die sich insbesondere in den ersten zwölf Therapiewochen entwickelte. Patienten der Eculizumab-Gruppe hatten gegenüber denjenigen mit Placebo deutlich seltener klinische Verschlechterungen und benötigten seltener eine Therapieintensivierung mit der hochdosierten Gabe eines Glucocorticoids, i. v. Gabe von Immunglobulinen beziehungsweise eine Apherese-Behandlung [16].
Therapierisiken
Eine Eculizumab-Therapie bedingt die Gefahr von Meningokokken-Infektionen. Das Risiko einer Meningokokken-Infektion erhöht sich durch Eculizumab um den Faktor von 1000 bis 2000 [22]. In den USA wurden 16 Meningokokken-Infektionen unter Eculizumab identifiziert, die im Zeitraum von 2008 bis 2016 auftraten. Überwiegend bestand eine Meningokokken-Sepsis ohne Zeichen einer Meningitis. Eine dieser 16 Infektionen verlief tödlich. Elf dieser Infektionen waren durch nicht genauer bestimmbare Neisseria-meningitidis-Stämme verursacht und 14 Patienten hatten vorab zumindest einmal einen Meningokokken-Impfstoff erhalten.
Bei den Meningokokken werden nach der Zusammensetzung der Kapselpolysaccharide zwölf Serogruppen unterschieden (A, B, C, X, Y, Z, E, W, H, I, K, L). Eine Impfung gegen sämtliche Meningokokken-Stämme steht nicht zur Verfügung. Es gibt Impfstoffe gegen die Serogruppen A, C, Y, W 135 (in dieser Kombination als quadrivalenter Konjugatimpfstoff) und B. Gemäß Fachinformation ist eine Meningokokken-Impfung Voraussetzung für eine Eculizumab-Therapie, um Infektionen mit den besonders häufig pathogenen Meningokokken-Serogruppen entgegenzuwirken [1]. Liegt bei Beginn der Eculizumab-Therapie die Meningokokken-Impfung weniger als 14 Tage zurück, dann ist gemäß Fachinformation zunächst eine Antibiotika-Prophylaxe erforderlich.
Da durch die derzeit verfügbaren Meningokokken-Impfstoffe jedoch kein sicherer Schutz erreicht wird, wird teilweise eine Penicillin-Prophylaxe über den gesamten Zeitraum der Eculizumab-Therapie, also gegebenenfalls lebenslang, empfohlen [22]. Essenziell ist das frühzeitige Erkennen einer Meningokokken-Infektion. In der Fachinformation wird gefordert, dass die behandelnden Ärzte die Patienten über Nutzen und Risiken der Eculizumab-Therapie aufklären und ihnen die Soliris®-Informationsbroschüre zusammen mit einer Patientenkarte aushändigen [1].
Therapiedauer
Die Daten der REGAIN-Studie sprechen dafür, dass in der Regel innerhalb der ersten zwölf Therapiewochen mit Eculizumab ein klinisches Ansprechen erreicht wird [16]. Wenn sich nach zwölf Wochen kein therapeutischer Nutzen zeigt, sollte erwogen werden, die Behandlung abzubrechen. Zur Dokumentation des Therapieeffekts bieten sich die in der REGAIN-Studie eingesetzten Myasthenie-Scores an. Sinnvoll erscheint es, zumindest vor Therapiebeginn und dann monatlich den Behandlungseffekt mit dem MG-ADL und dem QMG-Score zu erfassen. Unklar ist, wie lange die Eculizumab-Therapie fortgeführt werden muss. Patienten mit einer schwer verlaufenden Myasthenie sind nicht selten lebenslang auf eine Immunsuppression angewiesen. Trotzdem erscheint es als notwendig, in regelmäßigen Abständen, beispielsweise jährlich, die Notwendigkeit der Eculizumab-Therapie durch Absetzversuche zu überprüfen. Insgesamt fehlen jedoch bislang Langzeiterfahrungen zum Einsatz von Eculizumab bei der Myasthenie.
Kritische Wertung
Eculizumab ist eine aussichtsreiche Therapieoption für die kleine Gruppe der Patienten mit therapierefraktärer, generalisierter anti-AChR-positiver Myasthenia gravis – also Patienten, denen bislang trotz aller Therapieanstrengungen nicht ausreichend geholfen werden konnte [21]. Teilweise kommt es durch Eculizumab sehr rasch innerhalb weniger Wochen zu einer maßgeblichen Besserung. Gerade für Myasthenie-Patienten in der Situation der schwer verlaufenden, myasthenen Krise ist Eculizumab eine aussichtsreiche Therapieoption [35].
Die Zahl der bislang mit Eculizumab behandelten Patienten mit Myasthenia gravis oder einer anderen Autoimmunerkrankung ist international noch klein. Folglich kann das Therapierisiko derzeit nicht abschließend beurteilt werden. Zwar blieben in der REGAIN-Studie erhebliche Komplikationen aus, jedoch ist es unter den weltweit bislang nur wenigen mit Eculizumab behandelten Myasthenie-Patienten zumindest zu einem Todesfall nach einjähriger Therapie durch Multiorganversagen im Rahmen einer Zytomegalie-Virus-assoziierten Hepatitis mit hämophagozytischer Lymphohistiozytose als hyperinflammatorisches Syndrom gekommen [16, 31]. Dies sei als „possibly related to study medication“ eingestuft worden (E-Mail-Kommunikation an den Autor: Alexion, Medical Information, USA, 05.04.2018). Die Hemmung des Komplementsystems durch Eculizumab bedingt ein um den Faktor 1000 bis 2000 erhöhtes Risiko für Meningokokken-Infektionen. Darüber hinaus wurde über eine schwerwiegende Pseudomonas-aeruginosa-Infektion unter Eculizumab berichtet [18, 20, 22].
In der REGAIN-Studie war der Einsatz von Rituximab in den letzten sechs Monaten vorab ein Ausschlusskriterium. Vielfach wird heute Rituximab außerhalb der Zulassung bei ansonsten nicht ausreichend therapeutisch beeinflussbarer, generalisierter Myasthenia gravis im Rahmen einer individuellen Therapieeskalation eingesetzt [32]. Rituximab ist ein chimärer Anti-CD20-Antikörper, der zu einer B-Zell-Depletion führt. Es ist ungewiss, inwieweit der duale Eingriff in das Immunsystem durch eine Kombination von Eculizumab mit Rituximab mit einem noch akzeptablen Therapierisiko einhergehen würde.
Nur ein Teil der mit Eculizumab behandelten Myasthenie-Patienten zeigt eine signifikante Besserung. Bei immerhin 30 bis 40 % der Behandelten bleibt ein günstiger Therapieeffekt aus. Folglich wird nach den ersten zwölf Wochen der Eculizumab-Therapie der Therapieeffekt überprüft und, sofern dieser ausgeblieben ist, die Behandlung abgebrochen. Unklar ist, was dieses individuell unterschiedliche Ansprechen auf Eculizumab bedingt. Insgesamt ist der Langzeiterfolg der Eculizumab-Therapie derzeit noch nicht ausreichend belegt, sodass individuell vor Anwendung zunächst alle gängigen Therapieoptionen konsequent eingesetzt werden sollten, wozu sicher auch Rituximab trotz der fehlenden Zulassung für die Myasthenie-Therapie gehört [11]. Für die therapierefraktäre Myasthenie werden weitere Therapieoptionen geprüft, wie der Einsatz des die B-Zell-Proliferation hemmenden Antikörpers Belimumab und die autologe Stammzelltransplantation [3, 21, 28].
Die Kosten einer Eculizumab-Therapie sind immens und bemessen sich auf mehrere 100 000 Euro jährlich. Die zunehmende Anzahl dieser extrem teuren Behandlungsoptionen gerade für seltene neurologische Erkrankungen belastet das Gesundheitssystem finanziell zunehmend. Es wird sich zeigen, inwieweit die krankheitsbedingt in ihrer Mobilität eingeschränkten Myasthenie-Patienten, die fern von einer Metropolregion leben, unter den Strukturbedingungen des deutschen Gesundheitssystems Zugang zu dieser aufwendigen Therapie mit zunächst im wöchentlichen und dann zweiwöchentlichen Rhythmus erfolgenden i. v. Infusionen erhalten werden.
Interessenkonflikterklärung
JPS hat Honorare für die Beratung oder Teilnahme an einem Expertenbeirat sowie für Vorträge, Stellungnahmen oder Artikel von Alexion Pharma Germany GmbH erhalten.
Literatur
1. Alexion Europe SAS. Fachinformation Soliris®. www.fachinfo.de/suche/fi/010559 (Zugriff am 04.03.2018).
2. Beecher G, Anderson D, Siddiqi ZA. Subcutaneous immunoglobulin in myasthenia gravis exacerbation. A prospective, open-label trial. Neurology 2017;89:1135–41.
3. Bryant A, Atkins H, Pringle CE, Allan D, et al. Myasthenia gravis treated with autologous hematopoietic stem cell transplantation. JAMA Neurol 2016;73:652–8.
4. Cortes-Vicente E, Gallardo E, Martinez MA, Diaz-Manera J, et al. Clinical characteristics of patients with double-seronegative myasthenia gravis and antibodies to cortactin. JAMA Neurol 2016;73:1099–104.
5. Delwaide PJ, Salmon J, Van CH. Premiers essais de traitement de la myasthénie par azathioprine. Acta Neurol Psychiatr Belg 1967;67:701–12.
6. Deutsche Gesellschaft für Neurologie. Diagnostik und Therapie der Myasthenia gravis und des Lambert-Eaton-Syndroms. https://www.dgn.org/leitlinien/3005-ll-68-ll-diagnostik-und-therapie-der-myasthenia-gravis-und-des-lambert-eaton-syndroms (Zugriff am 01.05.2018).
7. Dhillon S. Eculizumab: A Review in generalized myasthenia gravis. Drugs 2018;78:367–76.
8. Drachman DB, Jones RJ, Brodsky RA. Treatment of refractory myasthenia: „Rebooting“ with high-dose cyclophosphamide. Ann Neurol 2003;53:29–34.
9. Gasperi C, Melms A, Schoser B, Zhang Y, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014;82:1976–83.
10. Gemeinsamer Bundesausschuss. Arzneimittel-Richtlinie/Anlage VI: Off-Label-Use Mycophenolat Mofetil bei Myasthenia gravis. https://www.g-ba.de/informationen/beschluesse/1817 (Zugriff am 07.03.2018).
11. Gilhus NE. Eculizumab: a treatment option for mysthenia gravis? Lancet Neurol 2017;16:947–8.
12. Hehir MK, Hobson-Webb LD, Benatar M, Barnett C, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: Multicenter blinded prospective review. Neurology 2017;89:1069–77.
13. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011;69:418–22.
14. Howard JF, Jr. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann N Y Acad Sci 2018;1412:113–28.
15. Howard JF, Jr., Barohn RJ, Cutter GR, Freimer M, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 2013;48:76–84.
16. Howard JF, Jr., Utsugisawa K, Benatar M, Murai H, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 2017;16:976–86.
17. Kanth KM, Solorzano GE, Goldman MD. PML in a patient with myasthenia gravis treated with multiple immunosuppressing agents. Neurol Clin Pract 2016;6:e17–9.
18. Kawakami T, Nakazawa H, Kurasawa Y, Sakai H, et al. Severe infection of pseudomonas aeruginosa during eculizumab therapy for paroxysmal nocturnal hemoglobinuria. Intern Med 2018;57:127–30.
19. Köhler W, Sieb JP. Myasthenia gravis. 4. Auflage. Bremen: UNI-MED, 2012.
20. Lebel E, Trahtemberg U, Block C, Zelig O, et al. Post-eculizumab meningococcaemia in vaccinated patients. Clin Microbiol Infect 2018;24:89–90.
21. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord 2018;11:1756285617749134.
22. McNamara LA, Topaz N, Wang X, Hariri S, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. Am J Transplant 2017;17:2481–4.
23. Morren J, Li Y. Myasthenia gravis with muscle-specific tyrosine kinase (MuSK) antibodies – A narrative review. Muscle Nerve 2018; doi: 10.1002/mus.26107.
24. Myasthenia Gravis Foundation of America – MGFA. Schulungsmaterial. http://www.myasthenia.org/HealthProfessionals/EducationalMaterials.aspx (Zugriff am 04.03.2018).
25. Pasnoor M, He J, Herbelin L, Burns TM, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology 2016;87:57–64.
26. Paul F, Murphy O, Pardo S, Levy M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs 2018;27:265–71.
27. Pevzner A, Schoser B, Peters K, Cosma NC, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012;259:427–35.
28. Sieb JP. Myasthenia gravis: an update for the clinician. Clin Exp Immunol 2014;175:408–18.
29. Silvestri NJ, Wolfe GI. Start high, then go low: An effective strategy in the treatment of myasthenia gravis. Muscle Nerve 2017;55:773–4.
30. Sommer N, Tackenberg B, Hohlfeld R. The immunopathogenesis of myasthenia gravis. Handb Clin Neurol 2008;91:169–212.
31. Talan J. Eculizumab found safe and effective for myasthenia gravis. Neurology Today 2018;18:27–8.
32. Tandan R, Hehir MK, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017;56:185–96.
33. Vissing J, O‘Brien F, Wang JJ, Howard JF, Jr. Correlation between MG-ADL and QMG assessments of anti-acetylcholine receptor antibody-positive refractory generalized myasthenia gravis in the phase 3 REGAIN study. Muscle Nerve 2018; doi: 10.1002/mus.26152.
34. Wolfe GI, Kaminski HJ, Aban IB, Minisman G, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med 2016;375:511–22.
35. Yeo CJJ, Pleitez MY. Eculizumab in refractory myasthenic crisis. Muscle Nerve 2018;58:E13–5.
36. Yoshikawa H, Kiuchi T, Saida T, Takamori M. Randomised, double-blind, placebo-controlled study of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry 2011;82:970–7.
Prof. Dr. med. Jörn Peter Sieb, Neurologische Klinik, HELIOS Hanseklinikum Stralsund, Große Parower Straße 47–53, 18435 Stralsund, E-Mail: joern-peter.sieb@helios-gesundheit.de
Eculizumab: A new treatment option for refractory myasthenia gravis
Eculizumab is a new treatment option for therapy-refractory, generalized, anti-acetylcholine receptor-antibody (AChR)-positive myasthenia gravis in adults. It has been available since autumn 2017 for this indication.
Eculizumab is a recombinant humanized monoclonal antibody that prevents activation of the terminal complement. It binds with high affinity and specificity to the human complement protein C5. Eculizumab prevents endplate destruction by complement activation as part of the immune process in anti-AChR-positive myasthenia gravis.
Data from the REGAIN approval study suggest that clinical response is typically achieved within the first 12 weeks of therapy with eculizumab. However, some of the treated patients show no positive therapeutic effect. According to data from the United States, the eculizumab inhibition of the complement system causes a 1000- to 2000-fold increased risk of meningococcal infections. The therapy costs are in the range of several 100,000 Euros annually.
Key words: Acetylcholine, endplate, complement, neuromuscular transmission, rituximab
Psychopharmakotherapie 2018; 25(05):227-233