Gudrun Hefner, Friedrichsdorf, Gerd Laux, Soyen/Waldkraiburg/München, Pierre Baumann, Prilly-Lausanne, Niels Bergemann, Bad Mergentheim, Hans-Willi Clement, Freiburg, Andreas Conca, Bolzano, Jürgen Deckert, Würzburg, Katharina Domschke, Freiburg, Gabriel Eckermann, Kaufbeuren, Karin Egberts, Würzburg, Manfred Gerlach, Würzburg, Christine Greiner, Bonn, Gerhard Gründer, Mannheim, Ekkehard Haen, Regensburg, Ursula Havemann-Reinecke, Göttingen, Renate Helmer, Bielefeld, Ger Janssen, Mönchengladbach, Eveline Jaquenoud, Königsfelden, Thomas Messer, Pfaffenhofen, Rainald Mössner, Tübingen, Matthias J. Müller, Berlin, Michael Paulzen, Aachen, Bruno Pfuhlmann, Dresden, Peter Riederer, Würzburg, Alois Saria, Innsbruck, Bernd Schoppek, München-Haar, Georgios Schoretsanitis, Bern, Markus J. Schwarz, München, Margarethe Silva Gracia, Regensburg, Benedikt Stegmann, Regensburg, Werner Steimer, München, Julia C. Stingl, Bonn, Manfred Uhr, München, Sven Ulrich, Berlin, Stefan Unterecker, Würzburg, Roland Waschgler, Feldkirch, Gerald Zernig, Innsbruck/Hall in Tirol, Gabriela Zurek, Bremen, und Christoph Hiemke, Mainz – TDM-Gruppe der AGNP
Hintergrund
Für die Psychopharmakotherapie von psychiatrischen und neurologischen Patienten stehen derzeit mehr als 200 Arzneistoffe zur Verfügung, die während der letzten 60 Jahre entdeckt und entwickelt wurden. Trotz der damit verbundenen medizinischen und ökonomischen Fortschritte sind die erzielten Therapieerfolge für viele Patienten und deren behandelnde Ärzte nicht immer zufriedenstellend. Nachdem sich die klinische Forschung auf die Entwicklung neuer Medikamente fokussierte, ist inzwischen erkannt worden, dass eine verbesserte Anwendung aktuell verfügbarer Medikamente für Patienten erheblichen Nutzen bringen kann [7, 37]. Zudem besteht eine Lücke zwischen dem verfügbaren pharmakologischen Wissen und dessen Gebrauch in der klinischen Praxis. Die neueste Initiative, um diese Lücke zu überwinden, ist die Präzisionsmedizin („precision medicine“). Diese berücksichtigt die individuelle Variabilität eines Patienten, auf deren Basis in der klinischen Praxis evidenzbasiert behandelt wird. Therapeutisches Drug-Monitoring (TDM) ist ein Patienten-Management-Tool für die Präzisionsmedizin [24]. TDM ermöglicht eine patientenindividuelle Dosiseinstellung der Medikation, indem die Messung der Arzneistoff-Konzentrationen im Blut mit Informationen bezüglich Arzneistoff-Eigenschaften und Patientencharakteristika kombiniert wird. Ein Hauptgrund für den Einsatz von TDM für die Steuerung der Neuropsychopharmakotherapie ist die hohe interindividuelle pharmakokinetische Variabilität von Arzneistoff-Konzentrationen. Bei gleicher Dosis kann ein mehr als 20-facher interindividueller Unterschied in der Steady-State-Konzentration eines Arzneistoffs im Körper auftreten, da sich die Patienten in Absorption, Verteilung, Verstoffwechselung und Ausscheidung der Arzneistoffe unterscheiden. Ursachen sind Begleiterkrankungen (Komorbiditäten), Alter, Begleitmedikation oder genetische Besonderheiten [11, 45]. Verschiedene pharmazeutische Formulierungen des gleichen Arzneistoffs können ebenfalls den Grad und die zeitlichen Muster der Absorption und damit der Arzneistoff-Konzentration im Körper beeinflussen (Abb. 1).
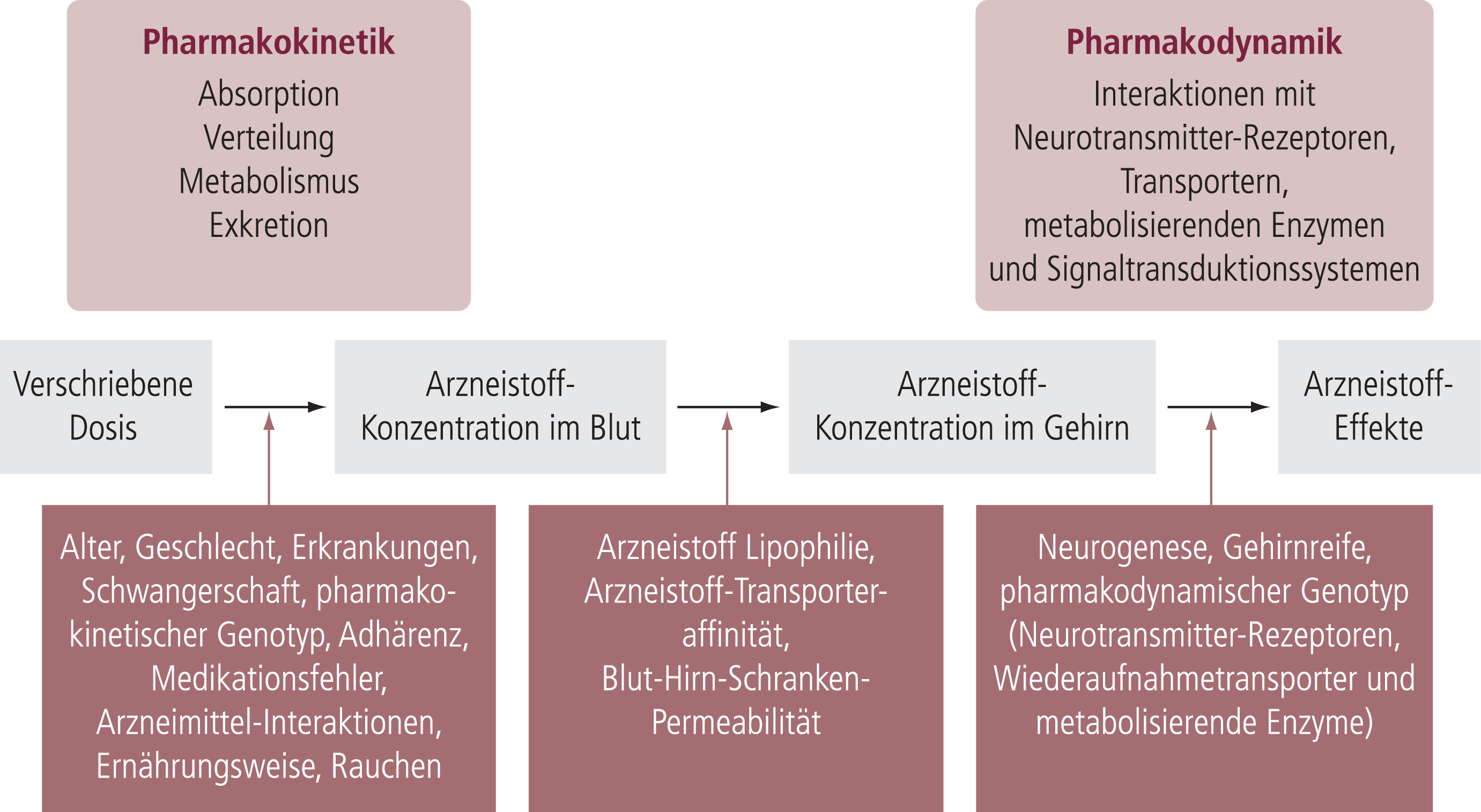
Abb. 1. Von der verschriebenen Dosis zur Arzneistoffwirkung, moduliert von verschiedensten Faktoren, die zu einer hohen interindividuellen pharmakokinetischen und pharmakodynamischen Variabilität führen
TDM nutzt die Quantifizierung der Arzneistoff-Konzentration im Blut (Plasma oder Serum), um die Dosis des individuellen Patienten zu titrieren, sodass eine Arzneistoff-Konzentration im Blut eingestellt wird, bei der höchstwahrscheinlich mit gutem Therapieansprechen bei minimalem Risiko für unerwünschte Arzneimittelwirkungen/Toxizität gerechnet werden kann. Darüber hinaus bietet TDM ein bislang weitgehend ungenutztes Potenzial, die Kosteneffektivität der Neuropsychopharmakotherapie zu verbessern [42]. Trotz des geschilderten Potenzials besteht jedoch ein erheblicher Widerspruch zwischen der Information über TDM in der offiziellen Produktinformation und der bestehenden medizinisch-wissenschaftlichen Evidenz von TDM. Selbst für gut untersuchte Arzneistoffe, beispielsweise Amitriptylin oder Clozapin, sind die Informationen über TDM in der Produktinformation/Packungsbeilage unzureichend. Für viele Neuropsychopharmaka ist die Quantifizierung der Arzneistoff-Konzentration im Blut für die Dosisanpassung jedoch klinische Routine geworden. Nachweise für den Nutzen einer TDM-geleiteten Dosiseinstellung existieren für Antikonvulsiva [30], trizyklische Antidepressiva, alte (erste Generation oder „typische“) und neue (zweite Generation oder „atypische“) Antipsychotika und Stimmungsstabilisierer. Für den Stimmungsstabilisierer Lithium ist aufgrund seines engen therapeutischen Bereichs ein regelmäßiges TDM obligater Bestandteil der Therapie geworden.
Die Vorteile von TDM zur Optimierung der Pharmakotherapie können allerdings nur erreicht werden, wenn die Methode adäquat in die klinische Behandlung integriert ist. Der Einsatz von TDM ist häufig suboptimal. Dies verschwendet Laborressourcen und birgt das Risiko irreführender Ergebnisse, die klinische Entscheidungen negativ beeinflussen können. Eine Studie über den klinischen Einsatz von TDM für trizyklische Antidepressiva in einer psychiatrischen Universitätsklinik zeigte, dass zwischen 25 und 40% der Anfragen für TDM nicht sinnvoll waren und die Interpretation der Ergebnisse bei etwa 20% der Patienten zu nicht angemessenen Dosisanpassungen führte. Andere typische Fehler sind fehlende Steady-State-Bedingungen zur Zeit der Blutentnahme und Übertragungsfehler auf dem Antragsformular. Studien über die Anwendung von TDM bei der Behandlung mit Antidepressiva oder stimmungsstabilisierenden Medikamenten ergaben weitere Informationen über den unangemessenen Einsatz von TDM [27]. Für Antikonvulsiva wurde festgestellt, dass die Hälfte sämtlicher TDM-Anforderungen inadäquat war.
Vor diesem Hintergrund hat die TDM-Gruppe der Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP) im Jahr 2004 Leitlinien für TDM in der Psychiatrie publiziert, inklusive Empfehlungen für eine Genotypisierung [3]. Im Jahre 2011 wurden die Leitlinien aktualisiert und um eine Vielzahl an Arzneistoffen erweitert, insbesondere neurologische [20]. Diese Leitlinien sind in vielen Laboren und Kliniken in die praktische Arbeit implementiert worden. Die ersten Leitlinien [3] wurden mehr als 300 Mal in der Literatur zitiert, die Leitlinien 2011 mehr als 400 Mal. Sie wurden übersetzt in Deutsch [21], Ungarisch, Französisch [4], Italienisch und Chinesisch. Seit 2011 sind das Wissen und die Akzeptanz bezüglich TDM weiter gestiegen. Deshalb hat die TDM-Gruppe der AGNP dieses zweite Update der Leitlinien erstellt.
Ziele der TDM Konsensus-Leitlinien
Das vorliegende Dokument befasst sich mit der Theorie und Praxis von TDM in Psychiatrie und Neurologie. Der erste Teil beschäftigt sich mit theoretischen Aspekten des Monitorings von Arzneistoff-Konzentrationen im Blut. Der zweite Teil beschreibt Indikationen für TDM und definiert Referenzbereiche für Arzneistoff-Konzentrationen zur Dosisfindung. Der dritte Teil beschreibt die praktische Anwendung von TDM, beginnend mit der Anforderung einer Blutspiegelmessung bis hin zur klinischen Entscheidung, eine bestehende Pharmakotherapie entweder unverändert weiterzuführen oder zu ändern.
Mit dem vorrangigen Ziel, die Anwendung von TDM zu verbessern, werden folgende Inhalte behandelt:
- Definition von Indikationen für TDM
- Definition von graduierten Empfehlungen zur Anwendung von TDM
- Definition therapeutischer und dosisbezogener Referenzbereiche
- Festlegung von Warnschwellenwerten für das Labor, wenn Blutkonzentrationen außergewöhnlich hoch sind
- Empfehlungen und Hilfen für die Interpretation der Laborbefunde
- Empfehlungen für die Kombination von TDM mit pharmakogenetischen Tests
- Auflistung von pharmakokinetischen Parametern, die für die Interpretation der TDM-Ergebnisse notwendig sind
Erstellung des Konsensus-Dokuments
Die aktualisierten Konsensus-Leitlinien wurden von der interdisziplinären TDM-Gruppe der AGNP erarbeitet. Die Gruppe besteht aus klinisch tätigen Psychiatern, Neurologen, Psychotherapeuten, Pharmakologen, Biochemikern, Pharmazeuten und Chemikern aus akademischen und nicht-akademischen Krankenhäusern und Institutionen in Deutschland, der Schweiz, Österreich und Italien.
Daten, die in den vorherigen AGNP-Konsensus-Leitlinien veröffentlicht wurden [3, 20], und andere Leitlinien für TDM von Antiepileptika [30] wurden berücksichtigt. Eine umfangreiche Literaturrecherche wurde durchgeführt, vor allem in PubMed und in Fachinformationen von Arzneistoffen, um TDM-bezogene Informationen zu identifizieren. Dabei wurde eine Suchliste (drug x AND concentration AND (blood OR plasma OR serum)) angewandt, um relevante Arbeiten zu finden und zu analysieren. Nach relevanter Literatur wurde auch von Hand in pharmakologischen und klinisch-chemischen Fachzeitschriften gesucht. Mehr als zweitausend Artikel wurden beurteilt. Schließlich wurden Daten von 1358 Artikeln als relevant für dieses zweite Update bewertet. Die Suche fokussierte sich auf therapeutische und dosisbezogene Arzneistoff-Konzentrationen in Serum, Plasma oder Blut. Für die Interpretation der TDM-Ergebnisse wurden Informationen über Cytochrom-P450-(CYP-)Substrat-Eigenschaften und metabolische Quotienten (Verhältnis Metabolit:Muttersubstanz) ermittelt oder berechnet. Darüber hinaus wurde nach CYP-induzierenden und -inhibitorischen Eigenschaften von Arzneistoffen und Nahrungsmitteln gesucht, welche klinisch relevant für pharmakokinetische Arzneimittel-Interaktionen sein könnten. Die finalen Entscheidungen über die Inhalte in diesem Update wurden während fünf Konsensus-Konferenzen und per E-Mail-Kommunikation gefällt.
Therapeutische Referenzbereiche sind in diesem Update für 154 Neuropsychopharmaka gelistet. Für 28 Arzneistoffe werden erstmalig Referenzbereiche gelistet (Levomilnacipran, Tianeptin, Vilazodon, Vortioxetin, Brexpiprazol, Cariprazin, Loxapin, Lurasidon, N-Desalkylquetiapin, Brivaracetam, Eslicarbazepin, Perampanel, Retigabin, Diphenhydramin, Doxylamin, Gamma-Hydroxybuttersäure, Medazepam, Modafinil, Promethazin, Zaleplon, Heroin, Morphin, Nalmefen, Nicotin und Rotigotin); geändert wurden Referenzbereiche für 18 Arzneistoffe (Bupropion, Milnacipran, Paroxetin, Aripiprazol, Asenapin, Flupentixol, Prothipendyl, Felbamat, Topiramat, Lorazepam, Temazepam, Zolpidem, Donepezil, Galantamin, Buprenorphin, Disulfiram, Methylphenidat und Methyldopa).
Umfänglich überarbeitet und erweitert wurde in diesem Update die Berechnung dosisbezogener Referenzbereiche. Die dosisbezogenen Referenzbereiche geben an, welche Wirkstoffkonzentration im Blut bei der verabreichten Dosis zu erwarten ist. Aus Abweichungen kann man unter anderem Adhärenzprobleme, individuelle pharmakokinetische Besonderheiten aufgrund Arzneimittel-Interaktionen, Gen-Polymorphismen oder Leber- oder Nierenfunktionsstörung eines Patienten identifizieren. Dieses Konzept wurde von Haen und Kollegen erstmalig vorgestellt [18] und in die vorherigen Konsensus-Leitlinien 2011 für 83 Neuropsychopharmaka aufgenommen [20]. Das Konzept wurde für dieses Update überarbeitet und auf 120 Neuropsychopharmaka erweitert, für 26 Arzneistoffe inklusive Metaboliten.
Theorie von TDM
Pharmakokinetische Aspekte
Absorption, Distribution und Elimination von Neuropsychopharmaka
Die meisten Neuropsychopharmaka zeichnen sich in ihren pharmakokinetischen Eigenschaften aus durch:
- Gute Resorption aus dem Magen-Darm-Trakt; die Blutkonzentration erreicht nach oraler Einnahme ihr Maximum innerhalb von einer bis sechs Stunden
- Sehr variable systemische Bioverfügbarkeit von 5 bis 100%
- Schnelle Verteilung aus dem Blut in das zentrale Nervensystem mit zumeist höheren Konzentrationen im Gehirn als im Blut
- Hohes Verteilungsvolumen (ca. 10 bis 50 l/kg)
- Niedrige Arzneistoff-Talspiegel im Blut unter Steady-State-Bedingungen (ca. 0,1 bis 500 ng/ml für psychiatrische Medikamente und bis zu 20 µg/ml für neurologische Medikamente)
- Elimination vorwiegend durch hepatischen Metabolismus
- Eliminationshalbwertszeit zumeist zwischen 12 und 36 Stunden
- Lineare Pharmakokinetik bei therapeutischen Dosen mit der Konsequenz, dass eine Verdoppelung der täglichen Dosis ungefähr zu einer Verdoppelung der Arzneistoff-Blutspiegel führt
- Enzyme der Cytochrom-P450-Familie (CYP) und UDP-Glucuronosyltransferasen (UGT) sind die wesentlichen Enzyme für die Metabolisierung
Es existieren jedoch zahlreiche Ausnahmen zu dieser Liste von pharmakokinetischen Eigenschaften. Zum Beispiel haben Agomelatin, Venlafaxin, Trazodon, Tranylcypromin, Moclobemid, Quetiapin, Rivastigmin oder Ziprasidon kurze (ca. 2 bis 10 Stunden) Eliminationshalbwertszeiten, während Aripiprazol und Fluoxetin sehr lange Eliminationshalbwertszeiten aufweisen (72 Stunden für Aripiprazol und 3 bis 15 Tage für Fluoxetin, unter Berücksichtigung seines aktiven Metaboliten Norfluoxetin). Amisulprid, Milnacipran, Memantin, Gabapentin oder Sulpirid werden nicht oder nur unwesentlich hepatisch metabolisiert und hauptsächlich über die Niere ausgeschieden, was für Patienten mit eingeschränkter Leberfunktion vorteilhaft sein kann. Paroxetin weist durch die Hemmung des eigenen Metabolismus eine nichtlineare Pharmakokinetik auf, da ein Stoffwechselprodukt irreversibel an das Enzym bindet und dieses inhibiert.
Viele Neuropsychopharmaka kommen als racemische Verbindungen zum Einsatz und ihre Enantiomere unterscheiden sich deutlich in ihrer Pharmakodynamik und Pharmakokinetik. Bisher ist jedoch nur für die racemischen psychotropen Substanzen Methadon und Methylphenidat eine TDM-Analytik der Enantiomere etabliert. Die wirksamen Enantiomere sind (R)-Methadon und L-Methylphenidat. Flupentixol ist verfügbar als 1:1-Mischung des geometrischen cis- und trans-Isomers (bzw. Z- und E-Isomers) für die orale Anwendung, während die Depot-Formulierung Flupentixoldecanoat ausschließlich cis-Flupentixol enthält. Nur das Letztere ist in Hinblick auf die Dopamin- (und Serotonin-) Rezeptoraffinität pharmakologisch aktiv. Dies wurde in klinischen Studien für die Wirksamkeit von cis-Flupentixol belegt. Für Forschungsprojekte und andere spezielle Fragestellungen sollte eine stereoselektive Analyse für Muttersubstanzen und/oder Metaboliten in Betracht gezogen werden (z.B. für Citalopram, Fluoxetin, Venlafaxin, Paliperidon oder Amitriptylin), nicht aber für die Routine.
Inter- und intraindividuelle Unterschiede von Neuropsychopharmaka-Konzentrationen im Blut (d.h. die pharmakokinetische Variabilität) werden wesentlich durch unterschiedliche Aktivitäten von Arzneistoff-metabolisierenden Enzymen verursacht. Die Enzymaktivität kann mit zunehmendem Alter abnehmen und aufgrund von Nieren- oder Lebererkrankungen verändert sein. Die meisten psychiatrischen oder neurologischen Arzneistoffe unterliegen einem Phase-I-Metabolismus durch oxidative (z.B. Hydroxylierung, Dealkylierung, Oxidation zu N-Oxiden, S-Oxidation, Sulfoxiden oder Sulfonen), reduktive (z.B. Carbonyl-Reduktion auf sekundäre Alkohole) oder hydrolytische Reaktionen. Die Phase-I-Reaktionen werden überwiegend durch Isoenzyme des Cytochrom-P450-(CYP-)Systems katalysiert. Sie sind Proteine einer Superfamilie, welche Häm als Kofaktor besitzen, und sie funktionieren als terminale Oxidasen in Elektronen-Transferketten. Der Begriff P450 ist abgeleitet vom spektrophotometrischen Absorptionsmaximum der CYP-Enzyme (450 nm) in ihrer reduzierten Form, komplexiert mit Kohlenmonoxid. CYP-katalysierte Phase-I-Reaktionen führen eine polare funktionelle Gruppe ein, was eine Konjugation mit stark polaren Molekülen wie Glucuronsäure oder Schwefelsäure in der Phase-II-Reaktion ermöglicht. Für Neuropsychopharmaka mit funktionellen Gruppen in der Ausgangsverbindung stellt die Glucuronidierung einer Hydroxyl- (z.B. Oxazepam oder Lorazepam) oder einer Amino-Funktion zur Bildung von N-Glucuroniden (z.B. Olanzapin) den wesentlichen Stoffwechselweg dar. Ausgehend von der Primärstruktur (Aminosäuresequenz) sind die Enzyme in 18 Familien von Cytochrom-P450-Genen und 43 Subfamilien klassifiziert. Im menschlichen Organismus sind 57 vermeintlich funktionale Gene und 58 Pseudogene von verschiedenen Genclustern kodiert. Die für den Metabolismus von Neuropsychopharmaka wichtigsten Isoenzyme sind CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 und CYP3A4/5 (Tab. 1). Viele CYP-Gene sind anfällig für Mutationen. Wie im Folgenden ausführlicher beschrieben, sind genetische Polymorphismen von CYP-Enzymen ein Grund für die hohe interindividuelle Variabilität von Arzneistoff-Konzentrationen im Körper, was eine Bestimmung der Konzentration im Blut notwendig macht.
Tab. 1. Enzyme und Effluxtransporter, die an der Metabolisierung und Verteilung von Neuropsychopharmaka beteiligt sind.
|
Arzneistoff |
Enzyme und Transporter |
|
Acamprosat |
Keine Metabolisierung |
|
Agomelatin |
CYP1A2, CYP2C19, CYP3A4 |
|
Alprazolam |
CYP3A4/5 |
|
Amantadin |
90% werden unverändert über die Niere ausgeschieden |
|
Amisulprid |
Mehr als 90% werden unverändert über die Niere ausgeschieden |
|
Amitriptylin |
CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A3, UGT1A4, UGT2B10, P-gp (ABCB1) |
|
Amitriptylinoxid |
Flavinmonooxygenase, CYP2C19, CYP2D6 |
|
Amphetamin (Dexamphetamin, Lisdexamfetamin) |
CYP2D6 |
|
Aripiprazol |
CYP2D6, CYP3A4, P-gp (ABCB1) |
|
Asenapin |
CYP1A2, UGT1A4 |
|
Atomoxetin |
CYP2C19, CYP2D6, P-gp (ABCB1) |
|
Benperidol |
Unklar |
|
Benserazid |
Hydroxylierung, COMT |
|
Biperiden |
Unklar |
|
Brexpiprazol |
CYP3A4, CYP2D6 |
|
Brivaracetam |
CYP2C8, renale Elimination |
|
Bromazepam |
CYP2C19, CYP3A4 |
|
Bromocriptin |
CYP3A4 |
|
Bromperidol |
CYP3A4 |
|
Brotizolam |
CYP3A4 |
|
Buprenorphin |
CYP2C8, CYP3A4, UGT1A3, UGT2B7 |
|
Bupropion |
CYP2C19, CYP2B6, CR |
|
Buspiron |
CYP3A4 |
|
Cabergolin |
Unklar, CYP3A4, P-gp (ABCB1) |
|
Carbamazepin |
CYP1A2, CYP2C8, CYP3A4/5, UGT2B7, P-gp (ABCB1), BCRP (ABCG2), Epoxidhydrolase |
|
Carbidopa |
Verlust der funktionellen Hydrazingruppe, ein Drittel wird nicht metabolisiert |
|
Cariprazin |
CYP2D6, CYP3A4 |
|
Chlordiazepoxid |
CYP3A4 |
|
Chlorpromazin |
CYP1A2, CYP2D6, P-gp (ABCB1) |
|
Chlorprothixen |
Wahrscheinlich CYP2D6, CYP3A4 |
|
Citalopram |
CYP2C19, CYP2D6, CYP3A4, P-gp (ABCB1) |
|
Clobazam (Norclobazam) |
CYP2C19, CYP3A4 |
|
Clomethiazol |
CYP2A6, CYP3A4 |
|
Clomipramin |
CYP1A2, CYP2C19, CYP2D6, CYP3A4, UGT2B10 |
|
Clonazepam |
CYP3A4 |
|
Clorazepat |
CYP2C19, CYP3A4 |
|
Clozapin |
CYP1A2, CYP2C19, CYP3A4, P-gp (ABCB1) |
|
Codein |
CYP2D6, CYP3A4, UGT2B4, UGT2B7 |
|
Cyamemazin |
CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4 |
|
Dapoxetin |
CYP2D6 |
|
Desipramin |
CYP2D6 |
|
Desvenlafaxin |
CYP3A4, CYP2C19, UGT |
|
Dextroamphetamin |
CPY2D6 |
|
Diacetylmorphin (Heroin) |
Carboxylesterase 2 und 1, UGT |
|
Diazepam |
CYP2B6, CYP2C19, CYP3A4, UGT2B7, P-gp (ABCB1) |
|
Dihydroergocryptin |
CYP3A4 |
|
Diphenhydramin |
CYP2D6, UGT1A4, UGT2B10, P-gp (ABCB1) |
|
Disulfiram |
CYP1A2, CYP2A6, CYP2B6, CYP2E1, CYP3A4 |
|
Donepezil |
CYP2D6, CYP3A4, P-gp (ABCB1) |
|
Dothiepin=Dosulepin |
CYP2C19, CYP2D6 |
|
Doxepin |
CYP2C9, CYP2C19, CYP2D6 |
|
Doxylamin |
Unklar |
|
Dronabinol |
CYP2C9, CYP3A4, UGT1A9, UGT1A7, UGT1A8, UGT1A10 |
|
Duloxetin |
CYP1A2, CYP2D6, P-gp (ABCB1) |
|
Entacapon |
UGT1A9 |
|
Escitalopram |
CYP2C19, CYP2D6, CYP3A4, P-gp (ABCB1) |
|
Ethanol |
Alkoholdehydrogenase, CYP2E1 |
|
Felbamat |
Renale Exkretion |
|
Flunitrazepam |
CYP2C19, CYP3A4 |
|
Flunarizin |
CYP2D6 |
|
Fluoxetin |
CYP2B6, CYP2C9, CYP2C19, CYP2D6, P-gp (ABCB1) |
|
Flupentixol |
CYP2D6 |
|
Fluphenazin |
CYP2D6, P-gp (ABCB1) |
|
Flurazepam |
CYP2C19, CYP3A4 |
|
Fluspirilen |
Renale Exkretion, CYP3A4 |
|
Fluvoxamin |
CYP2D6, CYP1A2, P-gp (ABCB1) |
|
Gabapentin |
Keine Metabolisierung, unveränderte renale Elimination |
|
Galantamin |
CYP2D6, CYP3A4 |
|
Gammahydroxybuttersäure (GHB) |
Beta-Oxidation |
|
Guanfacin |
CYP3A4, Epoxidhydratase, UGT |
|
Haloperidol |
CYP2D6, CYP3A4, AKR, UGT, P-gp (ABCB1) |
|
Heroin (Diacetylmorphin) |
Carboxylesterase 2 und 1, UGT |
|
Iloperidon |
CYP2D6, CYP3A4 |
|
Imipramin |
CYP1A2, CYP2C19, CYP2D6, CYP3A4, UGT1A4, UGT2B10 |
|
Koffein |
CYP1A2, CYP2A6, Xanthinoxidase, NAT |
|
Kokain |
Carboxylesterase 1 und 2, Pseudocholinesterase, CYP3A4 |
|
Lamotrigin |
UGT1A4, UGT3B7, P-gp (ABCB1), BCRP (ABCG2) |
|
Levetiracetam |
Keine Metabolisierung, P-gp (ABCB1) |
|
Levodopa |
DDC, COMT, MAO |
|
Levomepromazin |
CYP3A |
|
Levomilnacipran |
CYP3A4, P-gp (ABCB1) |
|
Levomethadon |
CYP2B6, CYP3A4, CYP2D6 |
|
Levosulpirid |
P-gp (ABCB1) |
|
Lisdexamfetamin |
Erythrozytpeptidase, CYP2D6 |
|
Lisurid |
CYP3A4, CYP2D6 |
|
Lithium |
Keine Metabolisierung, renale Clearance |
|
Lorazepam |
UGT2B15 |
|
Loxapin |
CYP3A4, CYP2D6, CYP1A2, CYP2C8, CYP2C19, FMO |
|
Lurasidon |
CYP3A4 |
|
Maprotilin |
CYP2D6, CYP1A2 |
|
Medazepam |
CYP2B6, CYP2C19, CYP3A4 |
|
Melatonin |
CYP1A2 |
|
Melperon |
Unklar |
|
Memantin |
Kaum metabolisiert |
|
Methadon |
CYP2B6, CYP3A4, CYP2D6, ABCB1 |
|
Methylphenidat |
Carboxylesterase 1 |
|
Mianserin |
CYP2D6, CYP1A2, CYP3A4 |
|
Midazolam |
CYP3A4, UGT1A4 |
|
Milnacipran |
CYP3A4, ABCB1, renale Exkretion |
|
Mirtazapin |
CYP3A4, CYP1A2, CYP2D6 |
|
Moclobemid |
CYP2C19, CYP2D6 |
|
Modafinil |
Amidhydrolyse, CYP3A4 |
|
Morphin |
CYP2C8, CYP3A4, UGT2B7 |
|
Nalmefen |
UGT |
|
Naloxon |
UGT2B7, AKR1C |
|
Naltrexon |
AKR1C4 |
|
Nicotin |
CYP2A6, UGT1A1, UGT1A2, UGT2B10 |
|
Nitrazepam |
CYP3A4 |
|
Nordazepam |
CYP3A4, CYP2C19 |
|
Nortriptylin |
CYP2D6, P-gp (ABCB1) |
|
Olanzapin |
UGT1A4, UGT2B10, Flavinmonooxygenase, CYP1A2, CYP2D6, P-gp (ABCB1) |
|
Opipramol |
CYP2D6 |
|
Oxazepam |
UGT1A9, UGT2B7, UGT2B15 |
|
Oxcarbazepin |
AKR, UGT2B15, P-gp (ABCB1) |
|
Paliperidon (=9-Hydroxyrisperidon) |
60% werden unverändert eliminiert, CYP3A4, UGT, P-gp (ABCB1), BCRP (ABCG2) |
|
Paroxetin |
CYP2D6, CYP3A4, P-gp (ABCB1) |
|
Perampanel |
CYP3A4, CYP2B6, UGT1A1, UGT1A4 |
|
Perazin |
CYP1A2, CYP2C9, CYP2C19, CYP3A4, Flavinmonooxygenase |
|
Pergolid |
CYP3A4 |
|
Perphenazin |
CYP1A2, CYP2C19, CYP2D6, CYP3A4 |
|
Phenytoin |
CYP2C9, CYP2C19, UGT2B15 |
|
Phenobarbital |
CYP2C19, UGT1A4 |
|
Pimozid |
CYP1A2, CYP2D6, CYP3A4 |
|
Pipamperon |
Unklar |
|
Piribedil |
Demethylierung, p-Hydroxylierung, N-Oxidation |
|
Pramipexol |
Keine Metabolisierung |
|
Prazepam |
CYP2C19, CYP3A4 |
|
Pregabalin |
Keine Metabolisierung, renale Exkretion |
|
Promazin |
CYP1A2, CYP2A6, CYP2C19, CYP3A4 |
|
Promethazin |
CYP2D6 |
|
Quetiapin |
CYP3A4, CYP2D6, ABCB1 |
|
Rasagilin |
CYP1A2 |
|
Reboxetin |
CYP3A4 |
|
Retigabin |
NAT, UGT |
|
Risperidon |
CYP2D6, CYP3A4, P-gp (ABCB1), BCRP (ABCG2) |
|
Rivastigmin |
Cholinesterase |
|
Ropinirol |
CYP1A2 |
|
Rotigotin |
CYP2C19, CYP1A1, CYP1A2, CYP2D6, CYP3A4, SULT1A1, SULT1A2, SULT1A3, SULT1B1, SULT1C4, SULT1E1, UGT |
|
Rufinamid |
Carboxylesterase |
|
Selegilin |
CYP2B6 |
|
Sertindol |
CYP2D6, CYP3A4 |
|
Sertralin |
CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, UGT1A1, P-gp (ABCB1) |
|
Sulpirid |
Keine Metabolisierung, renale Exkretion, P-gp (ABCB1) |
|
Temazepam |
CYP219, UGT2B7 |
|
Tetrahydrocannabinol, THC |
CYP2C9, CYP3A4 |
|
Thioridazin |
CYP1A2, CYP2D6, CYP3A4 |
|
Tianeptin |
Beta-Oxidation |
|
Tiaprid |
Keine Metabolisierung |
|
Tolcapon |
COMT, CYP2A6, CYP3A4, UGT |
|
Topiramat |
UGT, P-gp (ABCB1) |
|
Tranylcypromin |
MAO, unklar |
|
Trazodon |
CYP3A4, CYP2D6 |
|
Triazolam |
CYP3A4 |
|
Trifluoperazin |
UGT1A4 |
|
Trimipramin |
CYP2C19, CYP2D6, CYP2C9, CYP3A4, UGT2B10 |
|
Valproat |
UGT1A3, UGT1A6, UGT2B7, CYP2A6, CYP2B6, CYP2C9, CYP219, Beta-Oxidation |
|
Venlafaxin |
CYP2C19, CYP2D6, CYP2C9, CYP3A4, P-gp (ABCB1) |
|
Vilazodon |
CYP3A4, ABCB1 |
|
Vortioxetin |
CYP2D6, CYP3A4, CYP2A6, CYP2C9, P-gp (ABCB1) |
|
Zaleplon |
Aldehydoxidase, CYP3A4 |
|
Ziprasidon |
CYP3A4, Aldehydoxidase |
|
Zolpidem |
CYP1A2, CYP2C9, CYP3A4 |
|
Zopiclon |
CYP2C8, CYP3A4 |
|
Zotepin |
CYP1A2, CYP2D6, CYP3A4 |
|
Zuclopenthixol |
CYP2D6 |
ABC: ATP-binding cassette; AKR: Aldo-Keto-Reduktase; COMT: Catechol-O-Methyltransferase; CR: Carbonylreduktase; CYP: Cytochrom P450; DDC: Dopadecarboxylase (=aromatische Aminosäure-Decarboxylase); FMO: Flavinmonooxygenase; MAO: Monoaminoxidase; NAT: N-Acetyltransferase; SULT: Sulfotransferase; UGT: UDP-Glucuronosyltransferase
P-Glykoprotein (P-Gp) wird vom ABCB1-Gen und vom Brustkrebs-Resistenz-Protein (breast cancer resistance protein, BCRP) vom ABCG2-Gen kodiert. Die CYP-Substrateigenschaften basieren primär auf In-vivo-Studien, durchgeführt am Menschen, wohingegen ABC-Substrateigenschaften auf Tierstudien oder Studien an Zelllinien basieren. Wenn diese Substanzen mit starken oder moderaten Inhibitoren (siehe Tab. 2) oder Induktoren (siehe Tab. 3) kombiniert werden und Enzyme sind fett gedruckt, so kommt es zu einem Anstieg oder Abfall der Arzneistoff-Konzentration im Blut
Andere enzymatische Systeme sind ebenfalls am Phase-I-Metabolismus von Arzneistoffen beteiligt. Dazu gehören Aldo-Keto-Reduktasen (AKRs), die Aldehyd- oder Ketongruppen von endo- und exogenen Substanzen reduzieren. Im menschlichen Körper wurden 13 AKR-Proteine identifiziert. Die Enzyme reduzieren Ziprasidon zu seinem Dihydro-Derivat und Naltrexon zu Naltrexol. Die Monoaminoxidasen (MAO) MAO-A und MAO-B desaminieren Citalopram stereoselektiv zu einem offensichtlich inaktiven sauren Metaboliten. Arzneistoffe werden vorwiegend in der Leber und in geringem Maße in extrahepatischen Geweben, wie Darmschleimhaut oder Gehirn, metabolisiert.
Pharmakokinetische Arzneimittel-Interaktionen können auftreten, wenn Substanzen mit inhibitorischen oder induzierenden Eigenschaften an Arzneistoff-metabolisierenden Enzymen (Tab. 2 und 3) mit Arzneistoffen kombiniert werden, die Substrate des inhibierten oder induzierten Enzyms sind. Viele Interaktionen wurden anhand von TDM zufällig oder durch retrospektive Analyse von TDM-Datenbanken detektiert. Der Raucherstatus eines Patienten ist ebenfalls von hoher klinischer Relevanz für CYP1A2-Substrate, denn CYP1A2 wird dosisabhängig von Inhaltsstoffen des Zigarettenrauchs (polyzyklische aromatische Kohlenwasserstoffe, nicht Nicotin) induziert. Bei Konsum von 1 bis 5, 6 bis 10 und >10 Zigaretten pro Tag steigt die Aktivität von CYP1A2 1,2-, 1,5- bzw. 1,7-fach. Die gesteigerte Aktivität kehrt innerhalb von vier Tagen nach Zigarettenverzicht in den Grundzustand zurück. Effekte aufgrund des Rauchens sollten deshalb bei Konsum von mehr als zehn Zigaretten pro Tag berücksichtigt werden. Wenn ein starker Raucher zukünftig auf Zigaretten verzichten möchte, kann dies unter bestehender Therapie mit einem CYP1A2-Substrat (Tab. 1), zum Beispiel Clozapin, Duloxetin oder Olanzapin, eine Dosisreduktion notwendig machen, was durch TDM kontrolliert werden sollte.
Tab. 2. Inhibitoren von CYP-Enzymen, die am Abbau von Arzneistoffen beteiligt sind
|
Inhibierende Arzneistoffe |
Inhibierte Enzyme |
|
Amiodaron |
CYP2C9, CYP2D6, CYP3A4 |
|
Amprenavir |
CYP3A4 |
|
Aprepitant |
CYP3A4 |
|
Atazanavir |
CYP3A4 |
|
Boceprevir |
CYP3A4 |
|
Bupropion |
CYP2D6 |
|
Cimetidin |
CYP1A2, CYP2D6, CYP3A4 |
|
Ciprofloxacin |
CYP1A2, CYP3A4 |
|
Clarithromycin |
CYP3A4 |
|
Clomethiazol |
CYP2E1 |
|
Clopidogrel |
CYP2B6 |
|
Crizotinib |
CYP3A4 |
|
Diltiazem |
CYP3A4 |
|
Disulfiram |
CYP2E1 |
|
Duloxetin |
CYP2D6 |
|
Enoxacin |
CYP1A2 |
|
Erythromycin |
CYP3A4 |
|
Esomeprazol |
CYP2C19 |
|
Felbamat |
CYP2C19 |
|
Fluconazol |
CYP2C9, CYP3A4 |
|
Fluoxetin und Norfluoxetin |
CYP2D6, CYP2C19, CYP3A4 |
|
Fluvoxamin |
CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4 |
|
Fosamprenavir |
CYP3A4 |
|
Gemfibrocil |
CYP2C8 |
|
Grapefruitsaft |
CYP3A4 |
|
Indinavir |
CYP3A4 |
|
Isoniazid |
CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP3A4, MAO |
|
Itraconazol |
CYP3A4 |
|
Ketoconazol |
CYP3A4 |
|
Levomepromazin |
CYP2D6 |
|
Melperon |
CYP2D6 |
|
Metoclopramid |
CYP2D6 |
|
Miconazol |
CYP2C9, CYP3A4 |
|
Moclobemid |
CYP2C19, CYP2D6, MAO-A |
|
Nelfinavir |
CYP3A4 |
|
Norfloxacin |
CYP1A2 |
|
Omeprazol |
CYP2C19 |
|
Paroxetin |
CYP2D6 |
|
Perazin |
CYP1A2 |
|
Phenylpropanolamin |
CYP1A2 |
|
Posaconazol |
CYP3A4 |
|
Propafenon |
CYP1A2, CYP2D6 |
|
Quinidin |
CYP2D6 |
|
Ritonavir |
CYP2D6, CYP3A4 |
|
Saquinavir |
CYP3A4 |
|
Telaprevir |
CYP3A4 |
|
Telithromycin |
CYP3A4 |
|
Ticlopidin |
CYP2B6, CYP2C19 |
|
Tranylcypromin |
CYP2A6, MAO |
|
Valproat |
CYP2C9 |
|
Verapamil |
CYP3A4 |
|
Voriconazol |
CYP2B6, CYP2C9, CYP2C19, CYP3A4 |
|
Zileuton |
CYP1A2 |
Bei Arzneistoffen, die bevorzugt über ein inhibiertes Enzym metabolisiert werden, besteht das Risiko einer pharmakokinetischen Wechselwirkung (www.mediq.ch oder www.psiac.de). Bei Inhibition von fett gedruckten Enzymen (siehe Tab. 1) ist mit einem Anstieg der Arzneistoff-Konzentration im Blut um mehr als 50% zu rechnen.
Tab. 3. Induktoren von Enzymen und Effluxtransportern, die am Abbau und an der Verteilung von Arzneistoffen beteiligt sind
|
Induzierende Arzneistoffe |
Induzierte Enzyme oder ABC-Transporter |
Kommentare |
|
Bosentan |
CYP3A4 |
|
|
Carbamazepin |
CYP1A2, CYP2B6, CYP2C9, CYP3A4, P-gp (ABCB1), UGT |
Anstieg der CYP3A4-Aktivität innerhalb von 3 Wochen, Induktion des eigenen Metabolismus |
|
Efavirenz |
CYP2B6, CYP3A4 |
|
|
Ethanol |
CYP2E1 |
Induktion kann zu metabolischer Toleranz führen |
|
Isoniazid |
CYP2E1 |
Initiale Inhibition und dann Induktion von CYP2E1 |
|
Johanniskraut |
CYP3A4, CYP2C9, P-gp (ABCB1) |
|
|
Lamotrigin |
UGT |
|
|
Modafinil |
CYP1A2, CYP2B6, CYP3A4 |
|
|
Oxybutynin |
CYP3A4 |
|
|
Phenobarbital |
CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, UGT1A1 |
|
|
Phenytoin |
CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, UGT |
|
|
Primidon |
CYP2C9, CYP2C19, CYP3A4 |
|
|
Rauch |
CYP1A2 |
Maximaler Anstieg der Induktion bei Konsum von 10 oder mehr Zigaretten pro Tag, Abnahme der CYP1A2-Aktivität innerhalb von 3 Tagen nach Einstellung des Rauchens |
|
Rifabutin |
CYP3A4 |
Induktion des eigenen Metabolismus |
|
Rifampicin |
CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4 |
Nach Induktion durch Rifampicin verbleiben die CYP2C19- und CYP3A4-Aktivitäten nach Absetzen noch für 4 Tage gesteigert und erreichen wieder innerhalb von 8 Tagen normale Werte |
|
Ritonavir |
CYP2C9, CYP3A4 (high dose), UGT |
ABC: ATP-binding cassette transporter; CYP: Cytochrom P450; UGT: UDP-glucuronosyltransferase; P-Glykoprotein (P-Gp) wird vom ABCB1-Gen kodiert.
Die Induktion von fett gedruckten Enzymen führt zu einem Abfall der Arzneistoff-Konzentration im Blut um mehr als 50% (siehe Tab. 1), wenn der Arzneistoff bevorzugt über das induzierte Enzym abgebaut wird.
Neben Enzymen des Phase-I- und -II-Metabolismus spielen Arzneistoff-Transporter eine bedeutende Rolle bei der Pharmakokinetik von Arzneistoffen. Die „ATP binding cassette“-(ABC-)Proteine sind in Zellmembranen lokalisiert und fungieren als Efflux-Transporter für viele therapeutische Substanzen, um Organe vor schädlichen Stoffen zu schützen. Für viele Neuropsychopharmaka wurde insbesondere der ABC-Transporter P-Glykoprotein (P-gp; das Genprodukt von ABCB1), das „multidrug resistance protein“ (MRP; kodiert von ABCC1) und das „breast cancer resistance protein“ (BCRP; kodiert von ABCG2), als Hauptdeterminante für die Arzneistoff-Verteilung innerhalb des menschlichen Körpers identifiziert (Tab. 1). Arzneistoffe, die Substrate von ABC sind, werden durch passive Diffusion in die Zellen aufgenommen und dann via ABC-Transporter durch eine ATP-abhängige Konformationsänderung wieder in den extrazellulären Raum austransportiert. P-gp wird zumeist in der Blut-Hirn-Schranke sowie im Dünndarm exprimiert und spielt eine signifikante Rolle bei der Verteilung von Arzneistoffen in die Zielorgane. Tierstudien zeigten, dass P-gp die Verfügbarkeitsrate vieler Antidepressiva und Antipsychotika, beispielsweise Nortriptylin, Citalopram oder Risperidon im Gehirn kontrolliert. Es wird demnach angenommen, dass eine hohe P-gp-Funktion für ineffiziente Arzneistoff-Konzentrationen, und eine geringe P-gp-Funktion aufgrund hoher Arzneistoff-Konzentrationen für Unverträglichkeiten verantwortlich ist [44]. Ähnlich den CYP-Enzymen wurden multiple genetische Mutationen für ABC-Transporter identifiziert. Darüber hinaus wird die Expression von ABC-Transportern von einer Vielzahl von Faktoren kontrolliert, beispielsweise von pathophysiologischen Stressoren, Xenobiotika, Hormonen sowie diätetischen Faktoren.
Unterschiede bezüglich der Pharmakokinetik von Neuropsychopharmaka wurden ebenfalls zwischen den Geschlechtern beschrieben, höchstwahrscheinlich aufgrund der Effekte weiblicher Geschlechtshormone auf die pharmakokinetischen Prozesse der Absorption, Verteilung, Metabolismus und Exkretion. Bisherige Studienergebnisse sind jedoch bisher inkonsistent und die klinische Relevanz unklar. Auch das Körpergewicht sollte nach pharmakokinetischen Prinzipien eine wichtige Determinante der Arzneistoff-Konzentration im Blut darstellen. Viele Studien detektierten jedoch einen geringeren Einfluss des Körpergewichts auf die Pharmakokinetik von Arzneistoffen als erwartet. Systematische Forschung ist diesbezüglich weiterhin vonnöten.
Arzneistoff-Konzentration im Blut
Abbildung 2 zeigt die Konzentration-Zeit-Kurve eines hypothetischen Arzneistoffs nach oraler und intramuskulärer Depot-Applikation. Unter Steady-State-Bedingungen gleicht die Arzneistoff-Einnahme die Arzneistoff-Elimination über einen definierten Zeitrahmen aus. Konzentrationen insbesondere von Arzneistoffen mit kurzer Eliminationshalbwertszeit (<12 Stunden) fluktuieren im Tagesverlauf und sind abhängig vom Dosisschema, was bei der Interpretation der TDM-Ergebnisse berücksichtigt werden muss.
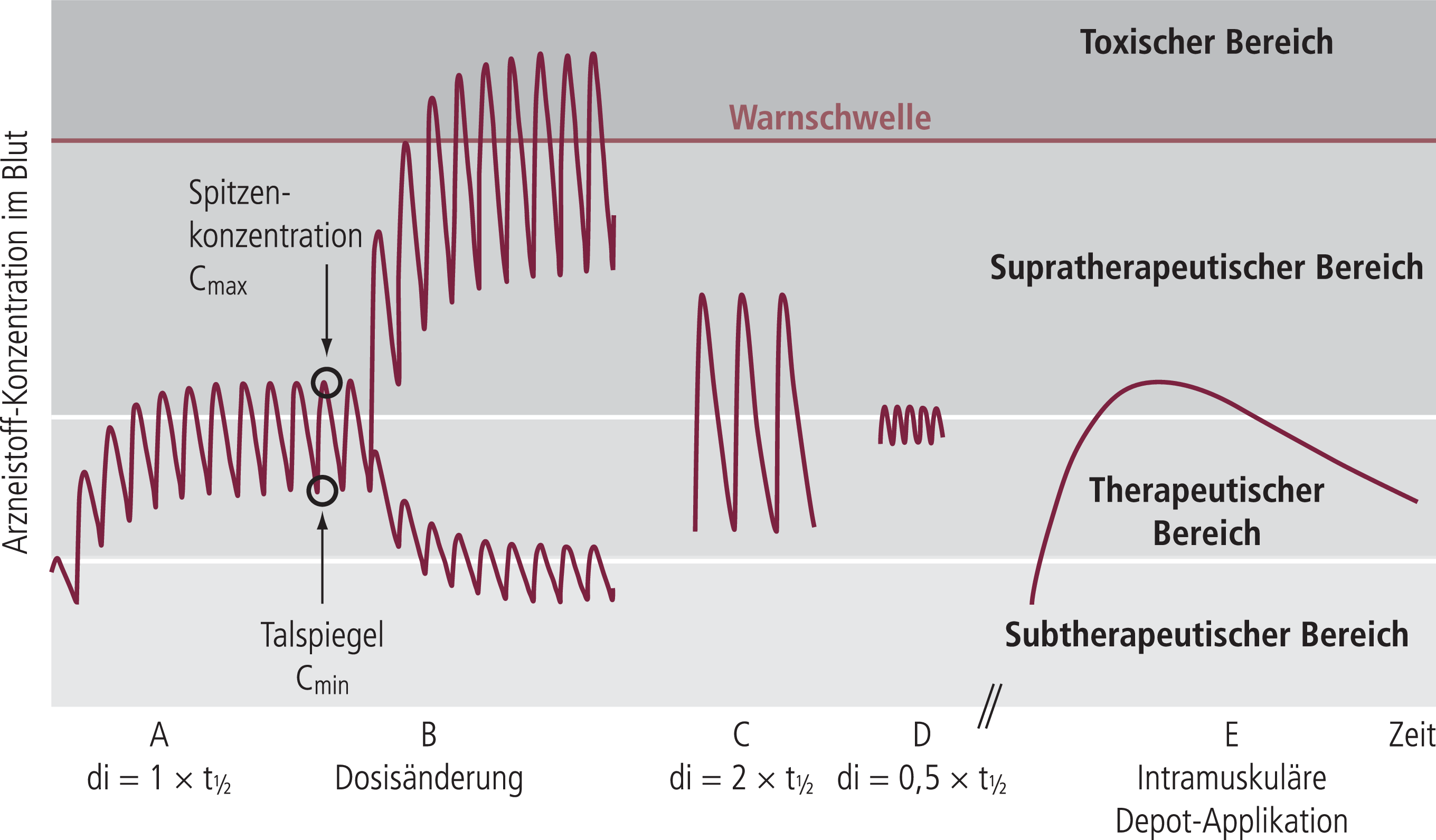
Abb. 2. Konzentration-Zeit-Kurve eines Arzneistoffs nach oraler Gabe oder intramuskulärer Depot-Applikation A: 94% des Steady-State (Therapie mit konstanter Dosis) ist nach vier Eliminationshalbwertszeiten (t1/2) des Arzneistoffs erreicht. Unter Steady-State-Bedingungen sind Arzneistoff-Aufnahme und -Elimination gleich. In der Regel werden für TDM Arzneistoff-Talspiegel (Cmin) unter Steady-State-Bedingungen gemessen und empfohlen. Die Abbildung zeigt einen hypothetischen Arzneistoff mit einem gleichen Dosisintervall (di) zu dessen Halbwertszeit (di =t1/2); eine Situation, die für viele Arzneistoffe betrachtet wird (z.B. t1/2=12 h, di =12 h, Kurve A). Arzneistoff-Talspiegel liegen genau in der Mitte des therapeutischen Bereichs, d.h., sie sind innerhalb des Zielbereichs, auch wenn die Arzneistoff-Konzentration während des Dosisintervalls den orientierenden therapeutischen Bereich manchmal überschreitet. B: Add-on-Gabe eines Arzneistoffs, der den Metabolismus des ersten Arzneistoffs inhibiert oder induziert. Die Inhibition oder Induktion resultiert in erhöhten oder reduzierten Konzentrationen des ersten Arzneistoffs. Eine solche pharmakokinetische Interaktion erfordert eine Dosisänderung. C: Eine Verdopplung des Dosisintervalls (di =2 × t1/2) und eine Verabreichung der Dosis einmal täglich ist in Kurve C dargestellt. Die Fläche unter der Blut-Konzentration-versus-Zeit-Kurve (Area under the blood concentration versus time curve, AUC) repräsentiert die totale Arzneistoff-Exposition und ist identisch für die Kurven A und C, auch wenn die Talspiegel in Kurve C (24 h nach der täglichen Dosis) signifikant niedriger sind als in Kurve A (12 h nach der halben täglichen Dosis). Hohe Tal- zu Spitzenspiegel können während Phasen hoher Arzneistoff-Konzentrationen mit Unverträglichkeiten assoziiert sein. D: Kurve D zeigt die Einnahme von vier gleichen Dosen pro Tag, insgesamt die gleiche tägliche Dosis wie bei den Kurven A und C. Die AUC ist identisch mit den Kurven A und C, aber höhere Talspiegel sind gegeben. Bei Anwendung dieser Applikationsform können niedrigere Dosen effektiv sein, da ausreichende Arzneistoff-Konzentrationen im Zielbereich verfügbar sind. E: Intramuskuläre Depot-Applikation: Spitzen-Konzentrationen des Arzneistoffs können in Abhängigkeit von der Formulierung frühestens nach einem Tag und spätestens nach vier Wochen erreicht werden. Talspiegel-Konzentrationen sind kurz vor der nächsten Applikation gegeben. Die Blutentnahme während der Eliminationsphase nach vollständiger Absorption (Maximum) resultiert trotz gleicher AUC in höheren Werten, im Vergleich zu der Messung der Talspiegel nach oraler Applikation. Die Zeitskala von Kurve E ist unterschiedlich zu der Zeitskala der Kurven A bis D.
Studien zur Beziehung zwischen Neuropsychopharmaka-Konzentrationen im Blut und klinischen Effekten untersuchten fast ausschließlich die Korrelaten klinischer Parameter mit Talspiegel-Konzentrationen im Blut, das heißt mit den minimalen Konzentrationen (Cmin) unter Steady-State (Therapie unter konstanter Dosis für mindestens 4 bis 6 Eliminationshalbwertszeiten). Als Konsequenz sind die therapeutischen Zielbereiche, die für TDM in der Neuropsychopharmakotherapie angewandt werden, Cmin-Bereiche unter Steady-State. Ausnahmen sind Antiparkinsonika und Arzneistoffe zur Behandlung der Aufmerksamkeits-Defizit-Hyperaktivität-Störung (ADHS), beispielsweise Methylphenidat oder Atomoxetin. Die meisten dieser Arzneistoffe besitzen kurze Eliminationshalbwertszeiten (2 bis 6 Stunden), deshalb wurden klinische Effekte mit maximalen Arzneistoff-Konzentrationen (Cmax) korreliert.
Arzneistoff-Konzentration in Gehirn und Liquor
Die pharmakologische Aktivität von Neuropsychopharmaka hängt von der Verfügbarkeit im Zielorgan, dem Gehirn, ab. Der Transport von Arzneistoffen vom Blut ins Gehirn führt über die Gehirnkapillaren der Endothelzellen, in Gesamtheit über die Blut-Hirn-Schranke (BHS). Die BHS kontrolliert die Gehirnumgebung, unter anderem durch Kontrolle des Austauschs von gelösten Substanzen, indem sie den Influx von potenziell schädlichen Xenobiotika, inklusive vieler Arzneistoffe, in das Gehirn verhindert. Die Permeabilität der BHS für ein bestimmtes Molekül definiert die Rate, in welcher ein Arzneistoff die interstitielle Flüssigkeit (ISF) des Gehirns erreicht, von wo aus die Moleküle weiter gleichmäßig über die Gehirnzellen verteilt werden. Der Arzneistoff-Transport vom Blut in den Liquor (CSF [cerebrospinal fluid]) und vice versa findet an der Blut-CSF-Schranke statt, unterstützt durch einen Austausch zwischen CSF und Gehirn-ISF. Der CSF ist empfänglich für die Messung der Konzentrationen von ungebunden vorliegenden Arzneistoffen. Zwei systematische Studien zu 39 Substanzen von Fridén et al. und 25 Substanzen von Kodaira et al. zeigten eine gute Korrelation zwischen Arzneistoff-Konzentrationen im CSF und ISF für Substanzen, die eine hohe Permeabilität und eine geringe oder keine Affinität zu Effluxtransporten aufwiesen. Die Rolle des CSF zur Messung von Arzneistoff-Konzentrationen ungebundener Substanzen im Gehirn ist allerdings noch immer umstritten.
Arzneistoffe, die effizient vom Gehirn an der BHS eliminiert werden, sind primär P-gp-Substrate wie Risperidon, Aripiprazol oder Venlafaxin [44]. Für diese Substanzen sind Gehirn-Konzentrationen viel geringer als Blut-Konzentrationen. Das Gehirn-zu-Blut-Konzentrationsverhältnis von P-gp-Substraten variiert stark für Substanzen mit ähnlichen physikochemischen Eigenschaften. Trotz der sehr unterschiedlichen Verhältnisse von Gehirn zu Blutspiegel der verschiedenen Psychopharmaka haben Tierversuche gezeigt, dass Steady-State-Blutspiegel von Psychopharmaka gut mit den Konzentrationen im Gehirn korrelieren. Diese Korrelationen sind besser als die zwischen Dosis und Gehirn-Konzentration. Dies wurde beispielsweise für trizyklische Antidepressiva oder Olanzapin gezeigt. Anhand einer Magnetresonanzspektroskopie wurde bei Patienten gezeigt, dass Konzentrationen von Fluoxetin und Norfluoxetin im Gehirn mit Konzentrationen im Blut korrelieren. Für Carbamazepin und sein Epoxid wurde ein linearer Zusammenhang zwischen Gehirn- und Blut-Konzentration bei Patienten gefunden, bei denen eine Gehirn-Operation vorgenommen wurde. Für Neuropsychopharmaka kann die Blut-Konzentration daher als ein zuverlässiger Surrogat-Marker für die Konzentration im Gehirn betrachtet werden.
Die Positronen-Emissions-Tomographie (PET) ermöglicht die Analyse der Rezeptor-Besetzung des zentralen Nervensystems in vivo. PET-Studien haben gezeigt, dass Arzneistoff-Konzentrationen im Blut gut mit der Besetzung der Zielrezeptoren im Gehirn korrelieren [15]. Antipsychotika wirken vorwiegend über die Blockade von D2-artigen Dopamin-Rezeptoren. Die Blockade der D2-Rezeptoren durch Antipsychotika reduziert die Bindung von radioaktiven PET-Liganden. Mit diesem Ansatz und in Verbindung mit der Quantifizierung der Verdrängung von Dopamin-Rezeptor-Radioliganden wurde gezeigt, dass die Rezeptor-Besetzung besser mit Blut-Konzentrationen von Antipsychotika als mit der Tagesdosis korreliert. Es ist sogar möglich, die Dopamin-D2-Rezeptorbesetzung anhand der Konzentration von Antipsychotika im Blut vorherzusagen. Eine optimale klinische Wirksamkeit wurde bei 70 bis 80% D2-Rezeptor-Besetzung gesehen, wobei ab einer Rezeptor-Besetzung von 80% die Schwelle für das Auftreten von extrapyramidalen Nebenwirkungen definiert wurde. Die PET-Analyse wurde ebenfalls verwendet, um in vivo die Serotonin-Transporter (SERT oder 5HTT) Besetzung mit selektiven Serotonin-Wiederaufnahmehemmern (SSRI) zu charakterisieren. Anhand eines Serotonin-Transporter-Radioliganden wurde gezeigt, dass die Blutkonzentrationen von Citalopram, Paroxetin, Fluoxetin und Sertralin gut mit der Serotonin-Transporter-Besetzung korrelieren. Dabei zeigte sich, dass für eine optimale klinische Wirkung eine Belegung von mindestens 80% erreicht werden sollte. PET-Untersuchungen haben somit von einer beträchtlichen Zahl an Psychopharmaka äußerst relevante Informationen für die Bestimmung der optimalen Blut-Konzentration erbracht [15].
Pharmakogenetische Aspekte
Die klinische Wichtigkeit von pharmakogenetischen Faktoren für die Pharmakokinetik und Pharmakodynamik von Neuropsychopharmaka ist zunehmend erkannt worden. Wie bereits erwähnt, weisen Arzneistoff-metabolisierende Enzyme, insbesondere CYP-Isoenzyme, eine hohe genetische Variabilität auf. Extensive Metabolisierer (EM) sind definiert als Wildtyp mit zwei aktiven Allelen. Langsame Metabolisierer (Poor Metabolizer, PM) weisen eine mangelnde Expression funktioneller Allele auf. Intermediate Metabolisierer (IM) sind entweder genetisch heterozygot, das heißt Träger eines aktiven und eines inaktiven Allels oder haben ein oder zwei Allele mit reduzierter Aktivität. Ultraschnelle Metabolisierer (UM) tragen Allele mit gesteigerter Aktivität oder Multiplikationen der funktionellen Allele. Die Genpolymorphismen der metabolisierenden Enzyme können klinisch äußerst relevant sein. Auf der einen Seite können bei PM unerwünschte Arzneimittelwirkungen und Toxizität aufgrund erhöhter Arzneistoff-Konzentrationen auftreten. Auf der anderen Seite kann bei UM unzureichendes Therapieansprechen aufgrund subtherapeutischer Arzneistoff-Konzentrationen auftreten. Prodrugs sind inaktive Vorstufen eines Arzneistoffs, die erst durch die Verstoffwechselung via CYP-Enzyme aktiviert werden, beispielsweise Codein zu Morphin und Tramadol zu Desmethyltramadol durch CYP2D6. Bei Einnahme solcher Arzneistoffe sind UM einem gesteigerten Risiko für Unverträglichkeiten ausgesetzt und PM sind nicht in der Lage, solche Prodrugs zu ihren pharmakologisch aktiven Metaboliten umzubauen. Ein neuer, vielversprechender Ansatz ist die Bestimmung der mRNA, welche CYP1A2, CYP2C9 und CYP2C19 in Leukozyten kodiert, denn mRNA-Level korrelieren gut mit hepatischen CYP-Aktivitäten, was sich bei Phänotypisierung der CYP-Enzyme mit Indikatorsubstanzen gezeigt hat.
Ursprünglich wurde der Metabolisiererstatus mit Indikatorsubstanzen (probe drugs) bestimmt, beispielsweise Coffein für CYP1A2, Omeprazol für CYP2C19, Metoprolol oder Dextromethorphan für CYP2D6 oder Midazolam für CYP3A4/5. Eine solche Phänotypisierung analysiert die metabolische Situation eines Patienten im Sinne eines „state markers“ und erlaubt die Detektion von metabolischen Besonderheiten, einschließlich genetischer Abweichungen. Sie kann auch angewendet werden, um den Einfluss von Umweltfaktoren, beispielsweise Rauchen oder Komedikation, auf die CYP-Aktivitäten zu untersuchen. Während der letzten Jahre wurde die CYP-Genotypisierung breit verfügbar. Der Vorteil einer Genotypisierung besteht darin, dass das Ergebnis nicht durch Umweltfaktoren beeinflusst werden kann, es repräsentiert einen „trait marker“. Eine Genotypisierung kann in jeder Situation durchgeführt werden und das Ergebnis gilt lebenslänglich. Obwohl die funktionelle Bedeutung genetischer Varianten von CYP-Enzymen sehr gut untersucht ist, ist eine Vorhersage der individuellen Enzymaktivität nur grob möglich, unter anderem durch seltene Genvarianten, die in der Praxis der Genotypisierung nicht erfasst werden.
Andere Enzymsysteme, wie UDP-Glucuronosyltransferasen (UGT), zeigen ebenfalls genetische Polymorphismen, aber ihre klinische Relevanz in der Pharmakotherapie und für Dosisanpassungen ist weniger gut untersucht als die CYP-Polymorphismen.
Für den ABC-Transporter P-gp wurde gefunden, dass der ABCB1-Genotyp das Therapieansprechen auf Antidepressiva und Antipsychotika beeinflussen kann. Patienten scheinen auf Antidepressiva, welche P-gp-Substrate sind, abhängig vom ABCB1-Genotyp, unterschiedlich zu respondieren. Eine ABCB1-Genotypisierung kann demnach hilfreich sein, um das Therapieansprechen auf Antidepressiva zu verbessern. Mittlerweile wurden über 30 Studien durchgeführt, welche untersuchten, ob genetische Varianten innerhalb ABCB1 die klinische Wirksamkeit und/oder Verträglichkeit von Antidepressiva vorhersagen können. Bei Trägern einer minoren Variante der SNPs rs2032583 und rs2235040 im ABCB1-Gen wurde wiederholt eine gesteigerte Empfindlichkeit für antidepressive Effekte entdeckt im Vergleich zu Nicht-Trägern [44]. Verschiedene weitere Studien beobachteten jedoch kein besseres Therapieansprechen oder häufigere Unverträglichkeiten zwischen Trägern und Nicht-Trägern. Eine klinische Studie mit Antidepressiva, die P-gp-Substrate waren, zeigte bei Dosen innerhalb des empfohlenen therapeutischen Bereichs eine gesteigerte Wirksamkeit in Trägern des minoren Allels von rs2235083. Die Strategie einer Dosissteigerung für Träger des majoren Allels war nicht effektiv. Andere Strategien, beispielsweise Wechsel auf ein Antidepressivum, welches kein P-gp-Substrat ist, wurden bisher nicht untersucht. Größere Studien sind vonnöten, bevor man sichere Schlussfolgerungen bezüglich der klinischen Relevanz von ABCB1-Genvariationen und den daraus folgenden Konsequenzen ziehen kann.
Zusätzlich zu den zuvor beschriebenen pharmakokinetischen Faktoren gibt es zunehmende Evidenz, dass auch pharmakodynamische Genvarianten, beispielsweise Rezeptoren, Enzyme oder Ionenkanäle, das Therapieansprechen bei psychiatrischen Erkrankungen beeinflussen. Bei affektiven Störungen ist das Serotonin-Transporter-Gen (5HTT; SLC6A4) das am häufigsten untersuchte Gen in diesem Kontext. Die Ergebnisse waren bisher jedoch nicht beweiskräftig.
Genomweite Assoziationsstudien (GWAS) mit hypothesenfreiem Ansatz wurden in den STAR*D, Munich Antidepressant Response Signature (MARS) und den Genome-based Therapeutic Drugs for Depression (GENDEP) Proben durchgeführt. Diese Studien fanden keine signifikanten genetischen Marker für antidepressives Therapieansprechen.
Therapieansprechen auf Lithium wurde per Metaanalyse in einer Kohorte von mehr als 2500 Patienten an 22 Zentren weltweit untersucht. Es fand sich kein Hinweis auf genetische Marker, die für die Therapieentscheidung in der klinischen Praxis nutzbar sind.
Bei psychotischen Erkrankungen wurde die Variation der Dopamin-Rezeptorgene DRD2, DRD3 und DRD4 intensiv bezüglich des antipsychotischen Therapieansprechens untersucht. Diese Studien brachten keine robusten Ergebnisse. Bei alkoholabhängigen Patienten wies eine kürzlich publizierte Metaanalyse auf eine Rolle des funktionalen A118G-Polymorphismus im µ-Opioid-Rezeptorgen (OPRM1) hin. Es konnte mit unterschiedlichem Therapieansprechen auf Naltrexon assoziiert werden. Weitere Untersuchungen sind notwendig zur Bestimmung der klinischen Validität (z.B. Sensitivität, Spezifität, positiver/negativer prädiktiver Wert) und des Nutzenprofils für pharmakogenetische Ansätze, basierend auf der OPRM1-Variation.
Die vielversprechendsten Ergebnisse in der Pharmakogenetik lieferten Analysen auf pharmakodynamischer Ebene bezüglich der Vorhersage von unerwünschten Arzneimittelwirkungen. Über den menschlichen Leukozyten-Antigen-Marker HLA-B*1502 wurde übereinstimmend berichtet, dass dieser bei asiatischen Patienten unter Behandlung mit Carbamazepin ein gesteigertes Risiko für die Entwicklung eines Stevens-Johnson-Syndroms anzeigt. Der PGxPredict: CLOZAPINE Test wurde für die HLA-DQB1-Genvariation für die Vorhersage des Agranulozytose-Risikos entwickelt. Der Test wurde jedoch aufgrund zwar hoher Spezifität (98,4%), aber geringer Sensitivität (21,5%) eingestellt. 5-HTR2C, Melanocortin-4-Rezeptor (MC4R), Neuropeptid Y (NPY), Cannabinoid-Rezeptor-1 (CNR1) und Leptin-Gen-Variationen scheinen prädiktiv für die Gewichtszunahme unter Antipsychotika zu sein. Gut replizierbare Gen-Variationen wurden bei Dystonie/tardiver Dyskinesie durch Antipsychotika beschrieben: Variationen im RGS2 (Regulator der G-Protein-2-Signalgebung), ein Gen, welches Dopamin-Rezeptor-Signal-Transduktionen moduliert, sowie Variationen im Serotonin-Rezeptor-Gen HTR2C und möglicherweise auch HTR2A. Eine Variation im Serotonin-Rezeptor-Gen HTR1A (rs6295; C-1019G) war durchgängig in Verbindung mit dem antipsychotischen Therapieansprechen bezüglich der Negativsymptomatik von schizophrenen Patienten beschrieben.
Arzneistoff-Konzentration im Blut zur neuropsychopharmakologischen Therapieleitung
Zur neuropsychopharmakologischen Therapieleitung berücksichtigt TDM pharmakodynamische und pharmakokinetische Aspekte. Es muss geprüft werden, (1) ob die Arzneistoff-Konzentration innerhalb des orientierenden therapeutischen Bereiches ist, sodass die therapeutische Wirksamkeit und eine akzeptable Verträglichkeit erwartet werden kann und (2) ob die Arzneistoff-Konzentration im Blut zu der verschriebenen Dosis passt um herauszufinden, ob die Medikation wie verschrieben eingenommen wurde und/oder ob pharmakokinetische Besonderheiten vorliegen. Deshalb muss zwischen therapeutisch effektiven und zu erwartenden dosisbezogenen Arzneistoff-Konzentrationen unterschieden, und beide bei der Beurteilung der TDM-Ergebnisse berücksichtigt werden [17, 18]. Des Weiteren ermöglicht die Bestimmung der Metabolit zu Muttersubstanz Ratio und eine Probearzneistoff-Phänotypisierung die Evaluation des individuellen pharmakokinetischen Phänotypen.
Der therapeutische Referenzbereich
Das Massenwirkungsgesetz besagt, dass alle pharmakologischen Effekte konzentrationsbezogen sind. TDM basiert auf dieser Annahme hinsichtlich therapeutischer Verbesserung und unerwünschten Arzneimittelwirkungen. TDM setzt auch voraus, dass es einen Blutkonzentrationsbereich des Wirkstoffs, den sogenannten „therapeutischen Referenzbereich“, gibt, der durch maximale Effektivität bei ausreichender Verträglichkeit und Sicherheit gekennzeichnet ist. Studien über die Beziehungen zwischen Arzneistoff-Konzentrationen im Blut und klinischer Besserung haben dieses Konzept seit den 60er-Jahren für Lithium, trizyklische Antidepressiva und Antipsychotika der ersten Generation unterstützt. Systematische Übersichtsarbeiten und Metaanalysen, die auf entsprechend angelegten Studien beruhen, haben überzeugend nachgewiesen, dass es eine signifikante Beziehung zwischen Blutkonzentration und klinischer Wirkung für Nortriptylin, Imipramin und Desipramin gibt [3]. Eine Metaanalyse von 45 Studien hat für Amitriptylin als Modellverbindung gezeigt, dass verschiedene statistische Ansätze zu nahezu identischen therapeutischen Referenzbereichen führten. Für neue Antipsychotika wie Aripiprazol, Olanzapin oder Risperidon wurde ebenfalls eine enge Beziehung zwischen Arzneistoff-Konzentration im Blut und klinischer Wirksamkeit detektiert.
Der therapeutische Referenzbereich ist ein essenzieller Zielbereich für die TDM-geleitete Pharmakotherapie. Seine Abschätzung erfordert die Bestimmung einer unteren und oberen Grenze von therapeutisch effektiven und verträglichen Arzneistoff-Konzentrationen im Blut. Eine generell akzeptierte Methode zur Abschätzung dieser Grenzen existiert nicht, und methodische Einschränkungen wie Therapieansprechen auf Placebo oder Therapieresistenz müssen berücksichtigt werden [35]. PET-Studien waren sehr hilfreich, um die Grenzen für Antipsychotika und Antidepressiva zu definieren. Diese Technik ist jedoch sehr teuer und nur in wenigen Zentren verfügbar. Studien mit fixer Dosis sind der angemessenste Weg, um therapeutische Referenzbereiche zu bestimmen. Die Angabe von therapeutisch wirksamen Arzneistoff-Konzentrationen im Blut ist jedoch bisher nicht gesetzlich für die Arzneistoff-Zulassung vorgeschrieben. Wir raten dringend, TDM im Entwicklungsprozess neuer Arzneistoffe während der klinischen Prüfung zu implementieren.
Für den Begriff „therapeutischer Referenzbereich“ (Kasten) existieren viele Synonyme, unter anderem „therapeutisches Fenster“, „therapeutischer Bereich“, „optimale Plasma-Konzentration“, „effektive Plasma-Konzentration“, „Zielkorridor“, „Zielkonzentration“ oder „orientierender therapeutischer Bereich“, der Begriff, der im ersten TDM-Konsensus angewandt wurde [3]. Die AGNP-TDM-Gruppe entschied sich 2011, den Begriff „therapeutischer Referenzbereich“, in Übereinstimmung mit den TDM-Leitlinien für Antiepileptika [30], und den Begriff „Arzneistoff-Konzentration im Blut“ zu nutzen, der gleichgesetzt werden kann mit den Begriffen „Plasma-Konzentration“, „Serum-Konzentration“, „Plasmaspiegel“, „Serumspiegel“ oder „Blutspiegel“.
Definition: Therapeutische Referenzbereiche
Die „therapeutischen Referenzbereiche“, die in dieser Leitlinie angegeben werden (Tab. 4), definieren Bereiche von Arzneistoff-Konzentrationen im Blut mit einer unteren Grenze, unterhalb derer eine durch das Medikament induzierte therapeutische Wirksamkeit relativ unwahrscheinlich ist, und einer oberen Grenze, ab der die Verträglichkeit abnimmt oder oberhalb derer es unwahrscheinlich ist, dass noch eine therapeutische Verbesserung erreicht werden kann. Der therapeutische Referenzbereich ist ein orientierender, populationsbezogener Bereich, der nicht unbedingt für alle Patienten gültig sein muss. Einzelne Patienten können ein optimales therapeutisches Ansprechen bei einer Arzneistoff-Konzentration zeigen, die außerhalb des therapeutischen Referenzbereichs liegt. Am besten sollte die neuropsychopharmakologische Therapie anhand der Identifikation der individuellen therapeutischen Konzentration des Patienten geleitet werden. Die therapeutischen Referenzbereiche, die von der TDM-Gruppe der AGNP empfohlen werden, sind in Tabelle 4 abgebildet.
Die in Tabelle 4 gelisteten therapeutischen Referenzbereiche sind evidenzbasiert ermittelt worden und wurden aus der Literatur durch die oben beschriebene strukturierte Überprüfung abgeleitet. Für nur 17 neuropsychiatrische Arzneistoffe wurden in der Literatur therapeutische Referenzbereiche aus randomisierten, klinischen Studien gefunden. Für die meisten Arzneistoffe wurden die Referenzbereiche aus Studien mit therapeutisch wirksamen Dosierungen abgeleitet. Die in Tabelle 4 gelisteten Referenzbereiche sind in der Regel jene für die primäre Indikation. Einige Medikamente sind jedoch für verschiedene Indikationen zugelassen. Zum Beispiel werden Antidepressiva auch für die Behandlung von Angstzuständen oder Zwangserkrankungen sowie chronischem Schmerz eingesetzt, und Antipsychotika sind zugelassen für die Behandlung von affektiven Störungen. Für diese Indikationen existieren bislang nur wenige Informationen bezüglich einer optimalen Arzneistoff-Konzentration im Blut. Ausnahmen sind Carbamazepin, Lamotrigin und Valproinsäure (Valproat), die deshalb zweimal in Tabelle 4 aufgeführt sind. Therapiebegleitende Studien sind angelaufen, um therapeutische Referenzbereiche für Kinder und Jugendliche zu evaluieren [11, 16]. Für Alterspatienten besteht dringender Bedarf, ähnliche Studien zukünftig durchzuführen.
Tab. 4. Empfohlene therapeutische Referenzbereiche (Konsensus) von Neuropsychopharmaka, Bereiche von Eliminationshalbwertszeiten (t1/2) und Warnschwellen für das Labor sowie Empfehlungsgrade* zur Anwendung von TDM in der klinischen Routine zur Dosisoptimierung ohne spezifische Indikation (siehe Tabelle 7). Sofern nicht anders vermerkt, beziehen sich die Referenzbereiche und die Warnschwellen auf Arzneistoff-Talspiegel (Cmin). Für die Interpretation der TDM-Ergebnisse muss geprüft werden, ob die gemessene Arzneistoff-Konzentration innerhalb des therapeutischen Referenzbereichs liegt. Konzentrationen unterhalb oder oberhalb des Bereiches sind Indikatoren für ein unzureichendes Therapieansprechen auf eine medikamentöse Behandlung oder für das Auftreten unerwünschter Arzneimittelwirkungen. CF: Konversionsfaktor
|
Arzneistoff und aktiver Metabolit |
Therapeutischer Referenzbereich |
t1/2 [h] |
Warnschwelle für das Labor |
Empfehlungsgrad* |
CF |
Kommentare |
|
Antidepressiva |
||||||
|
Agomelatin |
7–300 ng/ml (1–2 h nach Einnahme von 50 mg) |
1–2 h |
600 ng/ml |
4 |
4,11 |
Wegen der raschen Elimination sind keine messbaren Talspiegel unter Dauereinnahme zu erwarten; Messungen, bevorzugt von Cmax, sollten auf spezifische Fragestellungen begrenzt sein. |
|
Amitriptylin plus Nortriptylin |
80–200 ng/ml |
10–28 h 18–44 h |
300 ng/ml |
1 |
3,60 3,80 |
Gesteigerte Toxizität bei Kindern und PM von CYP2D6, konzentrationsabhängige Beeinträchtigung der Fahrtüchtigkeit |
|
Amitriptylinoxid Amitriptylin plus Nortriptylin |
80–200 ng/ml |
1,1–2,5 h 5–17 h 18–44 h |
300 ng/ml |
1 |
3,41 3,60 3,80 |
Prodrug, aktive Fraktion ist die Summe von Amitriptylin und Nortriptylin |
|
Bupropion Hydroxybupropion |
10–100 ng/ml 850–1500 ng/ml |
1–15 h 17–47 h |
– 2000 ng/ml |
2 |
4,17 3,91 |
Bupropion ist instabil, Hydroxybupropion ist die pharmakologisch aktive Hauptkomponente (50% der Bupropion-Aktivität), andere Metaboliten tragen mit maximal 20% zur pharmakologischen Aktivität bei; der therapeutische Referenzbereich bezieht sich ausschließlich auf Hydroxybupropion |
|
Citalopram |
50–110 ng/ml |
38–48 h |
220 ng/ml |
1 |
3,08 |
N-demethylierte Metaboliten scheinen sehr schwach zur pharmakologischen Wirkung beizutragen |
|
Clomipramin plus N-Desmethylclomipramin |
230–450 ng/ml |
16–60 h 37–43 h |
450 ng/ml |
1 |
3,18 3,32 |
Unterschiedliches pharmakologisches Profil der Muttersubstanz (bevorzugte Serotonin Wiederaufnahmehemmung) und des aktiven Metaboliten (bevorzugte Noradrenalin Wiederaufnahmehemmung) |
|
Desipramin |
100–300 ng/ml |
15–18 h |
300 ng/ml |
2 |
3,75 |
Metaboliten möglicherweise in vivo aktiv |
|
Desvenlafaxin |
100–400 ng/ml |
10–17 h |
800 ng/ml |
3 |
3,80 |
Keine aktiven Metaboliten |
|
Dosulepin=Dothiepin |
45–100 ng/ml |
18–21 h |
200 ng/ml |
2 |
3,39 |
Unerwünschte Arzneimittelwirkungen korrelieren mit der Arzneistoff-Konzentration im Blut |
|
Doxepin plus |
50–150 ng/ml |
15–20 h |
300 ng/ml |
2 |
3,58 3,77 |
|
|
Duloxetin |
30–120 ng/ml |
9–19 h |
240 ng/ml |
2 |
3,36 |
Keine aktiven Metaboliten, renale Unverträglichkeiten sind mit erhöhten Duloxetin Konzentrationen im Blut assoziiert |
|
Escitalopram |
15–80 ng/ml |
27–32 h |
160 ng/ml |
2 |
3,08 |
N-demethylierte Metaboliten scheinen schwach zur pharmakologischen Wirkung beizutragen |
|
Fluoxetin plus N-Desmethylfluoxetin |
120–500 ng/ml |
4–6 Tage 4–16 Tage |
1000 ng/ml |
3 |
3,23 3,39 |
Lange Eliminationshalbwertszeit von Norfluoxetin (durchschnittlich 14 Tage) und langanhaltende Hemmung von CYP2D6 |
|
Fluvoxamin |
60–230 ng/ml |
21–43 h |
500 ng/ml |
2 |
3,14 |
Inhibition von CYP1A2, CYP2C19 und altersabhängiger Anstieg der Arzneistoff-Konzentration im Blut, Maximale In-vivo-Inhibition von CYP1A2 und CYP2C19 bei 60 ng/ml erreicht |
|
Imipramin plus Desipramin |
175–300 ng/ml |
11–25 h 15–18 h |
300 ng/ml |
1 |
3,57 3,75 |
Hydroxylierte Metaboliten, CL vom Alter des Patienten beeinflusst |
|
Levomilnacipran |
80–120 ng/ml |
6–9 h |
200 ng/ml |
3 |
2,24 |
Referenzbereich bezieht sich auf Steady-State Konzentrationen, die unter einer therapeutischen Dosis von 100 mg/Tag zu erwarten sind |
|
Maprotilin |
75–130 ng/ml |
20–58 h |
220 ng/ml |
2 |
3,60 |
Aktiver Metabolit N-Desmethylmaprotilin |
|
Mianserin |
15–70 ng/ml |
14–33 h |
140 ng/ml |
3 |
3,78 |
|
|
Milnacipran |
100–150 ng/ml |
5–8 h |
300 ng/ml |
2 |
2,24 |
Referenzbereich bezieht sich auf zu erwartende Arzneistoff-Konzentrationen bei einer therapeutisch empfohlenen Dosis von 100 mg/Tag; optimale Konzentrationen könnten höher liegen, da Arzneistoff-Konzentrationen im Blut >200 ng/ml benötigt werden, um 80% der Serotonin- und Noradrenalintransporter zu besetzen |
|
Mirtazapin |
30–80 ng/ml |
20–40 h |
160 ng/ml |
2 |
3,77 |
Der N-demethylierte Metabolit scheint nicht zur pharmakologischen Wirkung beizutragen |
|
Moclobemid |
300–1000 ng/ml |
2–7 h |
2000 ng/ml |
3 |
3,72 |
Die Metaboliten tragen nicht zur pharmakologischen Wirkung bei |
|
Nortriptylin |
70–170 ng/ml |
18–44 h |
300 ng/ml |
1 |
3,80 |
Hydroxylierte Metaboliten, PM von CYP2D6 und geringe CYP3A4 Aktivität sind mit einem gesteigerten Toxizitätsrisiko assoziiert |
|
Paroxetin |
20–65 ng/ml |
12–44 h |
120 ng/ml |
3 |
3,04 |
Inhibition von CYP2D6 |
|
Reboxetin |
60–350 ng/ml |
13–30 h |
700 ng/ml |
3 |
3,19 |
|
|
Sertralin |
10–150 ng/ml |
22–36 h |
300 ng/ml |
2 |
3,27 |
Der N-demethylierte Metabolit hat eine zweifach längere Eliminationshalbwertszeit als Sertralin und 1/20 der pharmakologischen Aktivität von Sertralin; gleiche Arzneistoff-Konzentrationen bei Kindern und Jugendlichen |
|
Tianeptin |
30–80 ng/ml |
2,5–3 h |
160 ng/ml |
3 |
2,89 |
|
|
Tranylcypromin |
≤50 ng/ml |
1–3 h |
100 ng/ml |
4 |
7,51 |
Wegen der irreversiblen Hemmung von Monoaminoxidasen gibt es keine Korrelation zwischen Arzneistoff-Konzentrationen im Blut und pharmakologischen Effekten |
|
Trazodon |
700–1000 ng/ml |
4–11 h |
1200 ng/ml |
2 |
2,69 |
|
|
Trimipramin |
150–300 ng/ml |
23–24 h |
600 ng/ml |
2 |
3,40 |
Aktiver Metabolit N-Desmethyltrimipramin |
|
Venlafaxin plus O-Desmethylvenlafaxin |
100–400 ng/ml |
14–18 h 10–17 h |
800 ng/ml |
2 |
3,61 3,80 |
Für die meisten Patienten ist in vivo O-Desmethylvenlafaxin die hauptsächlich aktive Komponente; Konzentrationen über 222 ng/ml sind prädiktiv für ein Therapieansprechen; N-demethyliertes Venlafaxin scheint nicht zur pharmakologischen Wirkung beizutragen. Bei Arzneistoff-Konzentrationen im Blut (aktive Fraktion) unter 100 ng/ml wirkt die Substanz bevorzugt als SSRI; t1/2 ist angegeben für die Extended-Release-Formulierung (verzögerte Freisetzung) |
|
Vilazodon |
30–70 ng/ml |
18–32 h |
140 ng/ml |
3 |
2,26 |
Hauptmetaboliten repräsentieren 27% des insgesamt zirkulierenden Arzneistoffs, keine TDM Daten, Referenzbereiche beziehen sich auf Steady-State Konzentrationen bei therapeutischen Dosen |
|
Vortioxetin |
10–40 ng/ml |
57–66 h |
80 ng/ml |
2 |
3,35 |
Mindestens vier inaktive Metaboliten |
|
Antipsychotika |
||||||
|
Amisulprid |
100–320 ng/ml |
12–20 h |
640 ng/ml |
1 |
2,71 |
Keine Metabolisierung, einige Patienten könnten Arzneistoff-Konzentrationen über 320 ng/ml für einen ausreichenden Therapieeffekt benötigen |
|
Aripiprazol |
100–350 ng/ml |
60–80 h |
1000 ng/ml |
2 |
2,23 |
Dehydroaripiprazol-Konzentrationen betragen ca. 45% der Muttersubstanz Apparente Eliminationshalbwertszeit 30–47 Tage |
|
Asenapin |
1–5 ng/ml |
13–39 h |
10 ng/ml |
4 |
3,50 |
|
|
Benperidol |
1–10 ng/ml |
4–6 h |
20 ng/ml |
3 |
2,62 |
Unter Langzeitbehandlung mit hohen Dosen werden möglicherweise höhere Spiegel toleriert, wegen adaptiver Veränderungen. |
|
Brexpiprazol |
40–140 ng/ml |
91 h |
280 ng/ml |
3 |
Der Hauptmetabolit beträgt ca. 23–48% der Muttersubstanz und trägt nicht zum therapeutischen Effekt bei |
|
|
Bromperidol |
12–15 ng/ml |
20–36 h |
30 ng/ml |
2 |
4,38 |
|
|
Cariprazin |
10–20 ng/ml |
48–120 h |
40 ng/ml |
3 |
2,34 |
Aktive Metaboliten sind N-Desmethylcariprazin und N, N-Didesmethylcariprazin |
|
Chlorpromazin |
30–300 ng/ml |
15–30 h |
600 ng/ml |
2 |
3,14 |
|
|
Chlorprothixen |
20–300 ng/ml |
8–12 h |
400 ng/ml |
3 |
3,17 |
|
|
Clozapin |
350–600 ng/ml |
12–16 h |
1000 ng/ml |
1 |
3,06 |
Hauptmetabolit N-Desmethylclozapin mit ungeklärter antipsychotischer Aktivität; der therapeutische Referenzbereich ist bei pädiatrischen Patienten wahrscheinlich niedriger |
|
Flupentixol |
0,5–5 ng/ml |
20–40 h |
15 ng/ml |
2 |
2,30 |
Apparente t1/2 für Flupentixoldecanoat 17 Tage |
|
Fluphenazin |
1–10 ng/ml |
16 h |
15 ng/ml |
1 |
2,29 |
Apparente t1/2 für Fluphenazindecanoat 14 Tage |
|
Fluspirilen |
0,1–2,2 ng/ml |
7–14 Tage |
4,4 ng/ml |
3 |
2,10 |
|
|
Haloperidol |
1–10 ng/ml |
12–36 h |
15 ng/ml |
1 |
2,66 |
Unter Langzeitbehandlung mit hohen Dosen werden durch adaptive Rezeptorveränderungen möglicherweise höhere Spiegel toleriert; apparente t1/2 für Haloperidoldecanoat 17 Tage |
|
Iloperidon |
5–10 ng/ml |
18–33 h |
20 ng/ml |
3 |
2,34 |
|
|
Levomepromazin |
30–160 ng/ml |
16–78 h |
320 ng/ml |
3 |
3,04 |
|
|
Loxapin |
5–10 ng/ml |
6–8 h |
20 ng/ml |
3 |
3,05 |
Applikation als thermisch generiertes Aerosol |
|
Lurasidon |
15–40 ng/ml |
20–40 h |
120 ng/ml |
3 |
2,03 |
|
|
Melperon |
30–100 ng/ml |
4–6 h |
200 ng/ml |
3 |
3,80 |
QTc-Zeit korreliert wahrscheinlich mit Arzneistoff-Konzentration im Blut |
|
Olanzapin |
20–80 ng/ml |
30–60 h |
100 ng/ml |
1 |
3,20 |
Unter Olanzapinpamoat ist mit einem Postinjektionssyndrom zu rechnen, wenn die Arzneistoff-Konzentrationen auf über 100 ng/ml ansteigen; apparente t1/2 für Olanzapinpamoat 30 Tage |
|
Paliperidon (=9-Hydroxyrisperidon) |
20–60 ng/ml |
17–23 h |
120 ng/ml |
2 |
2,35 |
|
|
Perazin |
100–230 ng/ml |
8–16 h |
460 ng/ml |
1 |
2,95 |
|
|
Perphenazin |
0,6–2,4 ng/ml |
8–12 h |
5 ng/ml |
1 |
2,48 |
Apparente t1/2 für Perphenazinenanthat 4–6 Tage |
|
Pimozid |
15–20 ng/ml |
23–43 h |
20 ng/ml |
3 |
2,17 |
|
|
Pipamperon |
100–400 ng/ml |
17–22 h |
500 ng/ml |
3 |
2,66 |
Apparente t1/2 für Paliperidonpalmitat 25–49 Tage |
|
Prothipendyl |
30–80 ng/ml (12 h |
2–3 h |
500 ng/ml |
4 |
3,35 |
Für akute Sedierung |
|
Quetiapin N-Desalkylquetiapin |
100–500 ng/ml 100–250 ng/ml |
6–11 h 10–13 h |
1000 ng/ml |
2 |
2,61 3,39 |
Wenn die retardierte Formulierung am Vorabend eingenommen wurde und die Blutentnahme am Morgen danach erfolgt, sind die zu erwartenden Arzneistoff-Konzentrationen im Blut doppelt so hoch wie die Talspiegel vor der abendlichen Einnahme |
|
Risperidon plus 9-Hydroxyrisperidon |
20–60 ng/ml |
2–4 h 17–23 h |
120 ng/ml |
2 |
2,44 2,35 |
Unerwünschte Arzneimittelwirkungen korrelieren mit der Arzneistoff-Konzentration im Blut. Um neurologische unerwünschte Arzneimittelwirkungen zu vermeiden sollten Arzneistoff-Konzentrationen >40 ng/ml nur bei Fällen mit unzureichendem Therapieeffekt angestrebt werden; |
|
Sertindol |
50–100 ng/ml |
55–90 h |
200 ng/ml |
2 |
2,27 |
Aktiver Metabolit Dehydrosertindol (Konzentration bei therapeutischen Dosen 40–60 ng/ml); konzentrationsabhängige Zunahme des QT-Intervalls durch Blockade von kardialen Kaliumkanälen |
|
Sulpirid |
200–1000 ng/ml |
8–14 h |
1000 ng/ml |
2 |
2,93 |
Keine Metaboliten, renale Elimination |
|
Thioridazin |
100–200 ng/ml |
30 h |
400 ng/ml |
1 |
2,70 |
Kontraindiziert bei CYP2D6 PM |
|
Ziprasidon |
50–200 ng/ml |
4–8 h |
400 ng/ml |
2 |
2,55 |
Das Arzneimittel sollte mit einer Mahlzeit eingenommen werden, ansonsten ist mit niedrigeren Arzneistoff-Konzentrationen im Blut zu rechnen |
|
Zotepin |
10–150 ng/ml |
13–16 h |
300 ng/ml |
3 |
3,01 |
|
|
Zuclopenthixol |
4–50 ng/ml |
15–25 h |
100 ng/ml |
3 |
2,49 |
Apparente t1/2 für Zuclopenthixoldecanoat 19 Tage und 1–2 Tage für das -acetat |
|
Stimmungsstabilisierer |
||||||
|
Carbamazepin |
4–10 µg/ml |
10–20 h |
20 µg/ml |
2 |
4,23 |
Der aktive Metabolit Carbamazepin-10,11-epoxid trägt äquipotent zur Muttersubstanz zur klinischen Wirkung bei, und insbesondere zu unerwünschten Arzneimittelwirkungen |
|
Lamotrigin |
1–6 µg/ml |
14–104 h |
20 µg/ml |
2 |
3,90 |
Bisher kein spezifischer Referenzbereich für stimmungsstabilisierende Effekte; bei Patienten mit therapieresistenter Depression sollte die Arzneistoff-Konzentration im Blut über 3,25 µg/ml liegen; Valproat verlängert die Eliminationshalbwertszeit auf 45–75 h, Carbamazepin, Phenytoin oder Phenobarbital verringern diese auf 9–14 h aufgrund einer Induktion der UDP-Glucuronosyltransferase |
|
Lithium |
0,5–1,2 mmol/l Akut bis 1,2 mmol/l Chronisch 0,5–0,8 mmol/l |
14–30 h |
1,2 mmol/l (8 µg/ml) |
1 |
125,8 |
Altersabhänige Zunahme der Eliminationshalbwertszeit (30–36 h); die Lithium-Konzentration sollte in der Akutbehandlung bis 1,2 mmol/l betragen, in der Erhaltungstherapie 0,5–0,8 mmol/l |
|
Valproat |
50–100 µg/ml |
11–17 h |
120 µg/ml |
2 |
6,93 |
Manche Patienten benötigen 120 µg/ml in der akuten manischen Phase |
|
Antikonvulsiva |
||||||
|
Brivaracetam |
0,5–0,9 µg/ml |
7–11 h |
1,8 µg/ml |
3 |
4,72 |
Bei einer Dosis von 2-mal 50 mg/Tag |
|
Carbamazepin Carbamazepin-10,11-epoxid |
4–12 µg/ml |
10–20 h |
20 µg/ml |
2 |
4,23 |
Der aktive Metabolit Carbamazepin-10,11-epoxid trägt äquipotent zur Muttersubstanz zur klinischen Wirkung bei, und insbesondere zu unerwünschten Arzneimittelwirkungen |
|
Clobazam plus N-Desmethylclobazam |
30–300 ng/ml 300–3000 ng/ml |
36–42 h 71–82 h |
500 ng/ml 5000 ng/ml |
3 |
3,33 3,49 |
|
|
Clonazepam |
20–70 ng/ml |
30–40 h |
80 ng/ml |
3 |
3,17 |
Clonazepam akkumuliert nach wiederholter Einnahme; 7-Aminoclonazepam ist schwach pharmakologisch aktiv |
|
Ethosuximid |
40–100 µg/ml |
33–55 h |
120 µg/ml |
3 |
7,08 |
|
|
Eslicarbazepinacetat |
10–35 µg/ml |
20–40 h |
70 µg/ml |
3 |
3,37 |
Prodrug wird zum pharmakologisch aktiven Eslicarbazepin metabolisiert |
|
Felbamat |
30–80 µg/ml |
15–23 h |
100 µg/ml |
3 |
4,20 |
Clearance und Eliminationshalbwertszeit werden von einer eingeschränkten Nierenfunktion beeinflusst |
|
Gabapentin |
2–20 µg/ml |
5–7 h |
25 µg/ml |
3 |
5,84 |
|
|
Lacosamid |
1–10 µg/ml |
10–15h |
20 µg/ml |
3 |
2,66 |
|
|
Lamotrigin |
3–15 µg/ml |
14–104 h |
20 µg/ml |
2 |
3,90 |
Valproat verlängert die Eliminationshalbwertszeit auf 45–75 h, Carbamazepin, Phenytoin oder Phenobarbital verringern diese auf 9–14 h aufgrund einer Induktion der UDP-Glucuronosyltransferase |
|
Levetiracetam |
10–40 µg/ml |
6–8 h |
50 µg/ml |
4 |
5,88 |
Signifikante Abnahme der Clearance mit zunehmenden Lebensalter, was eine Dosisreduktion von 30 bis 50% erforderlich macht, altersabhängiger Anstieg der t1/2 |
|
Methosuximid N-Desmethylmethosuximid |
– 10–40 µg/ml |
1–3 h 36–45 h |
– 45 µg/ml |
2 |
4,92 5,29 |
Eliminationshalbwertszeit steigt bei schwerer Nierenfunktionsstörung; der Metabolit ist die aktive Komponente in vivo |
|
Oxcarbazepin 10-Hydroxycarbazepin |
– 10–35 µg/ml |
5 h 10–20 h |
– 40 µg/ml |
2 |
3,96 3,73 |
Der Metabolit ist die aktive Komponente in vivo |
|
Perampanel |
180–980 ng/ml |
48–105 h |
1000 ng/ml |
3 |
2,86 |
Carbamazepin und andere CYP3A4-Induktoren reduzieren die Eliminationshalbwertszeit auf 25 h |
|
Phenobarbital |
10–40 µg/ml |
80–120 h |
50 µg/ml |
1 |
4,31 |
|
|
Phenytoin |
10–20 µg/ml |
20–60 h |
25 µg/ml |
1 |
3,96 |
|
|
Pregabalin |
2–5 µg/ml |
6 h |
10 µg/ml |
3 |
6,28 |
|
|
Primidon Phenobarbital |
5–10 µg/ml 10–40 µg/ml |
14–15 h |
25 µg/ml 50 µg/ml |
2 |
4,58 |
Phenobarbital ist der aktive Metabolit von Primidon |
|
Retigabin |
0,45–0,90 µg/ml |
8–10 h |
1,8 µg/ml |
3 |
3,29 |
Therapeutischer Referenzbereich bezieht sich auf zu erwartende Arzneistoff-Konzentrationen im Blut unter einer therapeutischen Dosis von 600 mg/Tag |
|
Rufinamid |
5–30 µg/ml |
6–10 h |
40 µg/ml |
2 |
4,20 |
|
|
Stiripentol |
1–10 µg/ml |
4–13 h |
15 µg/ml |
2 |
4,27 |
|
|
Sultiam |
2–8 µg/ml |
3–30 h |
12 µg/ml |
2 |
3,46 |
|
|
Tiagabin |
20–200 ng/ml |
7–9 h |
300 ng/ml |
2 |
2,66 |
|
|
Topiramat |
2–10 µg/ml |
19–23 h |
16 µg/ml |
3 |
2,95 |
|
|
Valproat |
50–100 µg/ml |
17–30 h |
120 µg/ml |
2 |
6,93 |
|
|
Vigabatrin |
2–10 µg/ml |
5–8 h |
20 µg/ml |
4 |
7,74 |
|
|
Zonisamid |
10–40 µg/ml |
49–77 h |
40 µg/ml |
2 |
4,71 |
|
|
Anxiolytika/Hypnotika |
||||||
|
Alprazolam |
5–50 ng/ml 20–40 ng/ml (Panikstörung) |
12–15 h |
100 ng/ml |
3 3 |
3,22 |
|
|
Bromazepam |
50–200 ng/ml |
15–35 h |
300 ng/ml |
4 |
3,16 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme. Hohe Arzneistoff-Konzentrationen können auf Missbrauch hinweisen |
|
Brotizolam |
4–10 ng/ml |
3–6 h |
20 ng/ml |
4 |
2,53 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Buspiron plus Metabolit |
1–4 ng/ml |
1–5 h 4–7 h |
30 ng/ml |
3 |
2,59 2,49 |
Hauptmetaboliten sind 6-Hydroxybuspiron und 1-(pyrimidinyl)piperazin (1-PP) |
|
Chlordiazepoxid |
400–3000 ng/ml |
5–30 h |
3500 ng/ml |
4 |
3,48 |
|
|
Clonazepam |
4–80 ng/ml |
19–30 h |
100 ng/ml |
4 |
3,17 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme |
|
Diazepam plus N-Desmethyldiazepam, Temazepam, Oxazepam Aktive Fraktion |
– – – – 100–2500 ng/ml |
24–48 h 80–103 h 5–13 h 4–15 h |
– – – – 3000 ng/ml |
4 |
3,51 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme. Hohe Arzneistoff-Konzentrationen können auf Missbrauch hinweisen. In Blutproben von 1000 bzgl. der Fahrtüchtigkeit beeinträchtigten und inhaftierten Patienten war die durchschnittliche Arzneistoff-Konzentration von Diazepam plus N-Desmethyldiazepam im Blut 500 ng/ml (Bereich 110–7600 ng/ml) |
|
Diphenhydramin |
10–30 ng/ml |
7–12 h |
60 ng/ml |
4 |
3,92 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Doxylamin |
50–100 ng/ml 50–160 ng/ml |
9–17 h |
320 ng/ml |
4 |
2,57 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Flunitrazepam |
6–12 ng/ml 12–15 ng/ml |
10–30 h |
50 ng/ml |
4 |
3,20 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme. Wenn hohe Arzneistoff-Konzentrationen im Blut über 15 ng/ml für eine Sedierung oder Schlafinduktion benötigt werden, deutet dies auf einen Missbrauch hin |
|
Flurazepam |
0–4 ng/ml |
2–3 h |
330 ng/ml |
4 |
2,58 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut. Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme. Wenn höhere Arzneistoff Konzentrationen im Blut benötigt werden, deutet dies auf einen Missbrauch hin |
|
N-1-Desalkylflurazepam |
10–22 ng/ml 75–165 ng/ml |
47–100 h |
||||
|
Hydroxyethylflurazepam |
5–10 ng/ml |
2–3 h |
||||
|
Gammahydroxybuttersäure (GHB, Natrium Oxybat) |
0,5–1,0 µg/ml |
0,4–0,8 h |
200 µg/ml |
4 |
9,60 |
Endogene Konzentration im Blut |
|
50–100 µg/ml |
Für eine Sedierung oder Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|||||
|
100–200 µg/ml |
Arzneistoff-Konzentration im Blut, um Bewusstlosigkeit zu induzieren |
|||||
|
Lorazepam |
30–100 ng/ml |
12–16 h |
300 ng/ml |
4 |
3,20 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme |
|
Lormetazepam |
2–10 ng/ml (nach 1,5 h) |
8–14 h |
100 ng/ml |
4 |
2,98 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Medazepam Desmethyldiazepam, Temazepam plus Oxazepam |
– – – 200–2500 ng/ml |
– 24–48 h |
– – – 3000 ng/ml |
– 4 |
– 3,69 3,19 3,49 |
Prodrug, aktive Fraktion sind die Metaboliten Desmethyldiazepam, Temazepam und Oxazepam |
|
Midazolam |
6–15 ng/ml 60–80 ng/ml |
1–3 h |
1000 ng/ml |
4 |
3,07 |
|
|
Modafinil |
1000–1700 ng/ml nach 200 mg/Tag |
10–12 h |
3400 ng/ml |
3 |
4,21 |
|
|
Nitrazepam |
30–100 ng/ml (nach 0,5–2 h) |
18–30 h |
200 ng/ml |
4 |
3,56 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut. Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme. Hohe Arzneistoff-Konzentrationen können auf Missbrauch hinweisen |
|
Nordazepam |
120–800 ng/ml |
50–90 h |
1500 ng/ml |
4 |
3,69 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme |
|
Opipramol |
50–500 ng/ml |
11 h |
1000 ng/ml |
3 |
2,87 |
|
|
Oxazepam |
200–1500 ng/ml |
4–15 h |
2000 ng/ml |
4 |
3,49 |
Bei chronischer Einnahme können die Arzneistoff-Konzentrationen im Blut deutlich höher sein als bei Nichteinnahme |
|
Pregabalin |
2–5 µg/ml |
5–7 h |
10 µg/ml |
3 |
6,28 |
TDM bei schwangeren Frauen empfohlen; hohe Arzneistoff-Konzentrationen können auf Missbrauch hinweisen |
|
Prothipendyl |
5–20 ng/ml (12 h nach 40–80 mg) |
2–3 h |
500 ng/ml |
4 |
3,35 |
Für die Indikation Schlafstörung |
|
Promethazin |
2–18 ng/ml |
10–14 h |
100 ng/ml |
4 |
3,47 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Temazepam |
600–1100 ng/ml (nach 1 h) |
5–13 h |
2000 ng/ml |
4 |
3,19 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut, chronische Einnahme führt nicht zu einer Toleranzentwicklung |
|
Triazolam |
2–20 ng/ml |
1–5 h |
40 ng/ml |
4 |
2,91 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Zaleplon |
20–40 ng/ml (nach 1–2 h) |
1–2 h |
200 ng/ml |
4 |
3,28 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Zolpidem |
80–160 ng/ml |
1–4 h 1–3 h bei Kindern |
320 ng/ml |
4 |
3,25 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut |
|
Zopiclon |
55–85 ng/ml (nach 1,5– 2 h) |
2–6 h |
300 ng/ml |
4 |
2,57 |
Für eine Schlafinduktion benötigte Arzneistoff-Konzentration im Blut, instabil bei Raumtemperatur |
|
Antidementiva |
||||||
|
Donepezil |
50–75 ng/ml |
70–80 h |
75 ng/ml |
2 |
2,64 |
Positive Korrelation zwischen Donepezil Konzentration im Blut und der Inhibition der AChE-Aktivität von Erythrozytenmembranen und klinischer Verbesserung |
|
Galantamin |
10–40 ng/ml |
8–10 h |
90 ng/ml |
3 |
3,48 |
Berichtete unerwünschte Arzneimittelwirkungen unter 32 mg/Tag |
|
Memantin |
90–150 ng/ml |
60–100 h |
300 ng/ml |
3 |
5,58 |
Die meisten Patienten sind in der klinischen Routine unterdosiert |
|
Rivastigmin |
Oral 8–20 ng/ml (1–2 h nach oraler Dosis) 5–13 ng/ml (1 h vor Applikation eines neuen Pflasters) |
1–2 h |
40 ng/ml |
3 |
4,00 |
Wegen der kurzen Eliminationshalbwertszeit sollte Cmax nach oraler Applikation bestimmt werden. Bei der Pflasterapplikation sollten Talspiegel des Arzneistoffs bestimmt werden. Positive Korrelation zwischen der Rivastigmin-Konzentration im Blut und der Inhibition der AChE-Aktivität von Erythrozytenmembranen |
|
Arzneistoffe zur Behandlung substanzbezogener Störungen |
||||||
|
Acamprosat |
250–700 ng/ml |
3–33 h |
1000 ng/ml |
3 |
8,68 |
|
|
Buprenorphin |
1–3 ng/ml |
2–5 h |
10 ng/ml (Cmax) |
2 |
2,38 |
Effektive Konzentrationen im Blut variieren patientenindividuell. Patienten mit chronischer Opioid-Einnahme könnten höhere Arzneistoff-Konzentrationen im Blut benötigen, um Absetzsymptome zu vermeiden. Unter empfohlener Maximaldosis von 24 mg Buprenorphin pro Tag betragen die zu erwartenden Talspiegel 3–6 ng/ml für Buprenorphin und 6–15 ng/ml für Norbuprenorphin |
|
Bupropion Hydroxybupropion |
550–1500 ng/ml |
1–15 h 17–47 h |
2000 ng/ml |
2 |
4,17 3,91 |
Bupropion ist instabil, Plasma oder Serum muss nach Blutentnahme gefroren (unter −20°C) gelagert werden. In einer klinischen Studie war 300 mg die effektivste Dosis, die zu indizierten Arzneistoff-Konzentrationen im Blut führte |
|
Clomethiazol |
100–5000 ng/ml |
2–5 h |
4 |
6,19 |
In schwer alkoholabhängigen Patienten können viel höhere Arzneistoff-Konzentrationen im Blut benötigt werden. Detoxifikation sollte anhand der klinischen Symptomatik erfolgen |
|
|
Diacetylmorphin (Heroin) Morphin |
70–350 ng/ml 5–30 ng/ml |
8 min 2–5 h |
4 |
2,71 3,50 |
Wirkstoff Konzentration im Blut nach Inhalation oder Injektion von 600–900 mg Diacetylmorphin nach 1 h oder 24 h nach Injektion von 300–1000 mg Patienten, die keine Opioide einnehmen, wären bei diesen Konzentrationen intoxikiert. Bei Opioid-Gebrauch variieren effektive und toxische Konzentrationen patientenindividuell in Abhängigkeit vom Toleranzlevel. Cmax 60–110 ng/ml nach 1 h nach oraler Einnahme von 50 mg Diacetylmorphin in gesunden Probanden |
|
|
Disulfiram Diethylthiomethyl-carbamat |
50–400 ng/ml 270–310 ng/ml |
6–9 h |
500 ng/ml – |
3 |
3,37 |
Disulfiram (DSF) ist ein Prodrug, der aktive Metabolit Diethylthiomethylcarbamat (DDTC-Me) ist ein möglicher Marker für eine korrekte Dosistitration von Disulfiram; in einer pharmakokinetischen Studie unter 300 mg DSF war die durchschnittliche (±Standardabweichung) Steady-State-Konzentration von DSF im Blut 170±10 ng/ml, von DDTC-Me 290±20 ng/ml |
|
Levomethadon |
250–400 ng/ml |
14–55 h |
400 ng/ml |
2 |
3,23 |
Bei Patienten, die keine Opioide einnehmen, sind effektive oder toxische Wirkstoffkonzentrationen im Blut deutlich geringer (100 ng/ml) als bei Patienten, die Opioide konsumieren. Chronische Anwender könnten höhere Konzentrationen im Blut benötigen, um Absetzsymptome zu vermeiden |
|
Methadon |
400–600 ng/ml |
24–48 h |
600 ng/ml |
2 |
3,23 |
Bei Patienten, die keine Opioide einnehmen, sind effektive oder toxische Wirkstoffkonzentrationen im Blut deutlich geringer (300 ng/ml) als bei Patienten, die Opioide konsumieren. Das Risiko einer QT-Zeitverlängerung steigt mit der Wirkstoff-Konzentration im Blut. Bei Konzentrationen über 656 ng/ml besteht ein hohes Risiko für eine QTc-Zeit über 450 ms |
|
Morphin |
10–100 ng/ml (Schmerz) 50–200 ng/ml |
11–21 h |
100 ng/ml |
4 |
6,17 |
Zur Schmerzbehandlung. Bei Hospizpatienten lagen Arzneistoff-Konzentrationen im Blut zur Behandlung von Schmerzen bei Krebs zwischen 6 und 356 ng/ml; höhere Arzneistoff-Konzentrationen werden zur Opioid-Substitution benötigt, mit täglichen Dosen von 500 bis 800 mg Morphinsulfat |
|
Nalmefen |
10–20 ng/ml |
5–11 h |
200 ng/ml |
4 |
3,26 |
Akuter Gebrauch bei Bedarf, wenn ein erkennbares Risiko eines Rückfalls bzgl. eines Alkoholmissbrauchs besteht. TDM kann bei bestimmten Fällen nach einmaliger Einnahme sinnvoll sein. |
|
Naltrexon plus 6β-naltrexol |
25–100 ng/ml |
2–5 h 7–13 h |
200 ng/ml |
2 |
3,06 3,04 |
Therapeutische Effekte basieren primär auf dem aktiven Metaboliten |
|
Nikotin |
5–20 ng/ml |
2 h |
400 ng/ml |
4 |
6,16 |
Pflasterapplikation enthält 35 mg. Hoch variabel bzgl. oraler Applikation |
|
Vareniclin |
4–5 ng/ml |
23–39 h |
10 ng/ml |
3 |
4,73 |
|
|
Arzneistoffe zur Behandlung der Aufmerksamkeits-Defizit-Hyperaktivitäts-Störung |
||||||
|
Atomoxetin |
200–1000 ng/ml 60–90min nach der Einnahme von 1,2 (mg/kg)/Tag |
2–5 h |
2000 ng/ml |
3 |
3,91 |
Empfohlene Referenzbereiche beziehen sich auf Cmax, gemessen in remittierten Patienten Eliminationshalbwertszeit beträgt 21 h in PM von CYP2D6 |
|
Dexmethylphenidat |
13–23 ng/ml |
2 h |
44 ng/ml |
3 |
4,29 |
5,2–5,5 ng/ml sind assoziiert mit 50% Dopamintransporter-Blockade |
|
Methylphenidat |
Für Kinder und Jugendliche: 6–26 ng/ml 2 h nach 20 mg IR- oder 4–6 h nach 40 mg XR-Formulierung Für Erwachsene: 12–79 ng/ml 2 h nach 20 mg IR- oder 4–6 h nach 40 mg XR-Formulierung |
2 h |
50 ng/ml |
3 |
4,29 |
Methylphenidat ist bei Raumtemperatur instabil. Der Wirkstoff ist verfügbar als Nicht-Retard- (immediate release, IR) und Retard-Formulierung (XR). Retard-Formulierungen basieren z.B. auf dem OROS-Prinzip (Osmotic controlled release delivery systems) oder auf dem SODAS-Prinzip (Spheroidal drug absorption systems). Die Werte basieren auf Cmax Bereichen bei therapeutisch effektiven Dosen |
|
Antiparkinsonika |
||||||
|
Amantadin |
300–600 ng/ml |
10–14 h |
1 200 ng/ml |
3 |
5,98 |
Arzneistoff-Konzentrationen im Blut sind bei eingeschränkter Nierenfunktion erhöht. Konzentrationen ˃3000 ng/ml verursachen unerwünschte Arzneimittelwirkungen, z.B. Myoklonien, Delirium, Halluzinationen |
|
Biperiden |
1–6,5 ng/ml |
18–24 h |
13 ng/ml |
3 |
3,21 |
|
|
Bornaprin |
0,7–7,2 ng/ml |
30 h |
14 ng/ml |
3 |
3,04 |
|
|
Bromocriptin |
Niedrige Dosis (2,5 mg): Maximale Dosis (25 mg): |
38 h |
8 ng/ml |
3 |
1,53 |
Levodopa 250 mg + 25 mg DCI vermindert die Bromocriptin Konzentration um ca. 50% |
|
Cabergolin |
58–144 pg/ml (0,5–4 h nach der Einnahme für 4 Wochen) |
63–68 h |
390 pg/ml |
3 |
2,21 |
Instabil bei Raumtemperatur, Plasma oder Serum sollte gefroren gelagert werden (<−20°C) |
|
Carbidopa |
20–200 ng/ml |
2 h |
400 ng/ml |
3 |
4,42 |
Instabil bei Raumtemperatur, Plasma oder Serum sollte gefroren gelagert werden (<−20°C) |
|
Entacapon |
0,4–1,0 µg/ml (1 h) |
0,5 h |
2 µg/ml |
3 |
3,28 |
Instabil bei Raumtemperatur, Plasma oder Serum sollte gefroren gelagert werden (<−20°C) |
|
Levodopa 3-O-Methyldopa |
0,9–2,0 µg/ml 0,7–10,9 µg/ml 1 h nach 250 mg und kombiniert mit 25 mg Carbidopa |
1–3 h 2 h |
5 µg/ml 20 µg/ml |
3 |
5,07 3,28 |
Instabil bei Raumtemperatur, Plasma oder Serum sollte gefroren gelagert werden (<−20°C), Eliminationshalbwertszeit und Arzneistoff-Konzentration im Blut steigen bei einer Komedikation mit Carbidopa oder Benserazid |
|
Pramipexol |
0,4–7,2 ng/ml |
8–12 h |
15 ng/ml |
3 |
4,73 |
Optimale Arzneistoff-Konzentrationen können in Abhängigkeit von der Diagnose unterschiedlich sein oder unter Kombination mit Levodopa |
|
Ropinirol |
0,4–6,0 ng/ml |
3–10 h |
12 ng/ml |
3 |
3,84 |
|
|
Rotigotin |
0,1–0,7 ng/ml |
5–7 h |
2 ng/ml |
3 |
3,17 |
Transdermale Applikation. Multiple, pharmakologisch inaktive Metaboliten, die sehr schnell eliminiert werden |
|
Tolcapon |
3–6 µg/ml (nach 2 h) |
2 h |
12 µg/ml |
3 |
3,66 |
|
AUC: area under the curve; CL: Clearance; DCI: Dopadecarboxylase-Inhibitor; PD: Parkinson’s disease; PM: poor metabolizer
Die Arzneistoff-Konzentrationen sind in Masseneinheiten angegeben. Diese können in molare Einheiten durch Multiplikation mit dem Umrechnungsfaktor (CF) nmol/l=ng/ml × CF umgerechnet werden. Für Bupropion, Carbamazepin, Lamotrigin und Valproat sind zwei empfohlene therapeutische Referenzbereiche gelistet, in Abhängigkeit von den zwei verschiedenen Indikationen
Bestimmung der unteren Grenze des therapeutischen Referenzbereichs
Wann immer möglich sollte die untere Grenze des therapeutischen Bereichs eines Arzneistoffs auf Studien über die Beziehung zwischen der Blutkonzentration und der klinischen Wirksamkeit basieren. Unterhalb dieser Grenze werden im Vergleich zu einer Placebo-Behandlung keine signifikanten therapeutischen Effekte erwartet. Das optimale Studiendesign für die Bestimmung der unteren Grenze des therapeutischen Bereichs ist eine randomisierte, prospektive kontrollierte Doppelblind-Studie, in der Patienten mit Dosierungen, die zu einem definierten Blutkonzentrationsbereich des Arzneistoffs führen, behandelt werden. Ein nahezu optimales Studiendesign wurde von Van der Zwaag und Kollegen an mit Clozapin behandelten Patienten angewandt. Clozapin-Konzentrationen im Blut wurden in drei verschiedene Bereiche titriert, 50 bis 150 ng/ml, 200 bis 300 ng/ml oder 350 bis 450 ng/ml. In Vergleich zu niedrigen Clozapin-Konzentrationen wurde bei Patienten mit mittleren und hohen Blutkonzentrationen eine Überlegenheit der Wirksamkeit von Clozapin gefunden. Eine ähnliche Studie wurde mit Imipramin und Mirtazapin durchgeführt. Solche Studien sind jedoch eine beträchtliche logistische Herausforderung. Studien mit fixer Dosis werden daher für die Bestimmung der unteren Grenze des therapeutischen Referenzbereichs bevorzugt.
Für die Abschätzung der Schwellenwerte der therapeutischen Referenzbereiche hat sich die „Receiver Operating Characteristics“-(ROC-)Analyse als hilfreich erwiesen. Ein ROC-Plot ermöglicht die Identifizierung eines Cut-off-Wertes, der Responder von Non-Respondern unterscheidet und dabei die Sensitivität und Spezifität des Parameters „Arzneistoff-Konzentration im Blut“ schätzt. Die Nützlichkeit der ROC-Analyse wurde für eine Reihe antipsychotischer und antidepressiver Medikamente nachgewiesen.
Bestimmung der oberen Grenze des therapeutischen Referenzbereichs
In der ersten Studie über TDM in der Psychiatrie wurde für Nortriptylin eine U-förmige Beziehung zwischen Blutkonzentration und klinischer Wirkung detektiert. Dem Fehlen einer Wirkung bei hohen Konzentrationen wurde der Wirkungsmechanismus des trizyklischen Antidepressivums auf monoaminerge Neurone zugeschrieben. Gemäß aktuellem Wissen scheint es jedoch wahrscheinlicher, dass die reduzierte Besserung bei hohen Nortriptylin-Konzentrationen auf der höheren Wahrscheinlichkeit für Nebenwirkungen beruht. Die Obergrenze des therapeutischen Bereichs ist daher meist definiert aufgrund des gesteigerten Risikos für unerwünschte Arzneimittelwirkungen, auch in diesen Leitlinien. Ein Zusammenhang mit Arzneistoff-Konzentrationen im Blut wurde für motorische Symptome von Antipsychotika und für unerwünschte Effekte von trizyklischen Antidepressiva gezeigt. Für Paroxetin wurde ein Zusammenhang mit dem Auftreten eines Serotonin-Syndroms gefunden. Für Citalopram wurde nachgewiesen, dass unerwünschte Arzneimittelwirkungen invers mit der Arzneistoff-Clearance korrelieren. Für viele Neuropsychopharmaka, gelistet in Tabelle 4, fehlen jedoch verwertbare Daten über den Zusammenhang zwischen Blutkonzentration und der Inzidenz von Nebenwirkungen. Fallberichte über Verträglichkeitsprobleme oder über Vergiftungen können oft nicht verwendet werden, da Messungen der Arzneistoff-Konzentration im Blut fehlen. Sporadische Berichte über Todesfälle und Vergiftungen sind von begrenztem Wert. Die meisten letalen Arzneistoff-Konzentrationen im Blut, von denen berichtet wurde, liegen weit über den Arzneistoff-Konzentrationen, die mit einer maximalen therapeutischen Wirkung verbunden sind. Post-mortem-Umverteilung von Arzneistoffen aus dem bzw. in das Blut kann außerdem zu dramatischen Veränderungen der Blutkonzentrationen führen, wobei die Richtung der Veränderung nicht allgemein vorhersagbar ist. Aufgrund dieser Limitierungen ist die Abschätzung eines oberen Schwellenwerts, oberhalb dessen die Verträglichkeit abnimmt oder das Risiko einer Vergiftung erhöht ist, schwieriger als die Einschätzung der unteren Schwelle, vor allem für Arzneistoffe mit einem breiten therapeutischen Index, wie SSRI. Deshalb beziehen sich viele obere therapeutische Referenzbereiche, die in Tabelle 4 gelistet sind, auf Konzentrationen, bei denen eine maximale Wirksamkeit angenommen wird. In diesen Leitlinien wurden die oberen Bereiche zumeist durch Berechnung der zu erwartenden dosisbezogenen Arzneistoff-Konzentration im Blut unter zugelassenen Maximaldosen ermittelt.
Von bevölkerungsbezogenen zu subjektbezogenen Referenzwerten
Alle therapeutischen Referenzbereiche, die in Tabelle 4 gelistet sind, sind bevölkerungsbezogen. Die Populations-abgeleiteten Bereiche stellen deskriptive statistische Werte dar, die nicht notwendigerweise auf alle Patienten übertragbar sind. Eine optimale Neuropsychopharmakotherapie sollte versuchen, den „individuellen, optimalen therapeutischen Konzentrationsbereich“ eines Patienten für die Therapieleitung zu identifizieren [34], indem beispielsweise der Blutspiegel nach eingetretener Besserung gemessen wird. Darüber hinaus kann es vorkommen, dass der Schweregrad der psychischen Erkrankung die optimale Arzneistoff-Konzentration beeinflusst. Für Lithium wurde gezeigt, dass der empfohlene Konzentrationsbereich davon abhängt, ob der Patient in einer akuten manischen Phase ist oder ob er eine Erhaltungstherapie benötigt. Gaertner und Kollegen detektierten individuelle, optimale Clozapin-Konzentrationen im Blut in Phasen der stabilen Remission der Patienten. Damit war bekannt, welche Konzentration zur Erhaltungstherapie vonnöten ist. Sie fanden heraus, dass ein hohes Rückfallrisiko besteht, wenn die Arzneistoff-Konzentration während der Erhaltungstherapie um 40% oder mehr sinkt.
Empfehlung
Auch wenn der Patient den erwünschten Therapieeffekt erreicht hat, kann es sinnvoll sein, die Arzneistoff-Konzentration im Blut zu messen. Diese Konzentration kann als optimale Arzneistoff-Konzentration für den individuellen Patienten angesehen werden. Im Falle einer Symptomverschlechterung, eines Rückfalls oder bei Auftreten von unerwünschten Arzneimittelwirkungen ist der Wert hilfreich, um herauszufinden, ob pharmakokinetische Besonderheiten auftraten, welche die klinische Verschlechterung erklären könnten.
Abschätzung eines vorläufigen therapeutischen Referenzbereichs
Wenn neue Arzneistoffe zugelassen werden, ist es für die TDM-gestützte Therapieleitung von großem Nachteil, dass therapeutische Referenzbereiche unklar sind. Die Abschätzung von therapeutischen Referenzbereichen ist für eine Arzneistoff-Zulassung nicht gefordert, und deshalb werden sie so gut wie nie ermittelt. Um trotzdem ein aussagekräftiges TDM durchführen zu können, empfehlen wir bei unbekanntem therapeutischem Referenzbereich die Generierung eines vorläufigen Referenzbereichs.
Empfehlung
Solange valide Daten bezüglich eines therapeutischen Referenzbereichs eines neuropsychiatrischen Arzneistoffs fehlen, empfehlen wir die Bestimmung des Mittelwerts ± Standardabweichung (SD) der Arzneistoff-Konzentration im Blut von Respondern. Der Mittelwert ± SD-Bereich kann als vorläufiger therapeutischer Referenzbereich gelten. Weitere prospektive Studien oder Beobachtungs-Studien müssen dann diesen Bereich bestätigen oder korrigieren.
Warnschwelle für das Labor
Für die meisten in Tabelle 4 gelisteten Neuropsychopharmaka können die Blutkonzentrationen mit einem erhöhten Risiko für Toxizität in der Regel sehr viel höher als der obere Grenzwert des therapeutischen Referenzbereichs sein. Für die vorliegenden Leitlinien wurde jeweils eine „Warnschwelle für das Labor“ definiert (Kasten).
Definition: Warnschwelle für das Labor
Die in dieser Leitlinie aufgeführten „Warnschwellen für das Labor“ (Tab. 4) entsprechen Arzneistoff-Konzentrationen oberhalb des empfohlenen therapeutischen Referenzbereichs, ab denen das Labor verpflichtet ist, den behandelnden Arzt unmittelbar zu verständigen. Die Warnschwellen beruhen bei vielen Arzneistoffen auf Berichten über schwere unerwünschte Arzneimittelwirkungen oder Vergiftungen, bei denen Arzneistoff-Konzentrationen im Blut gemessen wurden. Meistens wurde die Warnschwelle jedoch willkürlich als Arzneistoff-Konzentration im Blut, die 2-fach höher als die obere Grenze des therapeutischen Referenzbereiches liegt, definiert. Die Meldung des Labors sollte zu einer Dosisreduktion führen, wenn der Patient Anzeichen von Intoleranz oder Toxizität zeigt. Wenn die hohe Wirkstoffkonzentration durch den Patienten toleriert wird und gleichzeitig bei Dosisreduktion die Gefahr der Symptomverschlechterung besteht, sollte die Dosis unverändert bleiben. Die klinische Entscheidung, insbesondere wenn die Dosis nicht geändert wird, muss in der Krankenakte dokumentiert werden.
Der dosisbezogene Referenzbereich
Für die Interpretation von TDM-Ergebnissen existiert neben dem therapeutischen Referenzbereich ein zweiter Konzentrationsbereich, der berücksichtigt werden sollte: der sogenannte dosisbezogene Referenzbereich. Die Anwendung des therapeutischen Referenzbereichs basiert auf einem pharmakodynamischen Ansatz, der dosisbezogene Referenzbereich auf einem pharmakokinetischen Ansatz. Der dosisbezogene Referenzbereich beschreibt den Arzneistoff-Konzentrationsbereich, der bei der verabreichten Dosis theoretisch zu erwarten ist. Ein einfaches Verfahren, um den Erwartungsbereich zu ermitteln, ist die Berechnung der mittleren Konzentration im Steady-State (Cav [av: average]) aus pharmakokinetischen Daten, die bevorzugt an einer Patientengruppe ohne Komedikation oder pharmakokinetische Besonderheiten („normale“ Patienten) erhoben wurden. Berechnet werden kann Cav aus täglicher Erhaltungsdosis (Dm), Dosisintervall (di), totaler Clearance (CL) und Bioverfügbarkeit (F):
Cav =(Dm/di) × (F/CL) (1)
Dosis und Dosisintervall kann man der Verschreibung entnehmen, und pharmakokinetische Parameter sind aus pharmakokinetischen Studien verfügbar. Anhand der täglichen Dosis (1 mg/24 Stunden=1000000 ng/1440 min) und der Standardabweichung (SD) der totalen, scheinbaren Clearance (ml/min), die ebenfalls in der Literatur beschrieben ist, kann man die durchschnittliche Cav±SD (ng/ml) anhand der Gleichung (1) berechnen. Für die Berechnung müssen die Dimensionen der verschiedenen Parameter berücksichtigt werden, und alle Dosen müssen in ng konvertiert werden, alle Volumina in ml und Zeitintervalle in Minuten. Beispiel: Wurde ein CL/F-Wert von 100±50 ml/min und somit ein Variationskoeffizient von 50% berichtet, so beträgt Cav 139±69 ng/ml für eine Dosis von 20 mg/Tag [(20000000 ng/1440 min) × 1/(100 ml/min)=139 ng/ml]. Der Cav±SD-Bereich erstreckt sich von 69 bis 208 ng/ml.
Bei einem Dosisintervall von 24 Stunden, das heißt einer einmal täglichen (quaque die, q.d.) Einnahme, wurde der Cav±SD-Bereich als dosisbezogener Referenzbereich von Haen und Kollegen vorgeschlagen [17, 18]. Der Durchschnitt–SD ist die untere und der Durchschnitt+SD die obere Grenze dieses Bereichs. Statistisch gesehen enthält der Bereich 68% der Konzentrationen, die unter normalen Bedingungen im Blut von normalen Personen (18 bis 65 Jahre) zu erwarten sind. Für die Leitlinien von 2011 [20] wurden Daten über die scheinbare totale Clearance (CL/F)±SD für 83 Neuropsychopharmaka aus der Literatur extrahiert. Dosisbezogene Referenzbereiche können anhand Multiplikation der berechneten Faktoren inklusive SD mit der täglichen Dosis berechnet und für die Interpretation der TDM-Ergebnisse verwendet werden (siehe Tab. 5).
Wenn die Arzneistoff-Konzentration eines Patienten, gemessen anhand TDM, innerhalb des dosisbezogenen Referenzbereichs liegt, wird die Konzentration als „wie erwartet“ definiert. Konzentrationen oberhalb oder unterhalb des Bereichs weisen auf mögliche Besonderheiten hin, unter anderem (partielle) Non-Adhärenz, Arzneimittel-Interaktionen, genetische Polymorphismen von Arzneistoff-metabolisierenden Enzymen oder Funktionsstörungen von Arzneistoff-abbauenden Organen.
Mit dem Konzept des dosisbezogenen Referenzbereichs wurden bisher unzureichend adhärente Patienten oder Patienten mit pharmakokinetischen Besonderheiten identifiziert [17]. Die Gleichung mit der durchschnittlichen Steady-State-Konzentration ist zuverlässig und brauchbar, wenn die Eliminationshalbwertszeit der Arzneistoffe (t1/2) lang im Vergleich zum Dosisintervall ist. Wenn die t1/2 jedoch kurz und das Dosisintervall länger als die t1/2 ist, sind die anhand von Gleichung (1) berechneten Werte nur unzureichend prädiktiv für die anhand von TDM gemessenen Cmin-Werte. Dieses Problem ist in Abbildung 3 für Valproinsäure dargestellt, die eine t1/2 von 14 Stunden besitzt und einmal oder zweimal pro Tag eingenommen wird.
Abb. 3. Konzentration-Zeit-Kurve von Valproinsäure, die im Blut unter Steady-State-Bedingungen nach einer täglichen Dosis von 900 mg mit Dosisintervallen von entweder 12 Stunden (hellrote Linie, Cmin1) oder 24 Stunden (dunkelrote Linie, Cmin2) zu erwarten ist. Die durchschnittliche Eliminationshalbwertszeit (t1/2) von Valproinsäure beträgt 14 Stunden. Kreise und Fehlerbalken zeigen die zu erwartenden Talspiegel (Cmin) und durchschnittlichen Konzentrationen (Cav) ±Standardabweichung (SD) an.
Unter einer täglichen Dosis von 900 mg beträgt der nach Gleichung (1) berechnete dosisbezogene Referenzbereich von Valproinsäure unabhängig vom Dosisintervall 94±35 µg/ml. Die Konzentration-Zeit-Kurven zeigen jedoch, dass die Talspiegelkonzentrationen niedriger als Cav, 49±15 µg/ml, sind, wenn das Dosisschema eine Einmalgabe von 900 mg/Tag ist. Wenn die Dosis zweimal täglich zu je 450 mg gegeben wird, beträgt die Konzentration 69±25 µg/ml. Cav-Bereiche passen zu den Cmin-Bereichen für Dosisintervalle <14 Stunden. Deshalb wurden Cav-Bereiche als passende Prädiktoren für eine zu erwartende Arzneistoff-Konzentration im Blut angesehen. Unter einer Einmaldosis ist die durchschnittliche Cmin 24 Stunden nach der letzten Dosis jedoch 54% geringer als die durchschnittliche Cav. Wie beispielhaft anhand von Valproinsäure erklärt wurde, muss diese Limitierung bei Anwendung von Gleichung (1) zur Berechnung des dosisbezogenen Referenzbereichs berücksichtigt werden. In Abhängigkeit von dem Dosisintervall kann diese Limitierung auch für Arzneistoffe wie Duloxetin, Paroxetin, Venlafaxin, Amisulprid, Paliperidon, Quetiapin, Lithium, Zopiclon, Atomoxetin oder Naltrexon relevant sein. Die berechneten Werte sind mehr als 30% niedriger für Cmin als für Cav, wenn die Dosisintervalle länger als die t1/2 sind. Insgesamt wurde dies für 32% der in Tabelle 5 gelisteten Arzneistoffe gefunden.
Zudem existiert eine weitere wichtige Limitierung bezüglich Cav-basierten Berechnungen. Die Validität der dosisbezogenen Referenzbereiche kann nicht einfacherweise durch Messungen geprüft werden, da TDM auf der Messung von minimalen Arzneistoff-Konzentrationen (Cmin) im Blut (Talspiegeln) beruht. Cav ist definiert als die Fläche unter der Blut-Konzentration-versus-Zeit-Kurve (Area under the time to concentration curve, AUC), geteilt durch das Dosisintervall. Cav kann also, anders als Cmin, nicht einem bestimmten Zeitpunkt zugeordnet werden, was notwendig für die Festlegung des Zeitpunkts der Venenpunktion wäre. Eine weitere Limitierung von Cav-basierten Berechnungen ist die Vernachlässigung von Fluktuationen von Arzneistoff-Konzentrationen über den Tag hinweg, was wichtig sein kann für die Effektivität und Verträglichkeit eines Arzneistoffs.
Aufgrund dieser Limitierungen wurde entschieden, die Berechnung der dosisbezogenen Referenzbereiche für dieses Update zu modifizieren. Die Details hierzu sind in der englischen Version beschrieben. Die Neuberechnungen basieren auf einer Erweiterung von Gleichung (1) und der Anwendung der Bateman-Funktion.
Abgeleitet vom ursprünglichen Konzept des dosisbezogenen Referenzbereichs für durchschnittliche Arzneistoff-Konzentrationen [18] ist der dosisbezogene Referenzbereich nun definiert als Bereich für minimale („Talspiegel“)-Konzentrationen.
Der dosisbezogene Referenzbereich dient der Identifikation von pharmakokinetisch auffälligen Patienten. Wenn ein TDM ausgeführt wird, sollte die vom Labor gemessene und berichtete Arzneistoff-Konzentration mit theoretisch zu erwartenden Werten, berechnet mit Angaben der Tabelle 5 in diesen Leitlinien (siehe auch Fälle im Folgenden), verglichen werden. Insofern die Arzneistoff-Konzentration eines Patienten innerhalb des zu erwartenden dosisbezogenen Referenzbereichs liegt, kann die Konzentration als „normal“ angesehen werden, das heißt, die Konzentration stimmt überein mit der verschriebenen Dosis. Konzentrationen oberhalb oder unterhalb des erwarteten durchschnittlichen ± SD-Bereichs sind Signale für potenzielle pharmakokinetische Besonderheiten, beispielsweise (partielle) Non-Adhärenz, Arzneimittel-Interaktionen, genetische Polymorphismen von Arzneistoff-metabolisierenden Enzymen oder Erkrankungen von Organen, die Arzneistoffe verstoffwechseln. Im klinisch pharmakologischen TDM-Kommentar (s. unten) sollten die vorgeschlagenen Gründe für die Abweichung erklärt und die Gründe hierfür abgeklärt werden.
Definition: Dosisbezogener Referenzbereich
Der „dosisbezogene Referenzbereich“ wird in den vorliegenden Leitlinien als zu erwartender Arzneistoff-Konzentrationsbereich unter Talspiegel- und Steady-State-Bedingungen angegeben. Er entspricht dem Bereich der durchschnittlichen Erwartungskonzentration – Standardabweichung (SD) bis zur durchschnittlichen Erwartungskonzentration + SD. Dieser Bereich umfasst 68% der Population „normaler“ Patienten, die den Arzneistoff eingenommen haben (18 bis 65 Jahre alt, mit einem Körpergewicht von 70 kg und ohne pharmakokinetisch relevante Komorbidität, Komedikation oder genetische Besonderheiten im Arzneistoff-Metabolismus). Die dosisbezogenen Referenzbereiche werden berechnet, indem man die DRC-Faktoren „niedrig“ und „hoch“ von Tabelle 5 mit der täglichen Dosis multipliziert.
Tab. 5. Apparente totale Clearance (d.h. Clearance/Bioverfügbarkeit, CL/F), Bioverfügbarkeit (F), durchschnittliche Eliminationshalbwertszeit (t1/2), Zeitintervall zwischen letzter Dosis und Blutentnahme (Δt) und DRC-Faktoren für die Berechnung der dosisbezogenen Referenzbereiche der Muttersubstanzen, Metaboliten und der aktiven Fraktion. DRC-Faktoren wurden, wie im Text beschrieben, berechnet. Entscheidungen für Δt erfolgten nach fachlichen Empfehlungen bezüglich der Arzneistoff-Applikation. Die dosisbezogenen Referenzbereiche werden berechnet, indem man die DRC-Faktoren niedrig und hoch mit der täglichen Dosis multipliziert. Ergebnis ist ein durchschnittlicher (± Standardabweichung, SD) Referenzbereich, welcher sich auf Arzneistoff-Talspiegel bezieht, insofern nicht anders beschrieben.
|
Arzneistoff und Metaboliten |
CL/F ± SD |
F |
t1/2 |
Δt |
DRC-Faktoren |
Kommentare |
|||
|
[ml/min] |
[%] |
[h] |
[h] |
Mittel |
Niedrig |
Hoch |
|||
|
Antidepressiva |
|||||||||
|
Agomelatin |
1100±500 |
3 |
1,5 |
2 |
2,78 |
1,52 |
4,04 |
Talspiegel bei ∆t =24 sind aufgrund schneller Metabolisierung nicht messbar, CL beeinflusst von CYP1A2 |
|
|
Amitriptylin Nortriptylin Aktive Fraktion |
1043±301 1435±609 |
50 |
19 30 |
12 |
0,65 0,48 1,12 |
0,46 0,28 0,73 |
0,83 0,68 1,51 |
CL beeinflusst von CYP2C19 und CYP2D6 und Alter |
|
|
Amitriptylinoxid Amitriptylin Nortriptylin Aktive Fraktion |
485±133 3947±1316 3488±969 |
64 |
2 19 31 |
12 |
0,20 0,17 0,20 0,37 |
0,14 0,11 0,14 0,26 |
0,26 0,23 0,25 0,48 |
Prodrug, aktive Substanzen sind Amitriptylin und Nortriptylin, CL beeinflusst von CYP2C19 und CYP2D6 |
|
|
Bupropion Hydroxybupropion |
2260±870 147±91 |
90 |
19 28 |
24 |
0,19 3,46 |
0,12 1,32 |
0,27 5,60 |
Bupropion ist bei Raumtemperatur instabil, der Metabolit ist die hauptaktive, pharmakologische Substanz, CL/F wird bei Patienten mit eingeschränkter Nierenfunktion beeinflusst, sowie durch CYP2B6 |
|
|
Citalopram N-Desmethylcitalopram |
360±105 622±384 |
80 |
36 50 |
24 |
1,52 0,94 |
1,07 0,36 |
1,96 1,52 |
CL beeinflusst von CYP2C19 und Alter |
|
|
Clomipramin N-Desmetylclomipramin Aktive Fraktion |
1120±667 622±384 |
50 |
21 36 |
12 |
0,60 1,11 1,71 |
0,24 0,42 0,67 |
0,96 1,79 2,75 |
CL beeinflusst von CYP2D6 und CYP2C19 |
|
|
Desipramin |
1750±1248 |
38 |
22 |
12 |
0,39 |
0,11 |
0,66 |
CL beeinflusst von CYP2D6 |
|
|
Desvenlafaxin |
315±82 |
80 |
14 |
24 |
1,15 |
0,85 |
1,45 |
CL beeinflusst von CYP2C19, nicht von CYP2D6 |
|
|
Dothiepin=Dosulepin |
2450±1867 |
30 |
20 |
24 |
0,18 |
0,04 |
0,32 |
CL beeinflusst von CYP2C19 |
|
|
Doxepin N-Desmethyldoxepin Aktive Fraktion |
1706±938 1750±940 |
27 |
17 51 |
12 |
0,39 0,40 0,79 |
0,18 0,18 0,36 |
0,61 0,61 1,22 |
CL beeinflusst von CYP2C19 und CYP2D6, Alter und Geschlecht |
|
|
Duloxetin |
750±264 |
60 |
12 |
24 |
0,43 |
0,28 |
0,58 |
CL beeinflusst vom Raucherstatus des Patienten aufgrund CYP1A2-Induktion, höher bei asiatischen Patienten |
|
|
Escitalopram N-Desmethylescitalopram |
495±218 622±384 |
80 |
30 52 |
24 |
1,05 0,95 |
0,59 0,36 |
1,51 1,53 |
CL beeinflusst von CYP2C19, Alter und Geschlecht |
|
|
Fluoxetin N-Desmethylfluoxetin Aktive Fraktion |
126±93 111±72 |
90 |
120 240 |
24 |
5,14 6,04 11,18 |
1,35 2,12 3,47 |
8,93 9,96 18,89 |
CL wurde berechnet aus Talspiegelkonzentrationen (Steady-State) im Blut |
|
|
Fluvoxamin |
1907±504 |
53 |
20 |
24 |
0,23 |
0,17 |
0,29 |
CL beeinflusst von CYP2D6 |
|
|
Imipramin Desipramin Aktive Fraktion |
1733±578 933±117 |
39 |
12 21 |
12 |
0,37 0,73 1,10 |
0,25 0,63 0,88 |
0,49 0,82 1,31 |
CL beeinflusst von CYP2D6 und CYP2C19 |
|
|
Levomilnacipran |
176±43 |
90 |
21 |
12 |
1,98 |
1,29 |
2,67 |
CL reduziert bei Nierenfunktionsstörung, aber nicht beeinflusst durch CYP Enzyme |
|
|
Maprotilin |
741±410 |
70 |
40 |
12 |
0,93 |
0,42 |
1,45 |
CL beeinflusst von CYP2D6 und Alter |
|
|
Mianserin |
664±258 |
30 |
32 |
12 |
1,03 |
0,63 |
1,44 |
CL beeinflusst von CYP2D6 |
|
|
Milnacipran |
592±95 |
85 |
8 |
12 |
0,99 |
0,83 |
1,14 |
CL vergleichbar in Asiaten und Kaukasiern, nicht beeinflusst durch CYP Enzyme |
|
|
Mirtazapin |
261±80 |
50 |
30 |
12 |
2,63 |
1,82 |
3,43 |
CL beeinflusst von Alter, Geschlecht und Raucherstatus und geringer in Asiaten |
|
|
Moclobemid |
208±82 |
70 |
2,5 |
12 |
0,80 |
0,48 |
1,11 |
CL beeinflusst von CYP2C19 |
|
|
Nortriptylin |
970±242 |
50 |
30 |
12 |
0,71 |
0,53 |
0,88 |
CL beeinflusst von CYP2D6 |
|
|
Paroxetin |
724±274 |
64 |
19 |
24 |
0,60 |
0,37 |
0,83 |
CL beeinflusst von CYP2D6, nichtlineare Pharmakokinetik aufgrund CYP2D6-Inhibition |
|
|
Reboxetin |
58±26 |
60 |
10 |
12 |
10,8 |
5,94 |
15,6 |
||
|
Sertralin N-Desmethylsertralin |
1167±450 822±278 |
66 |
26 70 |
24 |
0,42 0,75 |
0,26 0,50 |
0,58 1,00 |
CL nimmt während der Schwangerschaft ab und steigt bei Patienten>60 Jahre |
|
|
Tianeptin |
222±58 |
100 |
3 |
12 |
1,09 |
0,80 |
1,38 |
Metabolismus ohne Beteiligung von CYPs |
|
|
Tranylcypromin |
1152±607 |
100 |
3 |
12 |
0,21 |
0,10 |
0,32 |
Metabolismus ohne Beteiligung von CYPs |
|
|
Trazodon |
115±35 |
100 |
7 |
12 |
4,82 |
3,35 |
6,29 |
CL nimmt mit zunehmenden Alter ab, beeinflusst von CYP3A4 |
|
|
Trimipramin |
1113±330 |
41 |
24 |
12 |
0,61 |
0,43 |
0,79 |
CL beeinflusst von CYP2D6 und CYP2C19 |
|
|
Venlafaxin IR O-Desmethylvenlafaxin Aktive Fraktion N-Desmethylvenlafaxin |
1250±433 300±67 – 367±267 |
40 |
6 11 – 7 |
24 |
0,10 0,99 1,09 0,46 |
0,06 0,77 0,83 0,13 |
0,14 1,21 1,35 0,80 |
Daten für Retard- und Non-Retard-Formulierungen (XR und IR) unterscheiden sich in tmax, 1 h für IR und 6 h für XR Für die XR-Formulierung wurden CL/F von Cmin berechnet; CL beeinflusst von CYP2D6 und CYP2C19 und Alter |
|
|
Venlafaxin XR O-Desmethylvenlafaxin Aktive Fraktion N-Desmethylvenlafaxin |
1196±576 422±107 – 704±264 |
40 |
11 20 – 7 |
24 |
0,24 1,04 1,28 0,24 |
0,12 0,78 0,90 0,15 |
0,36 1,30 1,67 0,33 |
||
|
Vilazodon |
415±129 |
70 |
32 |
24 |
1,28 |
0,88 |
1,67 |
CL beeinflusst von CYP3A4 |
|
|
Vortioxetin |
550±83 |
80 |
66 |
24 |
1,11 |
0,94 |
1,28 |
CL beeinflusst von CYP2D6 |
|
|
Antipsychotika |
|||||||||
|
Amisulprid |
586±174 |
50 |
16 |
24 |
0,67 |
0,47 |
0,87 |
Metabolismus ohne Beteiligung von CYPs, renale Exkretion |
|
|
Aripiprazol Dehydroaripiprazol Aktive Fraktion |
53±16 132±49 |
90 |
70 94 |
24 |
11,72 4,82 16,54 |
8,15 3,04 11,19 |
15,29 6,60 21,89 |
CL beeinflusst von CYP2D6 |
|
|
Asenapin |
2761±1783 |
35 |
24 |
12 |
0,25 |
0,09 |
0,41 |
CL ähnlich in Patienten älter oder jünger als 65 Jahre |
|
|
Benperidol |
1266±513 |
50 |
8 |
12 |
0,45 |
0,27 |
0,64 |
||
|
Brexpiprazol |
23±13 |
95 |
91 |
24 |
27,4 |
11,6 |
43,2 |
CL beeinflusst von CYP2D6 |
|
|
Bromperidol |
1598±607 |
30 |
20 |
12 |
0,42 |
0,26 |
0,58 |
||
|
Cariprazin N-Desmethylcariprazin N-Didesmethylcariprazin |
279±67 869±178 125±32 |
100 |
44 37 446 |
24 |
2,05 0,63 5,45 |
1,56 0,50 4,06 |
2,54 0,76 6,85 |
CL beeinflusst von CYP3A4 |
|
|
Chlorpromazin |
623±203 |
30 |
30 |
24 |
0,83 |
0,56 |
1,10 |
||
|
Chlorprothixen |
2507±478 |
50 |
10 |
12 |
0,25 |
0,20 |
0,30 |
||
|
Clozapin – N-Desmethylclozapin |
637±367 – 667±283 |
50 |
12 – 8 |
12 24 12 24 |
1,01 0,50 0,87 0,31 |
0,43 0,21 0,50 0,18 |
1,59 0,79 1,25 0,44 |
CL bei Rauchern aufgrund CYP1A2-Induktion erhöht und kann während entzündlicher Erkrankungen erniedrigt sein, CL/F zweifach höher in Asiaten in Vergleich zu Kaukasiern. Die Eliminationshalbwertszeit von Clozapin ist bei intoxikierten Patienten zu 30 h verlängert |
|
|
Flupentixol |
2148±814 |
50 |
35 |
12 |
0,32 |
0,20 |
0,44 |
CL assoziiert mit CYP2D6 |
|
|
Fluphenazin |
9990±2 820 |
35 |
16 |
12 |
0,07 |
0,05 |
0,09 |
CL beeinflusst von CYP2D6 |
|
|
Fluspirilen |
Keine Daten |
||||||||
|
Haloperidol |
826±203 |
60 |
18 |
12 |
0,81 |
0,61 |
1,01 |
CL beeinflusst von CYP2D6 |
|
|
Iloperidon |
1258±425 |
100 |
18 |
12 |
0,53 |
0,35 |
0,71 |
CL beeinflusst von CYP2D6 |
|
|
Levomepromazin |
2630±1580 |
50 |
28 |
12 |
0,26 |
0,10 |
0,42 |
||
|
Levosulpirid |
425±140 |
30 |
8 |
12 |
1,35 |
0,90 |
1,79 |
CL beeinflusst von ABCB1 |
|
|
Loxapin |
807±138 |
30 |
7 |
4 24 |
1,51 0,21 |
1,26 0,17 |
1,78 0,25 |
Aerosol-Applikation |
|
|
Lurasidon |
3902±702 |
20 |
18 |
24 |
0,11 |
0,09 |
0,13 |
CL beeinflusst von der Nahrungsaufnahme (Kalorien) |
|
|
Melperon |
2555±476 |
60 |
5 |
12 |
0,18 |
0,14 |
0,21 |
||
|
Olanzapin |
372±132 |
80 |
33 |
12 |
1,85 |
1,19 |
2,50 |
CL bei Männern höher in Vergleich zu Frauen und bei Rauchern aufgrund der CYP1A2-Induktion beschleunigt |
|
|
Paliperidon |
112±54 |
30 |
20 |
24 |
3,98 |
2,06 |
5,90 |
Extended-Release-Formulierung |
|
|
Perazin |
3671±2134 |
10 |
12 |
12 |
0,17 |
0,07 |
0,27 |
Daten basierend auf Einzeldosis-Studien |
|
|
Perphenazin |
12567±6417 |
40 |
10 |
12 |
0,05 |
0,02 |
0,08 |
CLbei Rauchern beschleunigt, beeinflusst von CYP2D6 |
|
|
Pimozid |
1400±467 |
40 |
33 |
12 |
0,49 |
0,33 |
0,65 |
CL beeinflusst von CYP2D6 |
|
|
Pipamperon |
644±207 |
100 |
15 |
12 |
1,02 |
0,70 |
1,35 |
||
|
Prothipendyl |
910±300 |
12 |
2,5 |
6 |
0,96 |
0,64 |
1,28 |
||
|
Quetiapin IR Desalkylquetiapin |
1072±461 2094±621 |
9 |
8 18 |
12 12 |
0,54 0,32 |
0,31 0,23 |
0,78 0,41 |
Daten für Nicht-Retard- und Retard-Formulierung (immediate und extended release, IR und XR), mit tmax von 1 bzw. 6 h; CL/F wurde bei der Retard-Formulierung mit Cmin berechnet, CL beeinflusst von Geschlecht und Alter |
|
|
Quetiapin XR – Desalkylquetiapin |
596±421 – 1137±646 |
9 |
8 – 18 |
12 24 12 24 |
0,97 0,34 0,59 0,37 |
0,29 0,10 0,25 0,16 |
1,66 0,58 0,92 0,58 |
||
|
Risperidon 9-Hydroxyrisperidon Aktive Fraktion |
1447±1038 140±47 |
70 |
3 20 |
12 |
0,57 4,82 5,39 |
0,34 3,20 3,54 |
0,80 6,44 7,24 |
CL beeinflusst von CYP2D6 und Alter, möglicherweise vermindert bei entzündlichen Erkrankungen |
|
|
Sertindol |
317±211 |
70 |
73 |
24 |
1,95 |
0,65 |
3,25 |
CL beeinflusst von CYP2D6 |
|
|
Sulpirid |
1186±240 |
35 |
8 |
12 |
0,49 |
0,39 |
0,59 |
CL bei eingeschränkter Nierenfunktion vermindert |
|
|
Thioridazin |
693±289 |
60 |
30 |
12 |
0,99 |
0,58 |
1,40 |
CL beeinflusst von CYP2D6 |
|
|
Ziprasidon |
350±98 |
60 |
7 |
12 |
1,58 |
1,14 |
2,03 |
F durch Nahrungsmittelaufnahme beeinflusst |
|
|
Zotepin |
5367±4900 |
90 |
15 |
12 |
0,12 |
0,01 |
0,24 |
||
|
Zuclopenthixol |
1584±717 |
50 |
18 |
12 |
0,42 |
0,23 |
0,61 |
CL beeinflusst von CYP2D6 |
|
|
Antikonvulsiva und Stimmungsstabilisierer |
|||||||||
|
Brivaracetam |
54±13 |
100 |
9 |
12 |
11,2 |
8,5 |
14,0 |
||
|
Carbamazepin |
132±39 |
70 |
15 |
12 |
4,99 |
3,52 |
6,47 |
CL steigt im Zeitverlauf aufgrund einer CYP3A4/5-Induktion an, t1/2 sinkt unter chronischer Behandlung von 36 h nach akuter Dosis auf 15 h |
|
|
Clobazam N-Desmethylclobazam |
42±25 13±6 |
90 |
32 57 |
12 |
16,6 53,6 |
6,8 28,4 |
26,3 78,8 |
CL beeinflusst von CYP2C19 |
|
|
Felbamat |
35±9 |
90 |
19 |
24 |
12,4 |
9,1 |
15,7 |
||
|
Lamotrigin |
35±13 |
100 |
14 |
24 |
10,3 |
6,50 |
14,17 |
CL wird deutlich durch Arzneistoffe (Komedikation) mit induzierenden Eigenschaften beeinflusst, Valproat steigert die durchschnittliche Eliminationshalbwertszeit zu 70 h und Carbamazepin, Phenytoin oder Phenobarbital senken diese zu 9 bis 14 h |
|
|
Levetiracetam |
62±10 |
99 |
7 |
12 |
8,94 |
7,50 |
10,39 |
||
|
Lithium |
25,0±9,5 |
100 |
24 |
12 24 |
27,2 19,3 |
16,9 11,94 |
37,6 26,6 |
Für die Berechnung des zu erwartenden dosisbezogenen Referenzbereichs müssen die DRC mit der täglichen Dosis in mmol multipliziert werden, resultierende Konzentrationen sind angegeben in µmol/l |
|
|
Oxcarbazepin 10-Monohydroxycarbamazepin |
3383±1680 40±8 |
100 |
2 9 |
12 |
0,03 15,1 |
0,01 12,1 |
0,04 18,1 |
||
|
Primidon Phenobarbital Phenylethylmalonamid |
38,5±8,5 |
90 |
20 |
12 |
17,5 |
13,7 |
21,4 |
Aktiver Metabolit Phenobarbital |
|
|
Rufinamid |
5,6±3,7 |
80 |
8 |
24 |
86,0 |
29,2 |
142,8 |
||
|
Topiramat |
26±5 |
90 |
8 |
12 |
22,4 |
18,1 |
26,8 |
||
|
Valproat |
6,65±2,45 |
100 |
14 |
12 24 |
98,5 54,4 |
62,2 34,4 |
134,8 74,4 |
CL reduziert in UGT1A3*5 Trägern |
|
|
Anxiolytika/Hypnotika |
|||||||||
|
Alprazolam |
58±13 |
80 |
13 |
10 |
12,5 |
9,7 |
15,2 |
CL beeinflusst von CYP3A4 |
|
|
Bromazepam |
57,6±12,7 |
85 |
23 |
10 |
12,5 |
9,8 |
15,3 |
CL beeinflusst von CYP3A4 und CYP2C19 und Geschlecht |
|
|
Brotizolam |
273±72 |
70 |
5 |
10 |
2,19 |
1,62 |
2,77 |
CL beeinflusst von CYP3A4 |
|
|
Buspiron 1-Pyrimidinylpiperazin 6-Hydroxybuspiron |
42409±11438 5574±2573 3149±880 |
10 |
3 5 6 |
10 |
0,01 0,11 0,21 |
0,00 0,06 0,15 |
0,01 0,16 0,26 |
CL beeinflusst von CYP3A4 |
|
|
Chlordiazepoxid |
16,1±3,1 |
100 |
22 |
10 |
44,9 |
36,1 |
53,6 |
CL beeinflusst in Patienten mit alkoholischer Hepatitis und von CYP3A4 |
|
|
Clonazepam |
76,5±13,5 |
80 |
25 |
10 |
9,42 |
7,76 |
11,08 |
CL beeinflusst von CYP3A4 |
|
|
Clorazepat (-dikalium) N-Desmethyldiazepam |
– 19±5 |
13 |
2 53 |
10 |
– 38,0 |
– 28,5 |
– 47,5 |
Prodrug, N-Desmethyldiazepam ist die aktive Substanz |
|
|
Diazepam N-Desmethyldiazepam Aktive Fraktion |
25±16 22±9 |
80 |
43 65 |
10 |
28,5 32,8 61,3 |
10,2 19,4 29,6 |
46,9 46,1 93,0 |
CL beeinflusst von CYP2C19 |
|
|
Diphenhydramin |
1631±658 |
50 |
10 |
10 |
0,44 |
0,26 |
0,61 |
CL beeinflusst von CYP2D6 und Alter |
|
|
Doxylamin |
232±69 |
100 |
10 |
10 |
3,07 |
2,16 |
3,99 |
||
|
Flunitrazepam |
245±56 |
85 |
26 |
10 |
2,94 |
2,27 |
3,61 |
||
|
Flurazepam N-Desalkylflurazepam Hydroxyethylflurazepam |
47881±15960 85±36 10085±3362 |
70 |
3 75 3 |
10 |
0,01 8,31 0,04 |
0,01 4,79 0,03 |
0,01 11,82 0,05 |
||
|
Hydroxyzin |
686±224 |
70 |
14 |
10 |
1,05 |
0,71 |
1,40 |
Reduzierte CL in Patienten mit primärer biliärer Zirrhose |
|
|
Lorazepam |
73±37 |
94 |
14 |
10 |
9,91 |
4,89 |
14,93 |
||
|
Lormetazepam |
216±36 |
80 |
12 |
10 |
3,34 |
2,78 |
3,89 |
||
|
Medazepam Diazepam N-Desmethyldiazepam Oxazepam Aktive Fraktion |
– 25±16 22±9 98±42 |
80 |
2 43 65 20 |
10 |
– 28,5 32,8 7,4 68,6 |
– 10,2 19,4 4,2 33,8 |
– 46,9 46,1 10,5 103,5 |
Prodrug wird metabolisiert zu den aktiven Metaboliten Diazepam, N-Desmethyldiazepam und Oxazepam; CL beeinflusst von CYP2C19 und CYP3A4 |
|
|
Midazolam |
380±61 |
70 |
2 |
10 |
0,48 |
0,40 |
0,56 |
CL beeinflusst von CYP3A4 |
|
|
Nitrazepam |
82±34 |
78 |
28 |
10 |
8,77 |
5,13 |
12,41 |
CL beeinflusst von CYP3A4 |
|
|
Opipramol |
4329±2473 |
94 |
11 |
10 |
0,17 |
0,07 |
0,26 |
CL beeinflusst von CYP2D6 |
|
|
Oxazepam |
98±42 |
85 |
20 |
10 |
7,38 |
4,22 |
10,54 |
||
|
Pregabalin |
75±14 |
90 |
6 |
10 |
8,61 |
6,99 |
10,24 |
||
|
Prazepam N-Desmethyldiazepam |
167±50 22±9 |
70 |
1 65 |
10 |
0,26 32,2 |
0,18 19,0 |
0,33 45,3 |
Prodrug metabolisiert zu dem pharmakologisch aktiven N-Desmethyldiazepam |
|
|
Promethazin |
1140±410 |
25 |
12 |
10 |
0,63 |
0,40 |
0,86 |
Orale Dosis, CL beeinflusst von CYP2D6 |
|
|
Temazepam |
767±312 |
95 |
8 |
10 |
0,90 |
0,53 |
1,27 |
||
|
Triazolam |
440±60 |
85 |
3 |
10 |
0,87 |
0,75 |
0,99 |
CL beeinflusst von CYP3A4 |
|
|
Zaleplon |
1099±231 |
70 |
1 |
10 |
0,01 |
0,01 |
0,01 |
CL beeinflusst von CYP3A4 |
|
|
Zolpidem |
315±49 |
70 |
2 |
10 |
0,57 |
0,48 |
0,66 |
CL beeinflusst von CYP3A4 |
|
|
Zopiclon |
567±317 |
70 |
4 |
10 |
0,91 |
0,40 |
1,43 |
CL beeinflusst von CYP3A4 |
|
|
Antidementiva |
|||||||||
|
Donepezil |
128±23 |
100 |
70 |
12 |
5,40 |
4,42 |
6,38 |
CL beeinflusst von CYP2D6 |
|
|
Galantamin |
334±66 |
90 |
9 |
12 |
1,81 |
1,45 |
2,17 |
||
|
Memantin |
125±34 |
100 |
64 |
24 |
4,86 |
3,55 |
6,17 |
||
|
Rivastigmin |
2214±2584 1341±1046 |
36 |
2 – |
12 24 |
0,04 0,69 |
0,00 0,36 |
0,09 1,02 |
Orale Applikation Transdermale Pflasterapplikation, DRC von TDM-Daten berechnet |
|
|
Arzneistoffe zur Behandlung substanzbezogener Störungen |
|||||||||
|
Acamprosat |
2981±1240 |
11 |
13 |
12 |
0,22 |
0,13 |
0,31 |
F durch Nahrungsmittelaufnahme reduziert |
|
|
Buprenorphin N-Desmethylbuprenorphin |
3201±3676 1470±1533 |
40 |
28 69 |
24 |
0,16 0,42 |
0,00 0,00 |
0,34 0,85 |
HCV- Infektion mit erhöhten Arzneistoff-Konzentrationen im Blut assoziiert, CL beeinflusst von CYP3A4 |
|
|
Bupropion Hydroxybupropion |
2250±870 147±91 |
90 |
20 28 |
24 |
0,20 3,46 |
0,12 1,32 |
0,28 5,60 |
Bupropion ist bei Raumtemperatur instabil, der Metabolit ist die hauptaktive Substanz, CL beeinflusst bei Patienten mit Nierenfunktionsstörung und von CYP2B6 |
|
|
Clomethiazol |
733±167 |
32 |
4 |
6 |
1,41 |
1,09 |
1,74 |
||
|
Diamorphin (Heroin) 6-Acetylmorphin Morphin |
15500 – 1227±503 |
100 |
0,1 0,4 2 |
6 |
– – 0,59 |
– – 0,35 |
– – 0,83 |
Diamorphin und 6-Acetylmorphin sind aufgrund rascher Metabolisierung nicht messbar |
|
|
Disulfiram Diethyldithiomethyl-carbamat |
829±169 585±138 |
80 |
7 22 |
24 |
0,20 0,79 |
0,16 0,61 |
0,24 0,98 |
||
|
Levomethadon |
161±68 |
82 |
35 |
24 |
3,37 |
1,95 |
4,79 |
CL assoziiert mit CYP3A4 |
|
|
Methadon |
182±43 |
80 |
34 |
24 |
2,96 |
2,26 |
3,66 |
CL assoziiert mit CYP2B6 und CYP3A4 |
|
|
Morphinsulfat, SR Morphin |
– 2577±1933 |
– 30 |
– 21 |
– 12 24 |
– 0,26 0,18 |
– 0,06 0,05 |
– 0,46 0,31 |
Daten für SR-Formulierung (slow release, SR), t1/2, und CL berechnet von Cmin-Berechnung der dosisbezogenen Konzentrationen muss für Morphin-Salze korrigiert werden (Sulfat, Sulfatpentahydrat oder -hydrochlorid) |
|
|
Nalmefen |
1007±188 |
41 |
13 |
2 6 12 |
0,79 0,26 0,19 |
0,65 0,21 0,16 |
0,94 0,31 0,23 |
Faktoren für Einzeldosen (als Bedarfsmedikation) |
|
|
12 |
0,64 |
0,52 |
0,76 |
Faktoren bei täglicher Einnahme |
|||||
|
Naltrexon 6β-Naltrexol |
2334±300 1083±157 |
35 |
4 11 |
24 |
0,02 0,27 |
0,02 0,23 |
0,02 0,31 |
CL bei alkoholabhängigen Patienten um ca. 20% gesteigert |
|
|
Nikotin |
1333±450 |
30 |
2 |
3 |
1,53 |
1,01 |
2,05 |
Nicotin Kaugummi |
|
|
– |
1,05 |
0,70 |
1,40 |
Während Pflaster-Applikation |
|||||
|
Vareniclin |
83,7±14,9 |
100 |
24 |
12 24 |
8,13 5,75 |
6,69 4,73 |
9,58 6,77 |
CL linear bezogen auf renale Clearance |
|
|
Arzneistoffe zur Behandlung der Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung |
|||||||||
|
Atomoxetin |
288±103 |
79 |
4 |
6 24 |
3,54 0,11 |
2,28 0,07 |
4,81 0,15 |
CL beeinflusst von CYP2D6 und CYP2C19 |
|
|
Dextroamphetamin |
428±107 |
89 |
9 |
6 24 |
2,24 0,56 |
1,68 0,42 |
2,80 0,70 |
||
|
Dexmethylphenidat |
3530±1072 |
23 |
3 |
6 |
0,27 |
0,19 |
0,36 |
Formulierung: bimodale Freisetzung |
|
|
Guanfacin |
630±198 |
81 |
18 |
6 |
1,34 |
0,92 |
1,76 |
CL beeinflusst von CYP3A4 |
|
|
Lisdexamfetamin d-Amphetamin |
20579±9245 1155±268 |
50 |
1 11 |
6 |
– 0,80 |
– 0,61 |
– 0,98 |
Aktiver Metabolit d-Amphetamin |
|
|
d,l-Methylphenidat, IR XR, Kinder, 6–12 Jahre XR, Erwachsene |
3177±1023 |
0,14 |
2 4,3 3,5 |
3 6 6 |
0,64±0,21 0,31 0,21 |
0,44 0,24 0,15 |
0,85 0,38 0,27 |
Faktoren beziehen sich auf Cmax der Nicht-Retard- und Retard-Formulierung (immediate und extended release, IR und XR) |
|
|
Modafinil |
51±11 |
33 |
14 |
6 |
17,3 |
13,6 |
21,0 |
||
|
Antiparkinsonika |
|||||||||
|
Pramipexol |
483±64 |
90 |
8 |
10 |
1,44 |
1,25 |
1,63 |
CL angegeben für Dexpramipexol |
|
|
Ropinirol |
956±412 |
50 |
6 |
10 |
0,68 |
0,39 |
0,97 |
CL bei Rauchern aufgrund der CYP1A2-Induktion beschleunigt |
|
|
Rotigotin |
5553±2600 |
– |
– |
– |
0,66 |
0,55 |
0,77 |
Faktoren angegeben für die transdermale Applikation und berechnet von TDM Daten, CL beeinflusst von CYP2C19 |
|
Pharmakokinetische Parameter wurden nach bestem Wissen zusammengestellt. Trotzdem wird keine Verantwortung für die Korrektheit dieser Daten übernommen. –: Kein Wert für diese Zeile
Verhältnis der Arzneistoff-Konzentration zur Dosis
Das Verhältnis der Arzneistoff-Konzentration zur Dosis (Cmin/D, abgekürzt als C/D) ist ein weiterer Parameter, um pharmakokinetische Besonderheiten zu identifizieren [19]. C/D kann leicht aus TDM-Daten berechnet werden, indem die Arzneistoff-Talspiegelkonzentration im Steady-State durch die eingenommene Dosis des Patienten geteilt wird. C/D-Quotienten stehen in inversem Zusammenhang zur totalen Clearance. Ein hoher C/D-Quotient deutet auf eine geringe, ein niedriger C/D-Quotient auf eine schnelle Arzneistoff-Clearance hin.
C/D-Quotienten werden angewendet, um Arzneimittel-Interaktionen zu identifizieren, indem verschiedene Patientengruppen miteinander verglichen werden. Jerling und Mitarbeiter analysierten intraindividuelle C/D-Quotienten von Amitriptylin und Nortriptylin und detektierten interagierende Effekte von Levomepromazin, Perphenazin und Carbamazepin. Sie zeigten dies anhand von Werten mit und ohne die entsprechenden Begleitsubstanzen und bekräftigen mit dieser Studie frühere C/D-Ergebnisse. Wiederholte Messungen der C/D-Quotienten im selben Patienten sind auch hilfreich zur Detektion von (partieller) Non-Adhärenz zur Medikation, wie anhand von Clozapin gezeigt wurde. Die intraindividuelle Variabilität der C/D sollte unter 20% liegen. Eine Variabilität über 20% weist auf Adhärenzprobleme, pharmakokinetische Alterationen aufgrund Arzneimittel-Interaktionen oder auf Interaktionen zwischen Nahrungsmitteln oder einer Erkrankung und einem Arzneistoff hin.
Der C/D-Quotient kann auch angewandt werden, um die benötigte Dosis zum Erreichen einer gewünschten Zielkonzentration des Arzneistoffs im Blut abzuschätzen. Wenn beispielsweise ein C/D-Quotient von 0,5 (ng/ml)/mg gemessen wurde und der therapeutische Referenzbereich des Arzneistoffs ist 30 bis 100 ng/ml, wird eine tägliche Dosis von 60 bis 200 mg benötigt, um den therapeutischen Referenzbereich zu erreichen.
Verhältnis Metabolit zu Muttersubstanz
Die Biotransformation von Neuropsychopharmaka anhand von Phase-I-Enzymen kann zu Metaboliten mit ähnlichen oder unterschiedlichen pharmakodynamischen Eigenschaften in Vergleich zur Muttersubstanz führen. Beispiele für Metaboliten mit ähnlichen Eigenschaften sind Nortriptylin (Muttersubstanz Amitriptylin), N-Desmethyldoxepin (Muttersubstanz Doxepin), Desipramin (Muttersubstanz Imipramin), Norfluoxetin (Muttersubstanz Fluoxetin), O-Desmethylvenlafaxin (Muttersubstanz Venlafaxin) oder 9-Hydroxyrisperidon (Muttersubstanz Risperidon). Für diese Arzneistoffe ist die Summe der Konzentrationen von Muttersubstanz und aktivem Metaboliten, das heißt die aktive Fraktion, für die Dosisfindung anhand von TDM relevant. Beispiele für Metaboliten mit unterschiedlichen pharmakodynamischen Eigenschaften in Vergleich zur Muttersubstanz sind Carbamazepin-10,11-epoxid (toxischer als Carbamazepin), N-Desmethylclomipramin (noradrenerge Aktivität), N-Desmethylclozapin (cholinomimetische Aktivität) oder N-Desalkylquetiapin (noradrenerge Aktivität). Die Hauptmetaboliten von Olanzapin, Sertralin oder Citalopram tragen wahrscheinlich nicht zur Wirksamkeit und Verträglichkeit der Muttersubstanz bei. Es ist umstritten, ob das Monitoring der Metaboliten sinnvoll ist, wenn die Metaboliten keine pharmakodynamische Aktivität besitzen. Für eine pharmakokinetische Beurteilung kann die Messung von aktiven und inaktiven Metaboliten jedoch informativ sein. Das Metabolit-zu-Muttersubstanz-Verhältnis (MPR [metabolite to parent compound ratio]) ist ein direktes Maß der Aktivität von metabolischen Enzymen in vivo [8]. Wenn ein bestimmtes CYP-Isoenzym hauptsächlich die Phase-II-Reaktion bestimmt, reflektiert das MPR den Phänotyp dieses CYP-Enzyms (Tab. 6 und Kasten). Das MPR erlaubt die Identifizierung eines abnormalen Metabolismus, der beispielsweise durch pharmakokinetische Interaktionen oder genetische Besonderheiten verursacht wird. Für Venlafaxin und Risperidon ist ein niedriges MPR indikativ für einen PM-Genotyp von CYP2D6. Der PM-Genotyp kann vom EM mit einer Sensitivität von 91% unterschieden werden. Eine hohe MPR ist indikativ für eine beschleunigte enzymatische Aktivität und somit für einen UM-Metabolisiererstatus. Des Weiteren können enzyminduzierende Effekte, beispielsweise von CYP1A2 aufgrund Zigarettenrauch, anhand eines MPR steigernden Effekts identifiziert werden. Für Sertralin wurde gezeigt, wie das MPR von N-Desmethylsertralin zu Sertralin genutzt werden kann, um die Adhärenz der Patienten zu der Medikation zu prüfen [36].
Definition: Metabolit-zu-Muttersubstanz-Verhältnis, MPR
Der Begriff „metabolische Ratio“ wird in der Literatur inkonsequent genutzt, entweder als Verhältnis der Konzentration von Muttersubstanz zu Metabolit oder vice versa Metabolit zu Muttersubstanz. Um Verwirrung zu vermeiden, verwenden wir den Begriff Metabolit-zu-Muttersubstanz-Verhältnis bzw. MPR (metabolite to parent compound ratio [Metabolit-zu-Muttersubstanz-Quotient]). MPR-Werte sind in Tabelle 6 für 37 Neuropsychopharmaka gelistet zur In-vivo-Abschätzung der enzymatischen Aktivität, die in den Metabolismus des jeweiligen Arzneistoffs involviert ist. Bei einer angenommenen normalen Verteilung wurden die durchschnittlichen MPRs ± Standardabweichung (SD) für Standarddosierungen berechnet. Ausreißer vom durchschnittlichen (±SD) Bereich können auf (partielle) Non-Adhärenz oder Besonderheiten im Arzneistoffmetabolismus hinweisen, die Gründe hierfür sollten abgeklärt werden.
Tab. 6. Metabolit-zu-Muttersubstanz-Verhältnis (MPR, metabolite to parent compound ratio) von Neuropsychopharmaka. MPR-Bereiche sind durchschnittliche Quotienten ± Standardabweichung unter Steady-State und Talspiegelbedingungen. Die angegebenen Bereiche beziehen sich auf Quotienten in „normalen“ Individuen ohne genetische Besonderheiten bezüglich Arzneistoff-metabolisierender Enzyme oder Komedikation mit Inhibitoren oder Induktoren von Arzneistoff-metabolisierenden Enzymen. Für die Interpretation der TDM-Ergebnisse von gelisteten Neuropsychopharmaka muss geprüft werden, ob der gemessene Quotient oberhalb oder unterhalb des durchschnittlichen Bereichs liegt. Ein metabolischer Quotient außerhalb dieses Bereiches ist indikativ unter anderem für Adhärenzprobleme oder pharmakokinetische Besonderheiten des Patienten, welche geklärt werden sollten.
|
Muttersubstanz |
Metabolit |
Quotient |
Hauptsächlich involvierte CYP-Enzyme |
Kommentare |
|
Amitriptylin |
Nortriptylin |
0,2–1,8 (n=83) |
CYP2C19 |
|
|
Aripiprazol |
Dehydroaripiprazol |
0,3–0,5 (n=283) |
CYP3A4, CYP2D6 |
Ähnlicher Quotient für die orale Formulierung und die Depot-Injektion |
|
Bromperidol |
Reduziertes Bromperidol |
0,11–0,51 (n=31) |
CYP3A4 |
|
|
Buprenorphin |
N-Desmethylbuprenorphin |
1,58–2,36 (n=29) |
CYP3A4 |
|
|
Bupropion |
Hydroxybupropion |
11,2–21,0 (n=10) |
CYP2B6 |
Bupropion ist bei Raumtemperatur instabil |
|
Buspiron |
6-Hydroxybuspiron |
25–53 (n=20) |
CYP3A4 |
|
|
Carbamazepin |
Carbamazepin-10,11-epoxid |
0,07–0,25 (n=14) |
CYP3A4 |
|
|
Cariprazin |
N,N-Didesmethylcariprazin |
3–6 (n=38) |
CYP3A4 |
|
|
Citalopram |
N-Desmethylcitalopram |
0,31–0,60 (n=2330) |
CYP2C19 |
|
|
Clobazam |
N-Desmethylclobazam |
2–10 (n>150) |
CYP3A4 |
|
|
Clomipramin |
N-Desmethylclomipramin |
0,8–2,6 (n=115) |
CYP1A2, CYP2C19 |
|
|
Clozapin |
N-Desmethylclozapin |
0,45–0,79 |
CYP1A2, CYP2C19 |
Quotienten niedriger bei Rauchern in Vergleich zu Nichtrauchern |
|
Diazepam |
N-Desmethyldiazepam |
0,94–1,92 (n=7) |
CYP2C19, CYP3A4 |
|
|
Dothiepin |
N-Desmethyldothiepin |
0–1,4 (n=50) |
CYP2C19 |
|
|
Doxepin |
N-Desmethyldoxepin |
0,6–1,6 (n=12) |
CYP2C9, CYP2C19, CYP2D6 |
|
|
Escitalopram |
N-Desmethylescitalopram |
0,3–1,0 (n=243) |
CYP2C19 |
|
|
Fluoxetin |
N-Desmethylfluoxetin |
0,7–1,9 (n=334) |
CYP2B6, CYP2C9, CYP2C19 |
|
|
Fluvoxamin |
Fluvoxaminsäure |
0–1,2 (n=49) |
CYP2D6 |
|
|
Haloperidol |
Reduziertes Haloperidol |
0,14–0,42 (n=5) |
CYP2D6 |
|
|
Imipramin |
Desipramin |
0,6–3,2 (n=14) |
CYP2C19 |
|
|
Maprotilin |
N-Desmethylmaprotilin |
1,1–3,7 (n=76) |
CYP2D6 |
|
|
Mianserin |
N-Desmethylmianserin |
0,5–0,8 (n=182) |
CYP2D6 |
|
|
Mirtazapin |
N-Desmethylmirtazapin |
0,2–1,2 (n=100) |
||
|
Moclobemid |
Moclobemid N-oxid |
0,8–2,5 (n= 6) |
||
|
Olanzapin |
N-Desmethylolanzapin |
0,1–0,3 (n=76, Nichtraucher) |
CYP1A2 |
|
|
Perazin |
N-Desmethylperazin |
1,1–3,3 (n=27) |
CYP2C19 |
|
|
Perphenazin |
N-Desalkylperphenazin |
0,6–2,8 (n=54) |
CYP2D6 |
|
|
Quetiapin |
N-Desalkylquetiapin |
0,54–3,10 (n=601) |
CYP3A4 |
Ähnlicher Quotient bei Kindern und Erwachsenen |
|
Reboxetin |
O-Desethylreboxetin |
<0,1 (n=38) |
CYP3A4 |
|
|
Risperidon |
9-Hydroxyrisperidon |
3,6–22,7 (n=168) |
CYP2D6 |
|
|
Risperidon |
9-Hydroxyrisperidon |
1,2–4,3 (n=30) |
CYP2D6 |
Intramuskuläre Depot-Injektion |
|
Sertindol |
Dehydrosertindol |
1,1–2,7 (n=6) |
CYP2D6 |
|
|
Sertralin |
N-Desmethylsertralin |
1,7–3,4 (n=348) |
CYP2B6 |
|
|
Trazodon |
m-Chlorophenylpiperazin (mCPP) |
0,04–0,22 (n=43, gesamter Bereich) |
CYP3A4 |
|
|
Trimipramin |
N-Desmethyltrimipramin |
0,26–0,56 (n=17) |
CYP2C19 |
|
|
Venlafaxin |
O-Desmethylvenlafaxin N-Desmethylvenlafaxin |
2,7–7,7 (n=217) 0,28–0,85 (n=145) |
CYP2D6 CYP2C19 |
Informationen in dieser Tabelle beziehen sich auf bestimmte metabolische Abbauwege eines Arzneistoffs, wohingegen Tabelle 1 metabolisierende CYP-Enzyme sämtlicher Abbauwege eines Arzneistoffs berücksichtigt.
Wenn das MPR zur Charakterisierung des metabolischen Phänotyps eines Patienten genutzt wird, müssen Störfaktoren gut kontrolliert werden, um falsche Schlussfolgerungen zu vermeiden. Insbesondere der korrekte Zeitpunkt der Blutentnahme ist entscheidend, wenn die Muttersubstanz und der Metabolit unterschiedliche Eliminationshalbwertszeiten besitzen.
Die Validität des MPR, um CYP-Genvarianten vorherzusagen, wurde für Risperidon und Venlafaxin in verschiedenen Studien bewiesen. Für Risperidon und seinen Metaboliten 9-Hydroxyrisperidon beträgt die Grenze des MPR zur Unterscheidung zwischen CYP2D6-EM und -PM 1,0. Für das Antipsychotikum war die Sensitivität 91%, die Spezifität 86% und der positive, prädiktive Wert 35%, während der negative prädiktive Wert 99% betrug. Ähnliche Ergebnisse wurden für Venlafaxin und den Hauptmetaboliten O-Desmethylvenlafaxin gefunden. Eine Grenze (Cut-off) des MPR von 1,0 hatte eine Sensitivität von 93%, eine Spezifität von 86%, einen positiven prädiktiven Wert von 40% und einen negativen prädiktiven Wert von 99%. Um UM und EM zu unterscheiden, ist der MPR-Wert weniger sensitiv. Phänotypen dieser Genotypen überlappen. Diesbezüglich sollte berücksichtigt werden, dass die CYP2D6-Genotypisierung nur 30% der UM-Phänotypen erklärt. Trotz solcher Limitierungen empfehlen wir die Bestimmung des MPR für die Charakterisierung des metabolischen Phänotyps eines Patienten.
Probe-Drug-Phänotypisierung
Der pharmakokinetische Phänotyp wird mithilfe eines Testsubstrats, einer sogenannten „probe drug“ gemessen. Die Tests wurden in der Vergangenheit vorgestellt, als beobachtet wurde, dass der Metabolismus von Arzneistoffen genetisch bestimmt wird. Dies wurde für zahlreiche Arzneistoffe herausgefunden, beispielsweise Debrisoquin, Mephenytoin, Spartein und auch für das Antidepressivum Nortriptylin. Die systematische Forschung identifizierte Substanzen, die bevorzugt von bestimmten CYP-Enzymen metabolisiert werden. Anhand dieser Kenntnisse wurden Phänotypisierungs-Tests entwickelt und mit spezifischen Testsubstraten validiert, beispielsweise Coffein für CYP1A2, Efavirenz für CYP2B6, Losartan oder Tolbutamid für CYP2C9, Omeprazol oder Mephenytoin für CYP2C19, Dextromethorphan, Debrisoquin oder Metoprolol für CYP2D6, Midazolam oder Erythromycin für CYP3A4 und Chlorzoxazon für CYP2E1 [22]. Personen nehmen diese Testsubstrate in einer möglichst pharmakodynamisch ineffektiven Dosis ein, und die Konzentrationen der Muttersubstanz und Metaboliten, metabolisiert durch die Indikatorreaktion, werden bestimmt. Die Konzentrationen oder Quotienten der Konzentrationen reflektieren die In-vivo-Aktivität der jeweiligen CYP-Enzyme. Fortschritte in der Arzneistoff-Analyse anhand der Massenspektrometrie ermöglichten die Anwendung von Cocktails, die sechs oder mehr Testsubstrate enthielten. Sie erlauben die Quantifizierung der Aktivität von verschiedenen Isoenzymen anhand eines Tests. Eine praktische Idee für Phänotypisierungs-Tests war die Messung der optimalen Dosis von einem bestimmten Arzneistoff. Solche Untersuchungen waren bisher jedoch nicht erfolgreich. Da nur wenige Arzneistoffe von einem einzigen Isoenzym metabolisiert werden, ist es schwierig, die optimale Dosis anhand von Phänotypisierungs-Tests zu berechnen. Angemessener ist die Analyse der Konzentration des verschriebenen Arzneistoffes, das heißt die Anwendung von TDM für die Dosisfindung. Phänotypisierungs-Tests sind jedoch gut etabliert, um pharmakokinetische Interaktionen zu untersuchen, bevorzugt während der Entwicklungsphase eines Arzneistoffs. Wenn In-vitro-Daten Hinweise für CYP-inhibitorische oder -induzierende Eigenschaften eines Arzneistoffs liefern, ist eine Phänotypisierung für dessen Klärung empfohlen. Darüber hinaus kann eine Phänotypisierung als Add-on hilfreich für TDM sein. Mit Coffein als Test-Substanz konnten die CYP1A2 induzierenden Effekte von Zigarettenrauch charakterisiert werden. Des Weiteren konnte gezeigt werden, dass der induzierende Effekt bereits vier Tage nach dem Konsum der letzten Zigarette verschwunden ist.
Indikationen für die Messung der Arzneistoff-Konzentration im Blut
Tabelle 7 präsentiert eine Liste der Indikationen für TDM in der Psychiatrie und Neurologie. Die Gültigkeit dieser Angaben muss im Einzelfall individuell geprüft und bewertet werden. Ähnlich wie bei allen diagnostischen Tests sollte TDM nur angefordert werden, wenn das Ergebnis eine Antwort auf eine klar definierte Fragestellung liefern kann.
Tab. 7. Typische Indikationen für die Messung von Arzneistoff-Konzentrationen im Blut von psychiatrischen oder neurologischen Patienten
|
Obligatorisches TDM (für Arzneistoffe mit hohem Empfehlungsgrad |
|
|
Spezifische Indikationen für TDM (für Arzneistoffe unabhängig vom Empfehlungsgrad für die Anwendung von TDM) |
|
Für Arzneistoffe mit gut definierten therapeutischen Referenzbereichen oder mit einem engen therapeutischen Index ist es sinnvoll, Blutspiegel für die Dosisfindung nach initialer Verschreibung und nach einer Dosisänderung zu messen. Auch ohne ein spezifisches Problem gibt es genügend Nachweise, dass TDM positive Effekte für Patienten erzielt, die mit folgenden Arzneistoffen behandelt werden: Lithium, trizyklische Antidepressiva, verschiedene Antipsychotika oder Antikonvulsiva (Tab. 4). Für Lithium ist TDM aus Gründen der Sicherheit zwingend geboten.
Probleme mit der Adhärenz (Non-Adhärenz, partielle Adhärenz) – ein politisch korrekterer Ausdruck als der Begriff „Compliance“, da „Adhärenz“ voraussetzt, dass der Patient hierarchisch gleicher Partner in der therapeutischen Entscheidung ist – sind in der Pharmakotherapie häufig und kostspielig. Im Durchschnitt werden 50% der Medikamente für chronische Erkrankungen nicht wie verschrieben eingenommen. Bei Patienten mit Schizophrenie und bei Patienten mit unipolaren oder bipolaren Störungen reicht die Häufigkeit von Non-Adhärenz von 10 bis 69% [48]. In einer großen Anzahl (n=15809) an Patienten mit Demenz, die mit Acetylcholinesterase-Inhibitoren behandelt wurden, waren während einer Beobachtungsphase von 12 Monaten nur 34% adhärent. Unvollständige oder totale Non-Adhärenz beeinträchtigt die Wirksamkeit der Behandlung. Ein Bericht der Weltgesundheitsorganisation legt nahe, dass eine Verbesserung der Adhärenz einen größeren Einfluss auf den therapeutischen Erfolg haben kann als irgendeine Verbesserung spezifischer medizinischer Behandlungen. Methoden zur Messung der Adhärenz beinhalten unter anderem Pillenzählen, Hinzufügen gefärbter, im Urin detektierbarer Substanzen, Interview mit Patienten oder Urteil des behandelnden Arztes bezüglich der Adhärenz. Studien haben jedoch gezeigt, dass Ärzte die Adhärenz ihrer Patienten nicht verlässlich vorhersagen können. Die Messung von Arzneistoff-Konzentrationen im Blut ist vorteilhaft gegenüber anderen Methoden, da diese Methode dem behandelnden Arzt anzeigt, ob das Medikament in einer Konzentration vorliegt, die wahrscheinlich für die zu erwartende Besserung der Symptome ausreicht. Bei Patienten mit Epilepsie bestätigte das Monitoring der Arzneistoff-Konzentration häufiger Non-Adhärenz als eine adäquate Kontrolle der Krampfanfälle. Für Antikonvulsiva wurden subtherapeutische Konzentrationen bei den meisten Patienten detektiert, die aufgrund von Krampfanfällen hospitalisiert wurden. Abweichungen vom erwarteten dosisbezogenen Referenzbereich (Tab. 5) geben Hinweise, ob der Patient seine Medikamente genommen hat und/oder ob er ein schneller oder langsamer Metabolisierer ist. Die gleichzeitige Bestimmung der Metaboliten kann zusätzlich klären, ob das Medikament auch kontinuierlich eingenommen wurde. Für die Interpretation müssen jedoch auch mögliche Arzneistoff-Interaktionen mit der Komedikation berücksichtigt werden (Tab. 2 und 3). Reis und Mitarbeiter [36] analysierten die Adhärenz von Sertralin-behandelten Patienten anhand wiederholter Messung der Serum-Konzentrationen von Muttersubstanz und Metaboliten. Variationen des Verhältnisses der Konzentrationen von Norsertralin zu Sertralin waren höchst bezeichnend für versteckte und (partielle) Non-Adhärenz.
Wie erwähnt, listet dieser Konsensus MPR-Werte für 35 Neuropsychopharmaka auf (Tab. 6). Indem man mehrere Blutproben pro Tag entnimmt und die zu erwartende Arzneistoff-Konzentration im Blut berechnet kann unterschieden werden, ob eine niedrige Arzneistoff-Konzentration aufgrund einer reduzierten Bioverfügbarkeit, einem beschleunigten Abbau oder aufgrund mangelnder Adhärenz vorliegt. Ein pharmakokinetisches Modelling der zu erwartenden zeitabhängigen Arzneistoff- und Metabolit-Konzentration im Blut ermöglicht die Identifizierung von verschiedenen Typen der Non-Adhärenz.
Rückfallprävention ist ein wichtiges Ziel der Erhaltungstherapie. Regelmäßiges TDM ist eine sehr kostengünstige Methode, um Rückfälle, die zur Hospitalisierung führen können, zu verhindern. Bei schizophrenen Patienten hat sich gezeigt, dass Schwankungen der Clozapin-Blutspiegel prädiktiv für Rückfälle sind. TDM kann helfen, das Risiko eines Rückfalls zu mindern, indem die Achtsamkeit des behandelnden Arztes bezüglich der Adhärenz des Patienten zur Medikation gesteigert wird.
Empfehlung – Monitoring
Wir empfehlen ein regelmäßiges Monitoring, mindestens alle 3 bis 6 Monate, der Arzneistoff-Konzentrationen im Blut unter Erhaltungstherapie, um Rückfälle und Rehospitalisierungen zu verhindern. Die Häufigkeit von TDM-Anforderungen sollte erhöht werden, wenn der Patient bekanntermaßen zu Non-Adhärenz neigt. Ebenso sollte TDM bei Änderungen der Komedikation oder des Rauchverhaltens eingesetzt werden, welche die Pharmakokinetik des verschriebenen Arzneistoffs beeinflussen können.
Wenn eine klinische Verbesserung unter den empfohlenen Dosen nicht ausreichend und das Medikament gut verträglich ist, kann TDM klären, ob die Arzneistoff-Konzentration zu niedrig ist und ob es sinnvoll ist, die Dosis zu erhöhen.
Falls unerwünschte Arzneimittelwirkungen mit einer klinischen Verbesserung unter den empfohlenen Dosen zusammenfallen, kann die Messung der Blutkonzentration klären, ob die Nebenwirkungen mit zu hohen Arzneistoff-Konzentrationen im Blut verbunden sind und daher die Dosis ohne Verlust der Wirksamkeit verringert werden sollte.
Bei der Kombination von Medikamenten, die Inhibitoren oder Induktoren Arzneistoff-metabolisierender Enzyme sind (Tab. 2 und 3), mit einem Arzneistoff, der Substrat des inhibierten oder induzierten Enzyms ist (Tab. 1), sollte die Dosis anhand von TDM bestimmt werden, um Wirkverlust oder Unverträglichkeiten/Intoxikationen aufgrund pharmakokinetischen Arzneimittel-Interaktionen zu vermeiden. Effekte aufgrund von Rauchen sollten berücksichtigt werden, wenn die Patienten mit einem CYP1A2-Substrat behandelt werden, beispielsweise Clozapin, Duloxetin, Olanzapin, Rasagilin oder Ropinirol (Tab. 1).
Bei Patienten mit genetischen Besonderheiten in der Ausstattung mit Enzymen des Arzneimittelabbaus müssen die Dosierungen des Arzneistoffs angepasst oder durch therapeutische Alternativen ersetzt werden. Kirchheiner (Stingl) und Kollegen [39, 40] berechneten aufgrund pharmakokinetischer und pharmakodynamischer Erkenntnisse Dosen für PM oder UM von CYP2D6. Diese Dosisanpassungen, basierend auf pharmakogenetischer Evidenz, wurden von internationalen Konsortien weiterentwickelt, zum Beispiel vom Pharmacogenetic Clinical Implementation Consortium (CPIC), und evidenzbasierte Leitlinien wurden entwickelt, wie die Therapie im Falle einer pharmakogenetischen Variante für Trizyklika und SSRI angepasst werden muss. Doch selbst im Falle eines bestätigten abnormen CYP-Genotyps wird TDM empfohlen, da die meisten CYP-Isoenzyme nicht substratspezifisch sind und die Genotypisierung nur grob vorhersagen kann, in welchem Umfang sich Blutspiegel beim individuellen Patienten ändern.
Bei einer Pharmakotherapie von schwangeren oder stillenden Frauen sollte sichergestellt werden, dass die Arzneistoff-Konzentration im Blut innerhalb des therapeutischen Referenzbereichs ist, um das Risiko eines Rückfalls auf der Seite der Mutter und auf der anderen Seite die Medikamenten-Exposition des Fötus bzw. des Kindes zu minimieren [9]. Die renale Clearance und die Aktivität der CYP-Isoenzyme 2A6, 2C9, 2D6 und 3A4 sowie der Uridin-5’-diphosphat Glucuronosyltransferase (UGT) 1A4 und 2B7 sind während der Schwangerschaft erhöht, während die Aktivitäten von CYP1A2 und 2C19 und N-Acetyltransferase 2 (NAT2) verringert sind. TDM sollte bei Schwangeren und/oder Müttern mindestens einmal pro Trimester und innerhalb von 24 Stunden nach der Geburt durchgeführt werden [5, 25].
Viele Neuropsychopharmaka sind nicht für den Einsatz bei Kindern und Jugendlichen zugelassen. Bis heute basieren therapeutische Referenzbereiche der meisten Neuropsychopharmaka auf Studien, die bei Erwachsenen durchgeführt wurden, und Daten über die Korrelation der Arzneistoff-Konzentration mit dem Therapieansprechen oder unerwünschten Arzneimittelwirkungen in der pädiatrischen Bevölkerung sind knapp [10, 47]. Der relative Mangel an klinischen Studien und der daraus resultierende Off-Label-Gebrauch kann zu einem höheren Risiko von Dosisfehlern und unerwünschten Arzneimittelwirkungen führen. Pharmakokinetische und pharmakodynamische Veränderungen während der körperlichen Entwicklung [11] legen nahe, dass Dosierungen und klinische Effekte bei Kindern und Jugendlichen nicht von den evidenzbasierten Daten, die bei Erwachsenen gewonnen wurden, extrapoliert werden können. Die steigende Anzahl an Verschreibungen für pädiatrische Patienten steht in Widerspruch zu diesen Unsicherheiten bezüglich Sicherheit und Wirksamkeit [10], und die Ärzte und Betreuer der Kinder und Jugendlichen tragen demzufolge eine große Verantwortung. Unter diesen Umständen ist TDM dringend für eine individuelle, optimal wirksame und verträgliche Pharmakotherapie empfohlen. Bei Jugendlichen, die unter psychotischen Störungen leiden, ist komorbider Medikamentenmissbrauch sehr verbreitet, und die Adhärenz zu einer antipsychotischen Behandlung ist in der Regel marginal. Daher ist TDM bei diesen Patienten besonders empfohlen.
Die Extrapolation der therapeutischen Referenzbereiche, die auf Daten von Erwachsenen basieren, auf pädiatrische Patienten, insbesondere auf junge Kinder, muss für jede einzelne Substanz untersucht werden, da vorläufige TDM-Studien in der pädiatrischen Neuropsychiatrie divergente Ergebnisse lieferten. Glücklicherweise haben jedoch einige Studien ähnliche therapeutische Referenzbereiche für Kinder und Jugendliche und Erwachsene gezeigt, beispielsweise für Sertralin, Aripiprazol [33] und Fluvoxamin. Für die meisten Arzneistoffe besteht bei Kindern und Jugendlichen nach Einnahme derselben Dosis eine hohe interindividuelle Variabilität der Arzneistoff-Konzentration. Ähnlich wie bei erwachsenen Patienten besteht im Allgemeinen ein Zusammenhang zwischen Dosis und Konzentration. Schließlich gibt es Evidenz, dass bezogen auf das Gewicht höhere Dosen benötigt werden, um Arzneistoff-Konzentrationen innerhalb der Referenzbereiche für Erwachsene zu erzielen. Des Weiteren scheinen sich für Arzneistoffe wie Quetiapin, Clozapin oder Risperidon die Referenzbereiche von denen für Erwachsene zu unterscheiden.
Die Implementierung von TDM für Kinder ist jedoch schwieriger als bei Erwachsenen, da insbesondere invasive Probenentnahmen die Kooperation des Patienten voraussetzen. In klinischen Forschungsprojekten untersucht man deshalb die Eignung alternativer Matrizes, beispielsweise Speichel, und schonendere Probennahmetechniken, beispielsweise die Blutfleck-Technik (dried blood-spot). Im Routine-TDM könnte dies Unannehmlichkeiten für pädiatrische Patienten minimieren.
Neben einem Plädoyer für mehr klinische Studien und mehr pharmakokinetisch-pharmakodynamische Studien in Kindern und Jugendlichen ist die aktive und standardisierte Überwachung (d.h. Patienten-Monitoring) von Kindern und Jugendlichen nach Beginn einer Neuropsychopharmakotherapie notwendig. Ein standardisiertes Register, das solche Beobachtungen, Bewertungen und Messungen inklusive TDM von vielen Patienten erfasst, wurde vom TDM-Kompetenznetzwerk für Kinder und jugendliche Patienten [siehe www.tdm-kjp.com] etabliert. Es erfasst Pharmakovigilanzdaten (Evidenz) bezüglich Dosisschemata, Arzneistoff-Konzentrationen im Blut und die Effektivität und Verträglichkeit von Neuropsychopharmaka unter naturalistischen Bedingungen. Dieser Ansatz soll das Risiko, dass pädiatrische Patienten einer ineffektiven oder schlecht verträglichen Pharmakotherapie ausgesetzt werden, minimieren [12].
Für ältere Patienten wird TDM ebenfalls dringend empfohlen [43]. Altern beinhaltet fortschreitende Beeinträchtigungen der funktionalen Reserven mehrerer Organe. Insbesondere die renale Exkretion und die Leberfunktion können sich signifikant verändern. Phase-I-Reaktionen sind wahrscheinlicher in ihrer Funktion beeinträchtigt als Phase-II-Reaktionen. Die glomeruläre Filtration, tubuläre Reabsorption und die Sekretion verändern sich mit zunehmendem Alter, aber auch das Körpergewicht und das Verteilungsvolumen. Die hepatische Clearance kann um bis zu 30% reduziert sein, was primär eher aufgrund eines verminderten hepatischen Blutflusses erklärt werden kann als durch eine Abnahme der Aktivität der metabolisierenden Enzyme. Einigen Autoren zufolge gibt es keine wichtigen, altersabhängigen Veränderungen in der Aktivität von CYP-Isoenzymen, während andere eine leichte Abnahme in der Aktivität von CYP2D6, aber nicht von CYP2C und CYP3A beschreiben. Alterspatienten reagieren häufig überempfindlich auf Arzneistoffe, und Gebrechlichkeit ist ein erhebliches Problem. Sie besitzen ein gesteigertes Risiko für einen Verlust der Homöostase nach Belastungen und eine verminderte Fähigkeit, in einen stabilen Zustand zurückzukehren. Beispielhaft scheint das cholinerge System bei alten Patienten supersensitiv zu sein. Viele Psychopharmaka, zum Beispiel Clozapin, trizyklische Antidepressiva oder Paroxetin, besitzen anticholinerge Eigenschaften. Ihr Gebrauch kann zur Entwicklung eines Delirs führen, zu einer Abnahme kognitiver Funktionen sowie zu weiteren, schweren unerwünschten Arzneimittelwirkungen. Wie für Nortriptylin gezeigt wurde, steigt die anticholinerge Aktivität mit zunehmender Arzneistoff-Konzentration im Blut an und tritt bereits bei therapeutischen Nortriptylin-Konzentrationen im Blut auf. Das erhöhte Risiko für unerwünschte Arzneimittelwirkungen hat dazu geführt, Kriterien für die Identifizierung von potenziell inadäquater Medikation für Alterspatienten zu entwickeln, beispielsweise die Beers-Kriterien, die PRISCUS-Liste [23], STOPP und andere. Auf der anderen Seite sind ältere Patienten häufig mit potenziell nützlichen Arzneistoffen unterversorgt, inklusive Antidepressiva. Darüber hinaus erhöht die Gebrechlichkeit das Risiko weiterer Komorbiditäten und somit auch das Risiko für Polypharmazie, was die Pharmakotherapie bei Alterspatienten zusätzlich erschwert. Schließlich scheinen Off-Label-Verschreibungen von Neuropsychopharmaka für ältere Patienten verbreitet zu sein. Zweifellos sind bisher nur unzureichend Daten bezüglich der Nützlichkeit von TDM in älteren Patienten verfügbar. Infolgedessen gibt es kaum evidenzbasierte Empfehlungen, TDM in dieser Bevölkerungsgruppe durchzuführen, um die medikamentöse Behandlung zu optimieren. „Monitoring“ wird regelmäßig empfohlen, aber diese Empfehlung bedeutet nicht zwangsläufig den Einsatz von TDM.
Bei Personen mit geistiger Behinderung werden häufig Antipsychotika der zweiten Generation angewandt. Praxis-Leitlinien empfehlen TDM für diese Patienten zumindest dann, wenn sie mit Risperidon oder Olanzapin behandelt werden. Aus ethischen und rechtlichen Gründen werden die Patienten mit geistiger Behinderung jedoch aus klinischen Studien ausgeschlossen, obwohl viele von ihnen eine Medikation benötigen. Bei diesen Personen kann es schwierig sein, zwischen krankheits- und medikamentenbedingten Gründen für eine Symptomverschlechterung zu unterscheiden. Deshalb wird TDM als objektives Instrument für die Pharmakotherapie dieser Patienten empfohlen.
Bei Patienten mit gesteigertem C-reaktiven Protein (CRP), was auf eine Entzündung oder Infektion hinweist, und unter Pharmakotherapie mit Clozapin oder Risperidon wird TDM empfohlen, um das Risiko von Intoxikationen aufgrund erhöhter Arzneistoff-Konzentrationen im Blut zu minimieren.
Bei Patienten mit substanzbezogenen Störungen und Abhängigkeitssyndromen sind die verfügbaren Arzneistoffe mit geprüfter Wirksamkeit Kandidaten für TDM [6]. Ihre Arzneistoff-Konzentrationen sind zwischen den Patienten hoch variabel [6]. Bei der Substitution mit Opioid-Agonisten kann eine Überdosis fatale Konsequenzen haben. Des Weiteren ist die Non-Adhärenz-Rate hoch. Die Art der Non-Adhärenz dieser Patienten unterscheidet sich jedoch von anderen Patientengruppen. Patienten mit substanzbezogenen Störungen akzeptieren gewöhnlich die Medikation zur Substitutionstherapie. Aber sie können den Eindruck haben, dass ihre Dosis unzureichend ist, und deshalb höhere Dosen als verschrieben konsumieren oder zusätzlich illegal erworbene Substanzen einnehmen. Andere Patienten brechen die Substitutionstherapie ab. In einer Studie mit Opioid-abhängigen Patienten war eine medizinische Behandlung nur effektiv, sofern sie adhärent mit der Medikation waren. Die Opiat-Agonisten, beispielsweise racemisches Methadon, R-(–)-Methadon (Levomethadon), Buprenorphin mit und ohne Naloxon und Formulierungen mit langsamer Freisetzung von Morphin werden zur oralen Anwendung für die Opioid-Erhaltungstherapie genutzt. In bestimmten Fällen kann i.v. Diacetylmorphin (Heroin) gegeben werden. TDM wird ausdrücklich für Methadon oder R-(–)-Methadon, Buprenorphin und möglicherweise auch für Formulierungen mit langsam freisetzenden Morphin empfohlen. Basierend auf den Arzneistoff-Eigenschaften und den Patienten-Charakteristika wurde die Nützlichkeit von TDM für die Behandlung der Alkoholabhängigkeit mit Arzneistoffen wie Acamprosat, Naltrexon oder Disulfiram und von Opioid-Abhängigen mit Naltrexon bezüglich einer Abstinenz-orientierten Behandlung untersucht [6]. TDM hat das Potenzial, die moderate Wirksamkeit der Therapie zu steigern, und ermöglicht die Detektion von pharmakokinetischen Besonderheiten aufgrund von Genvarianten von Arzneistoff-metabolisierenden Enzymen oder Arzneimittel-Interaktionen. Aufgrund der verschiedenen Arten von Adhärenz von Patienten mit substanzbezogenen Störungen sollte man sich bewusst sein, dass nicht nur erniedrigte, sondern auch erhöhte Arzneistoff-Konzentrationen auftreten können.
Bei forensischen psychiatrischen Patienten ist die Medikation in erster Linie wichtig, um das Risiko von Gewalt und aggressivem Verhalten zu reduzieren [28]. Erst an zweiter Stelle gilt es, psychiatrische Symptome zu mindern. Um diese Ziele zu erreichen, ist die Adhärenz zur Medikation, die meist aus Antipsychotika besteht, essenziell, da die meisten forensischen Patienten eine Pharmakotherapie ablehnen [28]. Castberg und Spigset analysierten Daten einer forensischen Hochsicherheits-Einheit und stellten fest, dass bei forensischen Patienten höhere Dosen als bei einer Kontrollgruppe verschrieben wurden. Die dosisabhängigen Blutkonzentrationen waren aber in der Gruppe der forensischen Patienten für Olanzapin signifikant niedriger als in der Kontrollgruppe, für Quetiapin jedoch höher. TDM wird für diese Patientengruppe dringend empfohlen, insbesondere wenn sie ambulant überwacht werden.
Bei Gerichtsverfahren, in denen mutmaßliche unerwünschte Arzneimittelwirkungen eine Rolle spielen, ist TDM für den Gerichtsgutachter hilfreich, um zu beweisen oder zu widerlegen, ob der Kläger die Medikation tatsächlich eingenommen und Arzneistoff-Konzentrationen im Blut erreicht hat, die glaubhaft das mutmaßliche Leiden ausgelöst haben [48]. Es wurde gezeigt, dass 55% der Kläger für Erwerbsunfähigkeitsrente, bei denen eine Depression diagnostiziert wurde und bei denen Arzneistoffe verschrieben wurden, keine detektierbaren Antidepressiva im Blut hatten. Weitere 11% hatten Antidepressiva-Konzentrationen im Blut, die nahezu null und weit unter der unteren Grenze des therapeutischen Referenzbereichs waren. Dementsprechend konnten insgesamt 66% der Kläger, die vor Gericht eine Erwerbsunfähigkeitsrente erstreiten wollten, nicht beweisen, dass sie tatsächlich ihre Antidepressiva wie behauptet einnahmen. Dies ist bedeutsam, da eine kranke Person eindeutig zu ihrer Rekonvaleszenz beitragen muss. Nur im Falle von Behandlungsfehlern besteht ein Anspruch auf Krankengeld oder Beufsunfähigkeitsleistungen. Dementsprechend erfüllten in der zuvor erwähnten Gruppe 66% der Kläger nicht die Kriterien, und weckten den Tatverdacht eines Betrugs der Krankenkassen.
Für die Indikation „Umstellung der Medikation von dem Originalpräparat auf ein Generikum oder Umstellung von einem Generikum auf ein Anderes“ sollte TDM angewandt werden, anstatt abzuwarten, wenn Probleme wie Wirkverlust oder Unverträglichkeiten auftreten. In einer Studie, die Originalpräparate und Generika bei Freiwilligen untersuchte, waren nach Einnahme des Generikums die Venlafaxin-Spiegel während der Absorptionsphase im Blut 50% höher als unter dem Originalpräparat. Demzufolge war die Anzahl an unerwünschten Arzneimittelwirkungen erhöht. Dies war nicht der Fall für Originalpräparat und Generikum von Citalopram. Generika können sich auch untereinander um bis zu 45% unterscheiden. Jedes Generikum darf eine AUC und eine Cmax im Bereich von 80 bis 125% des Originalpräparats (125%–80%=45%) aufweisen. Während der initialen Absorptionsphase können sogar größere Unterschiede auftreten, auch wenn die AUC und Cmax weiterhin innerhalb des 80- bis 125%-Bereichs liegen.
Andere Indikationen für TDM sind der Gebrauch von frei verkäuflichen Arzneimitteln und von gefälschten Arzneimitteln aus dem Internet. Die gefälschten Medikamente entsprechen oft nicht den Standards zu Reinheit und Dosis und erhöhen somit das Risiko für Wechselwirkungen.
In Pharmakovigilanz-Programmen wird die Sicherheit der Medikamenteneinnahme unter Alltagsbedingungen untersucht [13, 14, 17]. Im Falle beobachteter Nebenwirkungen ist die Messung der Arzneistoff-Konzentration im Blut höchst hilfreich, um die Ursachen zu klären.
Graduierte Empfehlungen für die Messung der Arzneistoff-Konzentration im Blut von Neuropsychopharmaka
Die Nützlichkeit von TDM hängt von der klinischen Situation und dem jeweiligen Arzneistoff ab. Bei Verdacht auf Non-Adhärenz oder ungenügender Adhärenz (Compliance) zur Medikation oder bei einer Intoxikation ist die Messung von Blutspiegeln generell für alle Arzneistoffe und jeden Patienten sinnvoll. Es ist jedoch immer noch umstritten, ob TDM in der klinischen Routine eingesetzt werden sollte. Basierend auf empirischen Daten wurden vier graduierte Empfehlungen für die Anwendung von TDM definiert, von „dringend empfohlen“ bis „potenziell nützlich“ (Kasten „Empfehlungen – Definitionen“).
Empfehlungen – Definitionen
Level 1: Dringend empfohlen
Evidenz: Die berichteten therapeutischen Referenzbereiche sind durch Studien validiert. Kontrollierte klinische Studien haben positive Effekte von TDM nachgewiesen. Es gibt Berichte über Unverträglichkeiten oder Vergiftungen bei hohen Arzneistoff-Konzentrationen im Blut.
Empfehlung: TDM ist für die Dosisfindung und für spezielle Indikationen dringend empfohlen. So ist TDM zum Beispiel für eine Behandlung mit Lithium oder Carbamazepin obligat.
Klinische Konsequenzen: Bei Arzneistoff-Konzentrationen im Blut innerhalb der berichteten therapeutischen Referenzbereiche besteht die höchste Wahrscheinlichkeit des Ansprechens auf eine Pharmakotherapie oder einer Remission. Bei subtherapeutischen Arzneistoff-Konzentrationen im Blut ist die Ansprechrate mit Placebo unter akuter Behandlung vergleichbar und es besteht ein erhöhtes Risiko für einen Rückfall unter chronischer Behandlung. Bei supratherapeutischen Arzneistoff-Konzentrationen im Blut besteht ein erhöhtes Risiko für Unverträglichkeiten oder einer Vergiftung.
Level 2: Empfohlen
Evidenz: Die berichteten therapeutischen Referenzbereiche wurden aus Untersuchungen gewonnen, bei denen Arzneistoff-Konzentrationen im Blut bei therapeutisch wirksamen Dosierungen und klinische Wirkungen korreliert wurden. Es gibt Berichte über verminderte Verträglichkeit oder unerwünschte Arzneimittelwirkungen bei supratherapeutischen Arzneistoff-Konzentrationen im Blut.
Empfehlung: TDM wird für die Dosisfindung und für spezielle Indikationen oder Problemlösungen empfohlen.
Klinische Konsequenzen: TDM erhöht die Wahrscheinlichkeit des Ansprechens auf eine Pharmakotherapie bei Therapieversagern. Bei subtherapeutischen Arzneistoff-Konzentrationen im Blut besteht ein erhöhtes Risiko für Therapieversagen. Bei supratherapeutischen Arzneistoff-Konzentrationen im Blut besteht ein erhöhtes Risiko für Unverträglichkeiten oder eine Vergiftung.
Level 3: Nützlich
Evidenz: Die berichteten therapeutischen Referenzbereiche wurden aus Arzneistoff-Konzentrationen im Blut unter zugelassenen Dosen errechnet. Ein Zusammenhang zwischen Arzneistoff-Konzentrationen und Therapieeffekten ist bisher nicht nachgewiesen. Es gibt nur retrospektive Analysen von TDM-Daten, einzelne Fallberichte oder nicht-systematische klinische Erfahrung.
Empfehlung: TDM ist nützlich für spezielle Indikationen oder bei spezifischen Problemen.
Klinische Konsequenzen: TDM kann angewendet werden, um zu kontrollieren, ob die Arzneistoff-Konzentrationen im Blut mit dem dosisbezogenen Referenzbereich übereinstimmen. Eine klinische Verbesserung könnte erreicht werden, indem die Arzneistoff-Konzentration bei Therapieversagern gesteigert wird, welche niedrige Arzneistoff-Konzentrationen aufweisen.
Level 4: Potenziell nützlich
Evidenz: Arzneistoff-Konzentrationen im Blut korrelieren aufgrund besonderer pharmakologischer Eigenschaften des Arzneistoffs nicht mit klinischen Effekten, beispielsweise bei irreversibler Blockade eines Enzyms, oder die Dosierung kann leicht anhand klinischer Symptome eingestellt werden, wie zum Beispiel Schlaf-Induktion durch Hypnotika.
Empfehlung: TDM wird nicht für die Dosisfindung empfohlen, kann aber für spezielle Indikationen oder besondere Probleme potenziell nützlich sein.
Klinische Konsequenzen: TDM sollte auf spezielle Indikationen beschränkt werden.
Nach Auswertung der Literatur wurde TDM für 19 der 154 untersuchten Neuropsychopharmaka „dringend empfohlen“ und für 39 „empfohlen“. Für 61 Neuropsychopharmaka gilt TDM als „nützlich“ und für 35 als „potenziell nützlich“ (Tab. 4). TDM wird für die meisten trizyklischen Antidepressiva „dringend empfohlen“. TDM reduziert das Risiko von Vergiftungen. Für SSRI besteht eine schwache aber signifikante Dosisabhängigkeit der klinischen Besserung, wohingegen die Verträglichkeit bei hohen Dosen abnimmt. Auch wenn die Akzeptanz von TDM in der klinischen Praxis begrenzt ist [1], nimmt die Evidenz für dessen Nutzen zu. Für Citalopram wurde gezeigt, dass es vorteilhaft ist, in der frühen Behandlungsphase ein TDM durchzuführen, das heißt eine Woche nach dem Beginn der medikamentösen Behandlung [29]. Eine andere Limitierung für die Etablierung von TDM für SSRI ist die schwache Methodik bei der Analyse von Korrelationen zwischen Arzneistoff-Konzentration im Blut und klinischen Effekten. Bei Anwendung einer adäquaten Methodik konnte bei einer Re-Analyse von Daten zu Paroxetin, mit denen zunächst kein Zusammenhang zwischen Konzentrationen und Therapieansprechen detektiert worden war, eine klare Korrelation gefunden werden, die fast identisch mit der In-vivo-Besetzung von Serotonin-Transportern war. Die Toxizität von SSRI ist in Vergleich zu den meisten vorher eingeführten Antidepressiva gering [2]. Evidenz für einen statistisch signifikanten Zusammenhang zwischen Arzneistoff-Konzentration und Therapieansprechen fehlt für die tetrazyklischen Antidepressiva Maprotilin, Mianserin und Mirtazapin und auch für Trazodon und Reboxetin sowie die MAO-Hemmer Moclobemid und Tranylcypromin.
TDM wird für die typischen Antipsychotika (erste Generation) Haloperidol, Perphenazin und Fluphenazin und für die atypischen Antipsychotika (zweite Generation) Amisulprid, Clozapin, Olanzapin und Risperidon dringend empfohlen (Tab. 4). Eine Überdosierung kann zu extrapyramidalen Nebenwirkungen führen. Im Falle von Clozapin existiert eine klare Korrelation zwischen Clozapin-Blutspiegel und der Häufigkeit von Krampfanfällen. Die Vermeidung einer Überdosierung von typischen Antipsychotika durch TDM ist für die Mehrzahl der Patienten eher eine Frage der Lebensqualität als der Arzneimittelsicherheit. TDM von Antipsychotika ist auch nützlich, wenn die Medikation von der oralen zur Depot-Form, oder umgekehrt, umgestellt wird.
Depot-Formulierungen von zahlreichen Antipsychotika der ersten und zweiten Generation (Risperidon, Paliperidon, Olanzapin, Aripiprazol) werden häufig empfohlen, um Non-Adhärenz bei Patienten mit Schizophrenie zu begegnen. Es wurde angenommen, dass stabile Konzentrationen von Depot-Antipsychotika im Blut mit einer überlegenen Verträglichkeit und Wirksamkeit assoziiert sind. Unterschiede in der Effektivität (d.h. Rückfall-Prävention) oder der Verträglichkeit von Depot- und oralen Antipsychotika konnten jedoch nicht klar bewiesen werden und scheinen mehr von dem spezifischen Arzneistoff und der Dosis oder der Arzneistoff-Konzentration im Blut abhängig zu sein. Spitzen- zu Talspiegel-Fluktuationen im Steady-State (siehe Abb. 2) sind bei Depot-Formulierungen nicht generell geringer (abhängig von tmax und t1/2), und nicht alle Studien fanden eine positive Korrelation zwischen großen Fluktuationen der Arzneistoff-Konzentrationen im Blut und einer gesteigerten Rate an unerwünschten Arzneimittelwirkungen. Für die verfügbaren Depot-Antipsychotika existieren kaum pharmakokinetische Studien. Empfohlene therapeutische Bereiche von Depot- und anderen Formulierungen sind fast identisch.
Im Hinblick auf die stimmungsstabilisierenden und/oder antimanischen Medikamente Lithium, Valproinsäure und Carbamazepin sind die therapeutischen Referenzbereiche und die toxischen Konzentrationen validiert. Deshalb wird TDM für diese Medikamente dringend empfohlen (Tab. 4). Für Lithium gilt TDM als Standard guter klinischer Praxis. Bei langfristigem Einsatz werden Blutkonzentrationen von 0,5 bis 0,8 mmol/l empfohlen. Für eine akute Behandlung mit Lithium kann es gerechtfertigt sein, die Konzentration auf bis zu 1,2 mmol/l zu erhöhen.
Arzneimittel mit antidementiven Eigenschaften sind Donepezil, Rivastigmin, Galantamin und Memantin. TDM wird nur selten bei der medikamentösen Behandlung von Demenz eingesetzt, obwohl es Hinweise für seine Nützlichkeit gibt. Für Donepezil wurde gezeigt, dass bei Patienten mit Konzentrationen über 50 ng/ml im Blut das klinische Ansprechen deutlich besser war als bei Patienten mit niedrigeren Wirkstoffkonzentrationen.
Die meisten Anxiolytika und Hypnotika gehören zur Klasse der Benzodiazepine. Für Alprazolam kann TDM hilfreich sein, um Panikattacken zu unterdrücken. Anxiolytische und hypnotische Effekte treten meist rasch ein. Die Behandlung kann daher eher durch unmittelbaren klinischen Eindruck als durch TDM gesteuert werden. Messungen können jedoch informativ sein, um chronischen Gebrauch der Arzneistoffe zu identifizieren. Im Falle eines mangelnden Therapieeffekts unter gewöhnlichen Dosen kann TDM klären, ob das Therapieversagen durch einen Arzneimittelmissbrauch, der zu einer Toleranzentwicklung geführt hat, begründet ist oder durch pharmakokinetische Besonderheiten. Aufgrund adaptiver Veränderungen bei chronischen Anwendern korrelieren die Blut-Konzentrationen der Benzodiazepine nur schwach mit der Fahrtüchtigkeit.
TDM ist aufgrund der Arzneimittelsicherheit hoch indiziert für die Opioid-Agonisten racemisches Methadon, R-(–)-Methadon (Levomethadon), Buprenorphin und Morphin. Es muss berücksichtigt werden, dass, ähnlich zu den Benzodiazepinen, aufgrund von verschiedenen Toleranzleveln die optimale Arzneistoff-Konzentration deutlich von Patient zu Patient variieren kann. Andererseits können Opioid-abhängige Patienten aufgrund des starken Verlangens (Craving) für Arzneistoffe nach höheren Dosen fragen, als sie tolerieren können, was fatale Konsequenzen aufgrund toxischer Arzneistoff-Konzentrationen haben kann. Für „Anti-Craving“-Arzneistoffe wie Acamprosat oder Naltrexon oder den Einsatz von Alkohol-aversivem Disulfiram zur Behandlung von alkoholbezogenen Störungen wird TDM empfohlen, um die moderate Wirksamkeit der Substanzen zu steigern, ebenso im Falle von Naltrexon für die Abstinenzbehandlung bei Opioid-Abhängigkeit [6]. TDM von Arzneistoffen zur Behandlung substanzbezogener Störungen sollte die zu erwartenden Arzneistoff-Konzentrationen berücksichtigen (Tab. 5), um Adhärenz-Probleme, die Toleranz zur Medikation oder pharmakokinetische Besonderheiten zu klären.
Für Antikonvulsiva ist TDM nicht nur für die alten, relativ toxischen Substanzen [30], sondern auch für die neuen Antikonvulsiva gut etabliert [25].
Für Antiparkinsonika ist TDM bisher nicht etabliert. Valide Referenzbereiche sind kaum vorhanden. Bezüglich Levodopa wird eine moderate Korrelation zwischen Blutkonzentration und kurzfristiger klinischer Reaktion beschrieben. Trotzdem wurden die pharmakokinetischen Eigenschaften dieser neurologischen Arzneistoffe in dieser Leitlinie eingeschlossen (Tab. 1 bis 6), da Antiparkinsonika konzentrationsabhängige sedierende Eigenschaften aufweisen. TDM kann eine Überdosis vermeiden.
Praktische Anwendung von TDM in Psychiatrie und Neurologie
TDM-Anforderung für die Quantifizierung von Arzneistoff-Konzentrationen im Blut
Voraussetzung für einen effektiven TDM-Service ist die Verfügbarkeit von geeigneten, analytischen Methoden, die Ergebnisse innerhalb einer vernünftigen Zeit produzieren, das heißt innerhalb von 48 Stunden von der Ankunft der Blutprobe im Labor bis zur Versendung der Ergebnisse. Der TDM-Service sollte eine Interpretation der Befunde durch einen Experten inkludieren, der über pharmakokinetisches und pharmakotherapeutisches Wissen verfügt. Wie in Abbildung 4 dargestellt, beginnt der TDM-Prozess mit der Anforderung und endet mit der Entscheidung, wie die Psychopharmakotherapie des einzelnen Patienten durch den behandelnden Arzt angepasst werden soll.
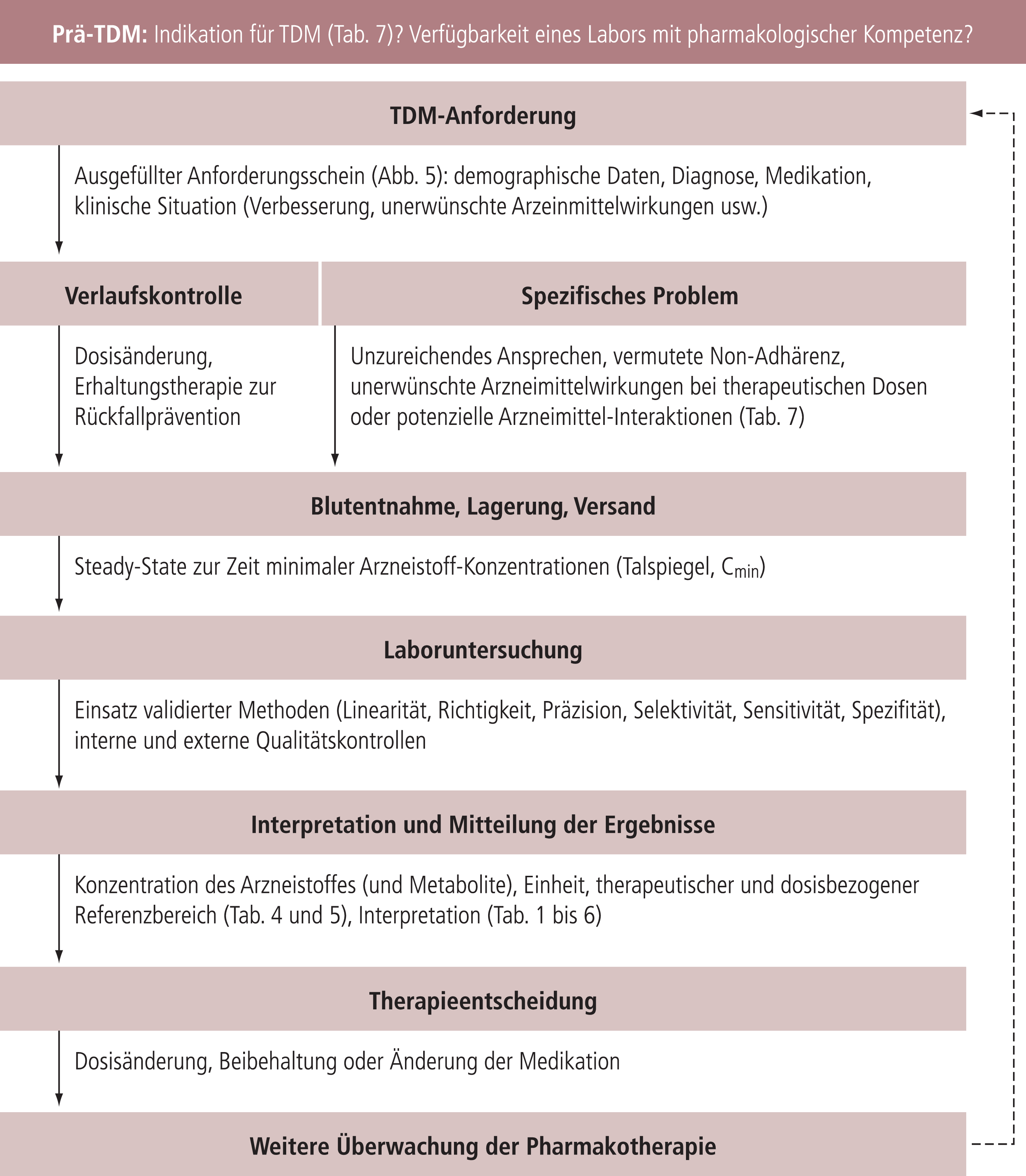
Abb. 4. Schematische Darstellung der Anwendung von TDM für die Neuropsychopharmakotherapie. Routine-TDM wird primär bei Arzneistoffen mit einem engen therapeutischen Fenster und einem gut definierten therapeutischen Referenzbereich durchgeführt. Trotzdem kann TDM hilfreich für jeden neurologischen oder psychiatrischen Arzneistoff sein, wenn spezielle therapeutische Probleme auftreten.
Wie bereits erwähnt, sollte eine Blutspiegelmessung nur angefordert werden, wenn es klare Hinweise gibt, dass das Ergebnis eine Antwort auf eine spezielle Fragestellung liefern kann. Typische Indikationen sind in Tabelle 7 aufgelistet. Zur Problemlösung reicht eine einzelne Messung oft nicht aus. Zum Beispiel kann eine Reihe von Messungen in angemessenen Zeitabständen erforderlich sein, um zu klären, ob ein niedriger Blutspiegel entweder durch schlechte Adhärenz, reduzierte Bioverfügbarkeit oder ungewöhnlich rasche Verstoffwechselung verursacht wird.
Eine TDM-Anforderung erfordert ein ausgefülltes Antragsformular (Abb. 5), das für eine effektive Laboranalyse und eine angemessene Interpretation der Ergebnisse unverzichtbar ist. Das Formular sollte den Patienten per Namen oder Code identifizieren und folgende Angaben enthalten: demographische Daten, Diagnose, Medikation, Grund für die Anfrage, den Wirkstoffnamen und möglichst auch den Handelsnamen des Medikaments sowie die Dosis, die Galenik, die Zeit der letzten Änderung der Dosis, den Zeitpunkt der Medikamenten-Einnahme und den Zeitpunkt der Blutentnahme. Ein kurzer Kommentar zur klinischen Situation des Patienten sollte ebenfalls angegeben werden. Wie in Abbildung 5 angezeigt, empfehlen wir die Verwendung von Ratingskalen, zum Beispiel die Clinical Global Impression-(CGI-)Skala, um die Schwere der Erkrankung (CGI-S) und die therapeutische Verbesserung oder Verschlechterung zu erfassen (CGI-I). Um auch die Häufigkeit und Schwere von Nebenwirkungen zu bewerten, hat sich die Verwendung der UKU-Skala bewährt. Dokumentierte Rückmeldungen von Fragebögen zeigen an, dass die behandelnden Kliniker häufig jedoch nicht so viele Informationen auf dem Bogen vermerken möchten. Deshalb sind die ausgefüllten Informationen auf dem Anforderungsschein oft unvollständig. Als Alternative kann interessierten Ärzten eine telefonische Beratung angeboten werden.
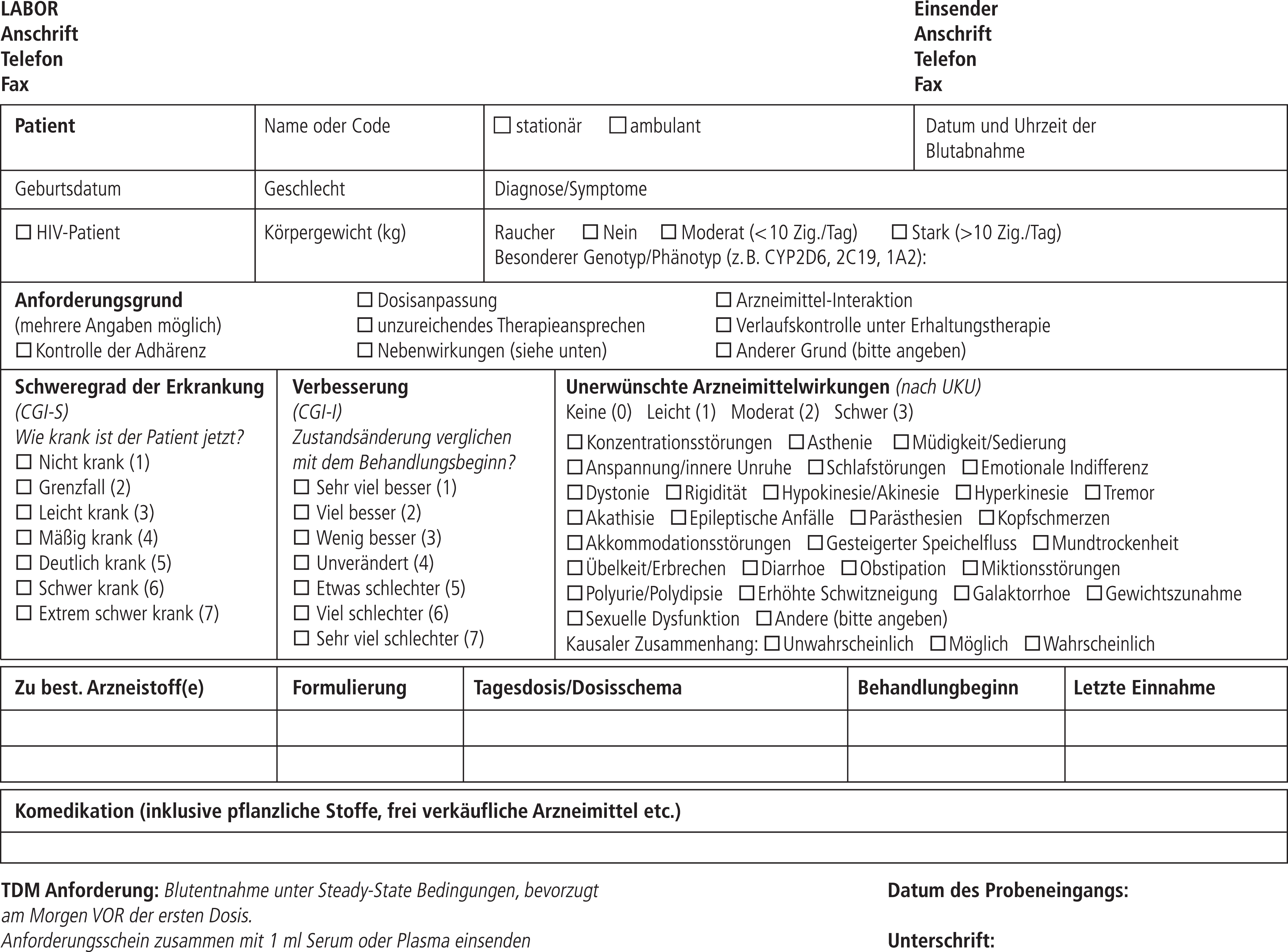
Abb. 5. Beispielformular zur Anforderung einer Blutspiegelmessung von Neuropsychopharmaka
Wenn eine Interpretation der Ergebnisse von dem Labor gewünscht ist, ist es notwendig, den Anforderungsbogen vollständig und adäquat auszufüllen. Die Online-Anforderung von TDM hat Vorteile. Sie ist kostengünstig und leitet den Arzt in einer komfortablen Art und Weise an, die für eine Interpretation notwendigen Informationen anzugeben.
Probensammlung
Blutentnahme
Üblicherweise werden für TDM die Arzneistoff-Konzentrationen in Plasma- oder Serum-Proben gemessen. Es existiert kein Konsens, ob Plasma oder Serum bevorzugt werden sollte. Experimentelle Daten zu Unterschieden in den Wirkstoffkonzentrationen zwischen Plasma und Serum fehlen weitgehend. Die wenigen verfügbaren Vergleichsuntersuchungen zeigen, dass aus Serum oder Plasma gewonnene Werte gleichermaßen verwendet werden können. Für die meisten Labore sollen die Proberöhrchen kein EDTA, Citrat, Heparin oder andere Zusatzstoffe enthalten. Eine Menge von einem Milliliter Plasma oder Serum ist für die meisten Labore ausreichend. Die in diesen Leitlinien angegebenen Konzentrationen neuropsychiatrischer Medikamente beziehen sich in Übereinstimmung mit der Literatur jeweils auf die Gesamtfraktion eines Arzneistoffs. Es gibt keine experimentellen Belege für die Hypothese, dass die Messung der freien Arzneistoff-Konzentration im Blut vorteilhaft wäre. Für Imipramin hat sich gezeigt, dass der Arzneistoff rasch und fast vollständig während einer einzigen Passage des Bluts durch das Gehirn ins Hirngewebe extrahiert wird. Die Extraktion wird dabei nicht signifikant durch Albumin, Lipoproteine oder Erythrozyten beeinflusst. Für Nortriptylin fand man keine statistisch signifikante Beziehung zwischen freier Wirkstofffraktion und klinischem Ansprechen. Deshalb scheint es zumindest für psychiatrische Arzneistoffe wahrscheinlich, dass das klinische Ansprechen von der gesamten Arzneistoff-Fraktion abhängt. Die Analyse von Neuropsychopharmaka in anderen Probenmaterialien wie Urin, Liquor, Tränen, Haare oder Muttermilch sind bislang nicht für TDM etabliert. Es stehen derzeit keine validierten Daten zu therapeutischen Konzentrationen zur Verfügung.
Die Probengewinnung via getrockneter Blutflecken (Dried-blood-spot-Methode) kann eine Alternative zur gängigen venösen Blutentnahme sein. Die Methode hat viele Vorteile: Sie ist minimalinvasiv, benötigt nur ein geringes Blutvolumen, der Transport und die Lagerung der Proben sind leicht und die Analyten besitzen eine gute Stabilität. Die hohe Sensitivität von modernen, analytischen Methoden wie Flüssig-Chromatographie-Tandem-Massenspektrometrie (LC-MS/MS) oder Ultra-high-Performance-Flüssig-Chromatographie-Tandem-Massenspektrometrie (UPLC-MS/MS) erlaubt die Verwendung von getrockneten Blutproben für TDM. Hierfür müssen jedoch einige Punkte berücksichtigt werden: Die Korrelation der Ergebnisse von getrockneten Blutproben mit Plasma- oder Serumkonzentrationen, der Einfluss des Hämatokrits, der Einfluss des gesammelten Blutvolumens und verschiedene Arten von Filterpapier. Volumendefinierende Techniken und automatisierte Techniken wie die Online-Desorption, die Papier-Spray-Analyse und vollautomatisierte Extraktion der getrockneten Blutproben sind bereits verfügbar. Trotzdem bedarf diese Methode einer weiteren klinischen Validierung, um eine geeignete und kosteneffektive Alternative in der klinischen Routine eines TDM-Labors zu werden.
In Hinblick auf den Zeitpunkt der Blutentnahme muss berücksichtigt werden, dass die TDM-geleitete Neuropsychopharmakotherapie zumeist auf minimalen Arzneistoff-Konzentrationen (Cmin) unter Steady-State-Bedingungen basiert. Steady-State wird unter konstanter Dosis nach mindestens 4 bis 6 Eliminationshalbwertszeiten erreicht (siehe Tab. 4), Cmin zum Zeitpunkt der Arzneistoffeinnahme nach dem längsten Dosisintervall. Aufgrund der Praktikabilität werden die Blutproben für die Bestimmung von Cmin in der Regel am Morgen vor der ersten Tagesdosis entnommen. Für die meisten Neuropsychopharmaka ist dies die Zeit minimaler Arzneistoff-Konzentrationen (tmin). Ein häufiges Problem ist jedoch die Blutentnahme zu verschiedenen Zeitpunkten während des Dosisintervalls. Dies führt zu Konzentrationen, die fehlinterpretiert werden könnten, wenn in der Realität Talspiegel niedriger oder höher sind. Für Antibiotika wurde berichtet, dass bis zu 55% der unangemessenen Konzentrationen auf einem ungeeigneten Zeitpunkt der Blutentnahme beruhten.
Für Antiparkinsonika und Arzneistoffe zur Behandlung der Aufmerksamkeits-Defizit-Hyperaktivität-Störung wie Methylphenidat wird das Blut bei tmax entnommen, zum Zeitpunkt der maximalen Arzneistoff-Konzentration (Cmax). Die meisten dieser Arzneistoffe besitzen eine kurze Eliminationshalbwertszeit und klinische Effekte korrelieren mit Cmax.
Blutentnahme unter Behandlung mit Depot-Formulierungen
Bei Patienten, die mit einer Depot-Formulierung eines Antipsychotikums behandelt werden, sollte das Blut kurz vor der nächsten Injektion entnommen werden. TDM kann natürlich zu jeder Zeit nach Arzneistoffeinnahme ausgeführt werden, falls unerwartete unerwünschte Arzneimittelwirkungen beobachtet werden. Es ist nicht notwendig, die Talspiegel zu messen, aber das Dosisschema sollte für die Interpretation angegeben sein.
Depot-Formulierungen von Antipsychotika wie von Haloperidol, Risperidon oder Aripiprazol sind durch eine langsame Absorption nach intramuskulärer Gabe charakterisiert. Maximale Konzentrationen von Depot-Antipsychotika der ersten Generation im Blut sind nach 1 bis 14 Tagen nach Injektion erreicht, die apparente Eliminationshalbwertszeit beträgt etwa zwei bis drei Wochen. Paliperidonpalmitat besitzt ähnliche Eigenschaften, mit einer Eliminationshalbwertszeit zwischen 25 und 49 Tagen. Für Risperidon-Mikrosphären beträgt die durchschnittliche Zeit bis zu Spitzenkonzentrationen vier Wochen und die Eliminationshalbwertszeit etwa vier bis sechs Tage. Die Olanzapinpamoat-Depot-Formulierung setzt Olanzapin langsam von der Injektionsstelle in das Muskelgewebe frei. Es löst sich jedoch schnell auf, wenn es mit Blut oder Plasma in Kontakt kommt. Letzteres führt zu hohen Konzentrationen im Blut und kann zu einer beträchtlichen Sedierung und Delirium führen, das sogenannte Post-Injektions-Syndrom. Die Absorption von Aripiprazol nach Gabe der Depot-Formulierung (einmal monatlich) ist aufgrund der geringen Löslichkeit langsam mit einer durchschnittlichen Absorptionshalbwertszeit von vier Wochen. Maximale Arzneistoff-Konzentrationen im Blut sind nach fünf bis sieben Tagen nach Injektion erreicht; die durchschnittliche terminale Eliminationshalbwertszeit nach 400 oder 300 mg Aripiprazol monatlich beträgt 47 bzw. 30 Tage.
Für andere Arzneistoffe, die nach oraler Einnahme verzögert freigesetzt werden (extended release), wie Quetiapin, muss für die korrekte Interpretation der Messwerte dem Zeitpunkt der Arzneistoffeinnahme besondere Aufmerksamkeit geschenkt werden (siehe Tab. 4). Bei diesen Formulierungen ist die Zeit von maximalen Arzneistoff-Konzentrationen im Blut verzögert, aber die Eliminationshalbwertszeit bleibt unverändert.
Speichel für TDM
Speichel bietet den Vorteil einer nichtinvasiven Probenahme. Die Methode wurde für die Optimierung der Behandlung von einigen antiepileptischen Arzneistoffen angewandt, in erster Linie zur Überprüfung der Adhärenz. Es wurde lange angenommen, dass die Arzneistoff-Konzentration im Speichel die freie Fraktion (d.h. nicht an Proteine gebunden) reflektiert, die im Blut zirkuliert. Diese Fraktion stellt für die meisten Psychopharmaka nur 10% oder weniger der gesamten Konzentration dar. Nachweisprobleme waren deshalb in der Vergangenheit ein Hauptproblem, wenn Speichel anstatt Blutplasma oder Serum verwendet wurde. Inzwischen gibt es verbesserte Methoden, um Speichelproben mit ausreichender Präzision und Genauigkeit zu analysieren. Bei Anwendung solcher Techniken wurde herausgefunden, dass sich das Verhältnis der Konzentration in Blut und Speichel von Arzneistoff zu Arzneistoff stark unterscheidet. Die Annahme, dass Konzentrationen im Speichel die freie Fraktion des Arzneistoffs im Blut widerspiegeln, wurde nicht bestätigt. Die Messung der Arzneistoff-Konzentration im Blut kann also nicht ohne Weiteres durch die Messung der Konzentration im Speichel ersetzt werden. Eine scheinbare, positive Korrelation existiert zwischen der Konzentration von Monohydroxyoxcarbazepin (MHD, dem Hauptmetaboliten von Oxcarbazepin) im Blutplasma und Speichel. Für Carbamazepin, Phenytoin und Phenobarbital war die Korrelation schwach, aber ebenfalls signifikant. Für Valproinsäure war die Korrelation jedoch nicht signifikant. Auch für Methadon wurde gefunden, dass Messungen im Speichel Messungen im Blut für das Monitoring nicht ersetzen können.
Für Amitriptylin und Nortriptylin konnte kein signifikanter Zusammenhang zwischen Konzentrationen im Speichel und Plasma gefunden werden. Viele Neuropsychopharmaka sind Basen, mit einem pKs-Wert >9. Die pKs-Werte sollten unter 5,5 liegen, um eine gute Vorhersage von Plasmakonzentrationen aus Speichelkonzentrationen zu treffen, da die Arzneistoffe in einer unionisierten Form vorliegen müssen. Der pH des Speichels steigt, wenn die Sekretion stimuliert wird. Für Methylphenidat wurde eine inverse Korrelation für das Verhältnis der Arzneistoff-Konzentrationen im Speichel zu Serum und dem pH-Wert der Speichelproben gefunden.
Eine Standardisierung und Optimierung der Probenahme ist erforderlich. In jedem Fall müssen mehr Daten für die Messung von Arzneistoff-Konzentrationen in Speichel als Matrixsubstanz gewonnen werden.
Lagerung und Versand von Blutproben
Wenn die Blutproben eingefroren gelagert oder versandt werden sollen, ist vor dem Einfrieren das Serum bzw. Plasma herzustellen und dieses einzufrieren, da es nicht möglich ist, Serum oder Plasma aus gefrorenem Blut zu gewinnen. Mit wenigen Ausnahmen können die Serum- bzw. Plasma-Proben im Dunkeln (bei 4°C) für mindestens 24 Stunden gelagert werden. Die meisten Proben können in nicht eingefrorenem Zustand versandt werden. Ausnahmen sind licht- und/oder sauerstoffempfindliche Substanzen. Für die Bestimmung von Bupropion oder Methylphenidat müssen die Proben sofort nach der Blutentnahme zentrifugiert und das Plasma oder Serum eingefroren oder extrahiert und stabilisiert werden (Tab. 4). Proben für Olanzapin-Bestimmungen müssen tiefgefroren (–20°C) gelagert werden, wenn sie nicht innerhalb von 72 Stunden analysiert werden. Das Labor sollte auf seiner Webseite oder auf dem Anforderungsschein angeben, wie Proben zu sammeln (Blutvolumen, Kennzeichnung der Proben), aufzubewahren und zu versenden sind.
Laboruntersuchung
Für die erfolgreiche Durchführung von TDM sind selektive und empfindliche Methoden für eine quantitative Bestimmung der Arzneistoffe und ihrer Metabolite essenziell. Die Methoden müssen validiert sein, das heißt, es muss nachgewiesen werden, dass eine verwendete Methode für eine quantitative Messung eines Analyten in einer gegebenen biologischen Matrix für den vorgesehenen Verwendungszweck zuverlässige und reproduzierbare Ergebnisse erzielt. Fundamentale Parameter für die Validierung sind (1) Genauigkeit, (2) Präzision, (3) Selektivität, (4) Empfindlichkeit, (5) Reproduzierbarkeit und (6) Stabilität. Die Validierung beinhaltet die Dokumentation, dass die Durchführungscharakteristika einer Methode geeignet und zuverlässig für die analytische Messung sind. Die Akzeptanz von Analysedaten entspricht direkt den Kriterien, die für die Validierung der Methode verwendet wurden (z.B. EMA Guidelines on validation of bioanalytical methods; EMEA/CHMP/EWP/192217/2009).
Für den Nachweis von Neuropsychopharmaka sind chromatographische Techniken (bevorzugt Hochleistungsflüssigkeitschromatographie, HPLC) in Kombination mit geeigneten Detektionsmethoden vorteilhaft. Sie sind ausreichend präzise, genau und robust und können auf die Analyse von fast jedem Neuropsychopharmakon angepasst werden. Ein Nachteil ist die Notwendigkeit einer Probenvorbereitung vor der chromatographischen Trennung und ein begrenzter Probendurchsatz. Der Durchsatz kann durch automatisierte Probenvorbereitung vor der HPLC erhöht werden. Einige Labors haben HPLC mit Säulenschaltung eingeführt, was die direkte Injektion von Plasma oder Serum in das HPLC-System erlaubt. Solche Verfahren sind für eine Reihe von Antidepressiva und Antipsychotika verfügbar. Eine chromatographische Hochdurchsatz-Methode ist Flüssig-Chromatographie (LC), gekoppelt mit Massenspektrometrie (LC-MS), vor allem gekoppelt mit Tandem-MS (LC-MS/MS). LC-MS/MS ist hochsensitiv und selektiv, insbesondere bei Verwendung der ultrahochauflösenden Flüssigkeitschromatographie (UPLC). Zudem kann diese Technik mit minimaler Probenvorbereitung wie Proteinfällung und Verdünnung angewendet werden. Viele Arzneistoffe können gleichzeitig analysiert werden. Ein hervorragendes Beispiel ist die LC-MS/MS-Methode von Kirchherr und Kühn-Felten, die für über 50 psychoaktive Medikamente validiert wurde. Hauptnachteil von LC-MS/MS-Methoden sind hohe Anschaffungskosten der Geräte und die Notwendigkeit von geschultem Personal. Darüber hinaus kann die Quantifizierung aufgrund von Matrixeffekten und Ionen-Suppression problematisch sein. Diese Effekte können durch gute chromatographische Trennung der Matrix und des Analyten minimiert werden, sowie durch den Gebrauch von stabilen Isotopen-markierten Standards für die interne Kalibrierung, bevorzugt deuterierte Analoga. Während der letzten Jahre wurden LC-MS/MS-Methoden zunehmend angewendet [31, 32]. Ihr großer Vorteil ist die Flexibilität, insbesondere in Vergleich zu immunologischen Methoden. Der Nachteil der hohen Kosten trifft nur noch bedingt zu. Die Kosten wurden allmählich auf einen akzeptablen Preis reduziert. Deshalb ist LC-MS/MS heutzutage in vielen spezialisierten Laboren die bevorzugte analytische Methode für TDM von Neuropsychopharmaka. HPLC mit UV- oder Fluoreszenzdetektion ist jedoch weiterhin eine zuverlässige Methode in vielen Laboren mit geringem bis mittleren Durchsatz, insbesondere aufgrund der Kosteneffektivität und der Robustheit.
Bei Verdacht auf Vergiftungen sollte eine Methode mit Analysezeit von ein bis zwei Stunden verfügbar sein. Zu diesem Zweck sind automatisierte Methoden von Vorteil. Die Anwendung von LC-MS/MS ist in dieser speziellen Situation aufgrund der hohen Selektivität der Massenspektrometrie bei der Identifizierung vorteilhaft.
Die Bestimmung der Enantiomere von chiralen Verbindungen erfordert entweder eine stereoselektive Derivatisierung der Arzneistoffe vor ihrer Quantifizierung oder die Trennung mittels chiraler Chromatographie über spezielle GC- oder HPLC-Säulen. Für die Detektion ist die Tandem-Massenspektrometrie die bevorzugte Methode. Beispielsweise kann der Nachweis der Enantiomere von Methadon, wenn er mit einem klassischen Nachweisverfahren wie Fluoreszenz- oder UV-Detektion geführt wird, durch Komedikation oder Beigebrauch von Drogen gestört werden. Diese Probleme können durch die Verwendung eines Massendetektors umgangen werden, vorzugsweise eines Tandem-Massenspektrometers.
Innerhalb des therapeutischen Referenzbereichs sollte die Präzision innerhalb eines Tages und zwischen verschiedenen Tagen nicht mehr als 15% betragen (Variationskoeffizient) und auch die Richtigkeit sollte nicht mehr als 15% vom Nominalwert abweichen.
Um die Qualität und Zuverlässigkeit der TDM-Methoden sicherzustellen, sind interne und externe Qualitätskontrollen obligatorisch. Die Proben müssen geeignete interne Standards enthalten und jede Probenserie muss interne Kontrollproben beinhalten. Wenn Standards nicht kommerziell erhältlich sind, sollten sie nicht vom selben Personal eingewogen und hergestellt werden, das für die Durchführung der Tests zuständig ist. Kommerzielle Proben für die Qualitätskontrolle sind heutzutage für eine breite Spanne von Neuropsychopharmaka zunehmend verfügbar. Die Weitergabe der TDM-Ergebnisse setzt voraus, dass die Ergebnisse der Qualitätskontrollen innerhalb des erwarteten Bereichs liegen. Wenn Qualitätskontrollen außerhalb des erwarteten Bereichs liegen, muss der Grund für den Ausreißer geklärt und dokumentiert werden.
Das Labor hat sich an einem externen Ringversuch zu beteiligen, obwohl dies nicht in allen Ländern gesetzlich vorgeschrieben ist. Für Neuropsychopharmaka wurde das erste externe Qualitätsprogramm von Cardiff Bioanalytical Services Ltd. im Jahre 1972 eingeführt. Der Service wurde von weiteren Anbietern externer Qualitätskontrollen übernommen, beispielsweise von LGC (www.lgcstandards.com) oder Instand e.V. (www.instand-ev.de). Darüber hinaus werden Ringversuche und externe Referenzmaterialien auch von der Deutschen Gesellschaft für Toxikologie und forensische Chemie angeboten (www.gtfch.org).
Berechnung von Talspiegel-Steady-State-Konzentrationen
Beim Vergleich einer gemessenen Arzneistoff-Konzentration im Blut mit der zu erwartenden Steady-State-Konzentration wird angenommen, dass die Blutentnahme zurzeit minimaler Arzneistoff-Konzentrationen (tmin) erfolgte. Für die Messung von Talspiegeln (Cmin) im Steady-State sollte das Blut nach mindestens vier Eliminationshalbwertszeiten nach Beginn der medikamentösen Therapie oder nach einer Dosisänderung und während der terminalen Beta-Eliminationsphase gesammelt werden. Die meisten Neuropsychopharmaka besitzen Eliminationshalbwertszeiten zwischen 12 und 36 Stunden (Tab. 4). Ausnahmen sind Quetiapin, Trazodon oder Venlafaxin, mit Eliminationshalbwertszeiten von etwa sechs Stunden. Fluoxetin, Donepezil und Aripiprazol besitzen längere Eliminationshalbwertszeiten. In der klinischen Praxis ist für die meisten Neuropsychopharmaka die angemessene Zeit für eine Blutentnahme eine Woche nach stabiler Dosiseinstellung und kurz vor der nächsten Einnahme der Morgendosis, was üblicherweise 12 bis 16 Stunden (oder 24 Stunden, wenn der Arzneistoff einmal täglich am Morgen eingenommen wird) nach der letzten Medikation ist. Wenn, aus logistischen Gründen, die Blutentnahme nur zu einem späteren Zeitpunkt als tmin stattfinden kann, sollte der Patient zuvor den Arzneistoff noch nicht einnehmen. Im ambulanten Bereich ist es für die Interpretation der Ergebnisse wichtig, die exakte Zeit der letzten Einnahme anzugeben. Dann ist es möglich, die zu erwartenden Talspiegel zu berechnen. Die Berechnung kann mit folgender Gleichung erfolgen (5):
Cmin=Ct × e–ke ∙ (tmin–t) Gleichung (5)
Ct stellt die Arzneistoff-Konzentration dar, die zum Zeitpunkt t gemessen wurde, tmin ist die Zeit von Cmin und ke ist die konstante Eliminationsrate (ke=ln2/t1/2).
Beispiel: Amisulprid, das eine durchschnittliche Eliminationshalbwertszeit von 16 Stunden besitzt (siehe Tab. 5, ke=0,0433 h–1), wurde einmal täglich verabreicht (um 08:00 Uhr); am Tag der Blutentnahme hat der Patient seine Medikation noch nicht eingenommen, da er diese erst nach der Blutentnahme für TDM einnehmen soll. Das Blut wurde um 11:00 Uhr morgens entnommen. Die gemessene Arzneistoff-Konzentration (Ct) war 351 ng/ml, daraus ergibt sich für Cmin zum Zeitpunkt 24 Uhr (=tmin):
351× e–0,0433 (24–27)=399 ng/ml
Gleichung (5) kann auch für die Schätzung von Cmin angewendet werden, wenn die Blutentnahme in der Postabsorptionsphase vor Erreichen von tmin erfolgte.
Beispiel: Lithium, das eine Eliminationshalbwertszeit von 24 Stunden (siehe Tab. 5) besitzt, wurde in einer Einmaldosis abends um 20:00 Uhr gegeben. Die Blutentnahme fand um 08:00 Uhr morgens statt (t=12 Stunden) und die gemessene Arzneistoff-Konzentration war 1,0 mmol/l; dann sollte Cmin zum Zeitpunkt 24 Uhr (=tmin) betragen:
1,0×e–ke (24–12)=0,71 mmol/l
Interpretation und Mitteilung der Ergebnisse
Die Konzentrationen der Neuropsychopharmaka und ihrer aktiven Metaboliten, die zu dem therapeutischen Effekt führen, sollten zusammen mit dem jeweiligen Referenzbereich (Tab. 4), in Massen- oder Mol-Einheiten, mitgeteilt werden. Wir empfehlen den Einsatz von Masse-Einheiten, um die gemessene Konzentration auf die ebenfalls in Masse-Einheiten angegebene Dosierung beziehen zu können. Die Laboratorien unterscheiden sich in der Darstellung ihrer Ergebnisse. Daher sollte der behandelnde Arzt auf die in der Ergebnismitteilung verwendeten Einheiten achten (z.B. ng/ml, µg/l, µmol/l oder nmol/l), in welchen die Analyseergebnisse präsentiert werden. Dies gilt insbesondere für Vergleiche von Befunden aus verschiedenen Laboratorien oder mit den in der Literatur publizierten Empfehlungen. Um molare Einheiten in Masseeinheiten umzuwandeln und umgekehrt, sind in Tabelle 4 die Umrechnungsfaktoren angegeben.
Bei Arzneistoff-Konzentrationen unterhalb der Bestimmungsgrenze (d.h. unter der niedrigsten Konzentration der Standardkurve, die mit mindestens 80 bis 120% Genauigkeit and 20% Präzision gemessen werden kann) sollte diese Grenze angegeben werden.
Die Ergebnisse sollten für die Entscheidungsfindung innerhalb eines klinisch sinnvollen Zeitfensters mitgeteilt werden. Für den TDM-Service sind 24 Stunden wünschenswert, eine Bearbeitungszeit von 48 Stunden ist in den meisten Fällen jedoch ausreichend. Bei Verdacht auf Vergiftungen muss das Ergebnis innerhalb weniger Stunden mitgeteilt werden. Um eine rasche Reaktion bei Patienten mit einem hohen Risiko für toxische Reaktionen oder verminderter Verträglichkeit zu ermöglichen, ist es wichtig, den behandelnden Arzt unmittelbar zu informieren (z.B. telefonisch), wenn Arzneistoff-Konzentrationen oberhalb der „Warnschwelle des Labors“ gemessen wurden (Tab. 4).
Wir empfehlen ausdrücklich, jedem Befundbericht einer Arzneistoff-Konzentration eine Interpretation des Befunds beizufügen. Die Interpretation der Arzneistoff-Konzentration durch einen Experten und die angemessene Umsetzung der Informationen sind unerlässlich, um den vollen klinischen Nutzen von TDM zu erzielen [3, 16, 18]. Die dem Ergebnisbericht beigefügten Kommentare und Empfehlungen zur Dosierung müssen auf der besten verfügbaren Evidenz beruhen. Um eventuelle Dosiskorrekturen zu berechnen oder Wechselwirkungen mit anderen Arzneimitteln zu analysieren, kann Expertenwissen notwendig sein. Für den Kliniker ist es daher vorteilhaft, ein Labor zu wählen, das diesen Service anbietet. Ansonsten hat der behandelnde Arzt, ein klinischer Pharmakologe oder ein ausgebildeter Spezialist der Klinik die Aufgabe, die Ergebnisse zu interpretieren. Der Zugriff auf fachliche Beratung ist auch nötig, wenn aufgrund des TDM-Ergebnisses eine Genotypisierung ratsam ist.
Manchmal ist es vorgeschrieben, einen klinischen Pharmakologen einzubeziehen. In der Schweiz kann ein Psychiater eine CYP-Genotypisierung verschreiben, aber diese wird von den Versicherungen nur erstattet, wenn der Test von einem Arzt verschrieben wurde, der in klinischer Pharmakologie spezialisiert ist.
Diagnose und Medikamentendosis sind wichtige Informationen für die Interpretation. So ist es möglich zu prüfen, ob ein Ergebnis plausibel ist oder nicht. Darüber hinaus muss kontrolliert werden, ob die Blutproben unter den nach Leitlinien empfohlenen Bedingungen gesammelt wurden. Dies ist vor allem bei ambulanten Patienten bedeutsam, wenn die Blutkonzentration unerwartet hoch ist. Wenn das Medikament wenige Stunden vor der Blutentnahme eingenommen wurde, kann die Wirkstoffkonzentration um ein Mehrfaches höher sein als der Talspiegel (Abb. 2). Der Talspiegel kann anhand Gleichung (5) berechnet werden, sofern Blut in der postabsorptiven Phase entnommen wurde.
Arzneistoff-Konzentrationen müssen unter Berücksichtigung der klinischen Situation des Patienten interpretiert werden. Empfehlungen bezüglich einer Dosisänderung stellen den größten Anteil an Empfehlungen dar.
Für die Interpretation der Ergebnisse muss geprüft werden, ob sich die Arzneistoff-Konzentration im Blut innerhalb des therapeutischen Referenzbereichs (Tab. 4) und des dosisbezogenen Referenzbereichs (Tab. 5) befindet. Wenn sich eine Arzneistoff-Konzentration außerhalb des therapeutischen Referenzbereichs befindet, sollte man berücksichtigen, welcher TDM-Empfehlungsgrad für den entsprechenden Arzneistoff vorliegt (Tab. 4). Eine Arzneistoff-Konzentration außerhalb des dosisbezogenen Referenzbereichs (Tab. 5) sollte das TDM-Labor alarmieren, nach möglichen Ursachen zu suchen: Arzneimittel-Interaktionen, Genpolymorphismen (UM, PM), eine eingeschränkte Leber- oder Nierenfunktion, Alter und/oder erkrankungsbedingte Änderungen der Pharmakokinetik eines Patienten, Adhärenzprobleme, fehlende Steady-State-Bedingungen oder auch eine Signalinterferenz mit einem weiteren Arzneistoff, den der Patient dem Arzt nicht genannt hat (z.B. Johanniskraut). Es sollte auch in Betracht gezogen werden, ob der Patient die tägliche Dosis als Einmaldosis oder mehrmals täglich eingenommen hat.
Häufig ist es notwendig, die pharmakokinetischen Eigenschaften der am Metabolismus beteiligten Enzyme sowie die Substrat- und Inhibitor-/Induktor-Eigenschaften aller Medikamente des Patienten in die Interpretation der Ergebnisse miteinzubeziehen. In den vorliegenden, aktualisierten Leitlinien werden unterstützende Informationen über die Eigenschaften der Arzneistoffe als Substrate (Tab. 1) und Inhibitoren bzw. Induktoren (Tab. 2 und 3) gegeben, um mögliche Wechselwirkungen zwischen Medikamenten zu erkennen.
Für die Behandlung von Schmerzen können relativ niedrige Blutspiegel trizyklischer Antidepressiva ausreichend sein. Sie können innerhalb des „dosisbezogenen Referenzbereichs“ (Tab. 5), aber außerhalb des „therapeutischen Referenzbereichs“ der Tabelle 4 liegen, die für die Indikation Depression zusammengestellt wurde.
Ein Labor kann empfehlen, dass eine weitere Probe nach einer bestimmten Zeit abgenommen werden sollte, wenn bei ungewöhnlich niedrigen oder hohen Blutspiegeln eine wiederholte Messung klären kann, ob der Patient das Medikament unregelmäßig einnimmt (unzureichende Adhärenz) oder ob der Patient ein schneller oder langsamer Metabolisierer ist.
Anwendung der TDM-Leitlinien für die Interpretation der Ergebnisse – Fallbeispiele
Um die Anwendung der Informationen der Konsensus-Leitlinien für die Interpretation der Laborergebnisse zu demonstrieren, werden im Folgenden drei repräsentative Fälle vorgestellt:
Fall 1:
Die Eckdaten sind in der Tabelle Fall 1 zusammengefasst.
Fall 1
|
Patient |
51 Jahre/männlich/stationär/Raucher (>10 Zigaretten/Tag) |
|
Diagnose |
Paranoide Schizophrenie |
|
Grund der Anforderung |
Unklare Adhärenz |
|
Schweregrad der Erkrankung |
Schwer krank (CGI-S-Score 6) |
|
Verbesserung |
Keine Änderung (CGI-I-Score 4) |
|
Unerwünschte Arzneimittelwirkungen |
Keine berichtet |
|
Probearzneistoff/Dosis |
Clozapin/250 mg/Tag |
|
Beginn der Medikation |
Vor 5 Wochen, letzte Dosisänderung: vor 2 Wochen |
|
Letzte Arzneistoffeinnahme |
Vor 12 Stunden |
|
Komedikation |
Acetylsalicylsäure, Simvastatin, Sertralin |
|
Laborergebnisse |
Clozapin: 224 ng/ml |
|
N-Desmethylclozapin: 175 ng/ml |
Interpretation
TDM war in Übereinstimmung mit den Konsensus-Leitlinien indiziert (Tab. 7). Unter einer therapeutischen Dosis von 250 mg verbesserte sich die klinische Symptomatik des Patienten nach Beurteilung durch die CGI-I-Skala nicht (siehe Abb. 5). TDM sollte abklären, ob der Patient nicht adhärent bezüglich seiner Medikation ist oder ob die Dosis für eine gesteigerte therapeutische Wirksamkeit erhöht werden sollte. Die Bestimmung von Clozapin im Blut offenbarte eine Konzentration von 224 ng/ml, also unterhalb des therapeutischen Referenzbereichs von 350 bis 600 ng/ml (siehe Tab. 4) und innerhalb des dosisbezogenen Referenzbereichs für Clozapin und seinen Metaboliten (Tab. 5). Bei einer Dosis von 250 mg/Tag ist für Clozapin eine Konzentration zwischen 108 und 398 ng/ml und für N-Desmethylclozapin zwischen 125 und 313 ng/ml zu erwarten (für die Berechnung des dosisbezogenen Referenzbereichs siehe DRC-Faktoren „niedrig“ und „hoch“ in Tab. 5, 250×0,43=108 bis 250×1,59=398 ng/ml für Clozapin, und 250×0,50=125 bis 250×1,25=313 ng/ml für N-Desmethylclozapin). Das Verhältnis der Konzentrationen N-Desmethylclozapin zu Clozapin war mit 0,78 erwartungsgemäß. Erwartet wurde ein Quotient zwischen 0,45 und 0,78 (siehe Tab. 6). Der Patient war Raucher. Tabelle 2 listet keinen Inhibitor bezüglich der Komedikation des Patienten, aber Tabelle 3 zeigt an, dass Rauchen CYP1A2 induziert, das in den Metabolismus von Clozapin involviert ist (Tab. 1).
Empfehlung
Eine Dosiserhöhung wird zur Steigerung des Therapieeffekts empfohlen. Anhand des Quotienten von Konzentration zur Dosis von 0,9 (ng/ml)/mg kann eingeschätzt werden, das 400 mg/Tag benötigt werden, um therapeutisch empfohlene Konzentrationen zu erreichen (350–600 ng/ml).
Fall 2:
Die Eckdaten sind in der Tabelle Fall 2 zusammengefasst.
Fall 2
|
Patient |
70 Jahre/weiblich/stationär/Raucher (>10 Zigaretten/Tag) |
|
Diagnose |
Schwere depressive Episode |
|
Grund der Anforderung |
Unerwünschte Arzneimittelwirkung und klinische Verbesserung |
|
Schweregrad der Erkrankung |
Mäßig krank (CGI-S-Score 4) |
|
Verbesserung |
Viel besser (CGI-I-Score 2) |
|
Unerwünschte Arzneimittelwirkungen |
Gastrointestinale Störungen |
|
Probearzneistoff/Dosis |
Venlafaxin XR/225 mg/Tag |
|
Beginn der Medikation |
Vor 3 Wochen, letzte Dosisänderung: vor 1 Woche |
|
Letzte Arzneistoffeinnahme |
Vor 24 Stunden |
|
Komedikation |
Levomepromazin |
|
Laborergebnisse |
Venlafaxin: 168 ng/ml |
|
O-Desmethylvenlafaxin: 251 ng/ml |
|
|
Aktive Fraktion: 419 ng/ml |
|
|
N-Desmethylvenlafaxin: 143 ng/ml |
Interpretation
TDM war in Übereinstimmung mit den Konsensus-Leitlinien indiziert. Unter einer therapeutischen Dosis von 225 mg entwickelte die 70 Jahre alte Patientin unerwünschte Arzneimittelwirkungen, nach Beurteilung durch CGI-I-Skala (siehe Abb. 5) verbesserte sich die klinische Symptomatik jedoch stark unter der gegebenen Dosis. TDM sollte abklären, ob die unerwünschten Arzneimittelwirkungen aufgrund hoher Konzentrationen der aktiven Fraktion von Venlafaxin zu begründen sind und ob die Dosis erniedrigt werden könnte, ohne einen Verlust der therapeutischen Wirksamkeit zu riskieren.
Die Summe der Konzentrationen der aktiven Fraktion von Venlafaxin plus O-Desmethylvenlafaxin war 419 ng/ml und somit leicht oberhalb des therapeutischen Referenzbereichs von 100 bis 400 ng/ml (siehe Tab. 4) und oberhalb des dosisbezogenen Referenzbereichs. Bei einer Dosis von 225 mg/Tag beträgt der zu erwartende dosisbezogene Referenzbereich für Venlafaxin 225×0,12=27 bis 225×0,36=81 ng/ml, für O-Desmethylvenlafaxin 225×0,78=176 bis 225×1,30=293 ng/ml (für die Berechnung siehe DRC-Faktoren in Tab. 5). Die zu erwartende Konzentration der aktiven Fraktion betrug demnach 203 bis 376 ng/ml. Tabelle 1 listet Venlafaxin als Substrat von CYP2D6 und CYP2C19. Das Verhältnis der Konzentrationen von O-Desmethylvenlafaxin zu Venlafaxin war 1,49 und somit unterhalb des zu erwartenden MPR-Bereichs von 2,7 bis 7,7 (siehe Tab. 6). Dies deutet auf einen PM-Phänotyp von CYP2D6 hin. Das Verhältnis der Konzentrationen für N-Desmethylvenlafaxin zu Venlafaxin war 0,85, was für einen normalen CYP2C19-Phänotyp spricht (siehe Tab. 6). Komedikation war Levomepromazin, und die Patientin war Raucherin. Tabelle 2 listet Levomepromazin als Inhibitor von CYP2D6, das den Metabolismus von Venlafaxin zu O-Desmethylvenlafaxin katalysiert, und Tabelle 3 zeigt, dass Rauchen CYP1A2 induziert, das nicht in den Metabolismus von Venlafaxin involviert ist (Tab. 1). Es ist somit wahrscheinlich, dass die unerwünschten Arzneimittelwirkungen auf den hohen Arzneistoff-Konzentrationen beruhen, die aufgrund der CYP2D6-Inhibition durch Levomepromazin zu erklären sind. Der PM-Phänotyp von CYP2D6 wird weiterhin bestätigt aufgrund der höheren als zu erwartenden Konzentration von N-Desmethylvenlafaxin (143 ng/ml anstelle von 34 bis 74 ng/ml).
Empfehlung
Die berichteten, unerwünschten Arzneimittelwirkungen sind aufgrund einer hohen Konzentration von Venlafaxin und O-Desmethylvenlafaxin zu erklären, am wahrscheinlichsten aufgrund einer Arzneimittel-Interaktion und des Lebensalters. Die Patientin ist wahrscheinlich ein PM-Phänotyp von CYP2D6 aufgrund der Inhibition durch Levomepromazin oder ein PM-CYP2D6-Genotyp. Eine Dosisreduktion kann hilfreich sein und die Verträglichkeit möglicherweise steigern, ohne einen Wirkverlust zu riskieren. Alternativ kann Levomepromazin durch einen nicht CYP-inhibierenden Arzneistoff ersetzt werden, beispielsweise Pipamperon, denn die berichteten gastrointestinalen Störungen könnten auch durch Levomepromazin induziert worden sein.
Fall 3:
Die Eckdaten sind in der Tabelle Fall 3 zusammengefasst.
Fall 3
|
Patient |
51 Jahre/männlich/stationär/Raucher (<10 Zigaretten/Tag) |
|
Diagnose |
Bipolare Störung, aktuell manisch |
|
Grund der Anforderung |
Unzureichende klinische Verbesserung/Unklare Adhärenz |
|
Schweregrad der Erkrankung |
Deutlich krank (CGI-S-Score 5) |
|
Verbesserung |
Keine Änderung (CGI-I-Score 4) |
|
Unerwünschte Arzneimittelwirkungen |
Nein |
|
Probearzneistoff/Dosis |
Valproat/900 mg/Tag, Olanzapin/10 mg/Tag |
|
Beginn der Medikation |
Vor >6 Wochen, letzte Dosisänderung: vor 2 Wochen |
|
Letzte Arzneistoffeinnahme |
Vor 12 Stunden |
|
Komedikation |
Keine |
|
Laborergebnisse |
Valproat: 37 µg/ml |
|
Olanzapin: 7 ng/ml (therapeutischer Referenzbereich für bipolare Störungen unklar; berücksichtigt man die Dosis von 10 mg, die für eine Kombinationstherapie empfohlen ist, sind 8 bis 23 ng/ml als orientierender therapeutischer Bereich vorgeschlagen) |
|
|
N-Desmethylolanzapin: 2 ng/ml |
Interpretation
TDM war in Übereinstimmung mit den Konsensus-Leitlinien indiziert. Die Symptomatik des Patienten hat sich nach dem CGI-I-Score von 4 nicht verändert (siehe Abb. 5). TDM kann abklären, ob der Patient seine Medikation wie verschrieben eingenommen hat und ob eine Dosiseskalation hilfreich sein könnte.
Die Valproinsäure-(Valproat-)Konzentration von 37 µg/ml befindet sich unterhalb des therapeutischen Referenzbereichs (Tab. 4) und auch unterhalb der zu erwartenden dosisbezogenen Konzentrationen. Die Berechnung des dosisbezogenen Referenzbereichs (siehe DRC-Faktoren in Tab. 5) ergibt Konzentrationen von 55980 bis 121320 ng/ml (=56–121 µg/ml) für eine Dosis von 900 mg Valproinsäure. Für Olanzapin und seinen Metaboliten betragen die Konzentrationen 7 ng/ml bzw. 2 ng/ml. Diese Konzentrationen können nicht mit therapeutischen Effekten in Zusammenhang gebracht werden, da ein therapeutischer Referenzbereich für die Indikation der bipolaren Störung bisher nicht etabliert wurde. Die zu erwartenden Konzentrationen bei einer Dosis von 10 mg/Tag können jedoch berechnet werden (siehe Tab. 5). Sie sollten 12 bis 25 ng/ml für Olanzapin betragen. Die gemessene Olanzapin-Konzentration von 7 ng/ml war demnach niedriger als die erwartete Cmin. Auf der anderen Seite war das Metabolit-zu-Muttersubstanz-Verhältnis 0,29 und somit innerhalb des zu erwartenden Bereichs (siehe Tab. 6). Der Patient war ein moderater Raucher. Tabelle 3 listet, dass Rauchen CYP1A2 induziert und Tabelle 1 zeigt an, dass Olanzapin ein Substrat von CYP1A2 ist. Niedrigere als zu erwartende Konzentrationen von Olanzapin und ein normaler MPR-Wert können am besten aufgrund von Adhärenzproblemen erklärt werden. Darüber hinaus könnte jedoch auch der Raucherstatus einen gesteigerten Abbau von Olanzapin erklären.
Empfehlung
Der mangelnde Therapieeffekt ist wahrscheinlich aufgrund der niedrigen Arzneistoff-Konzentration im Blut zu erklären. Die Adhärenz des Patienten sollte thematisiert und überprüft werden. Sofern der Patient adhärent mit der Medikation ist, kann eine Dosissteigerung hilfreich sein.
Die drei Fälle demonstrieren, wie die in den Tabellen 1 bis 6 aufgeführten Informationen für die Interpretation der Labordaten genutzt werden können. Anhand der Informationen können valide Schlussfolgerungen gezogen und bedeutsame Empfehlungen für eine rationale Pharmakotherapie gegeben werden. Dennoch beruht die Interpretation von TDM-Ergebnissen auf komplexen, quantitativen Beziehungen. Deshalb ist eine Schulung in klinischer Neuropsychopharmakologie, in Pharmakokinetik und in der Anwendung von TDM-Informationen wichtig. Regelmäßige Konferenzen mit Diskussion der Interpretation von echten Fallbeispielen sind hilfreich für das Erlernen dieser Thematik. Es wird auch empfohlen, junge Psychiater unter Aufsicht eines Experten die Interpretation der Ergebnisse durchführen zu lassen.
Pharmakogenetische Tests in Kombination mit TDM
Wenn vor dem Verschreiben eines bestimmten Arzneistoffs unter definierten Umständen ein pharmakogenetischer Test durchgeführt wird [7, 46], können Konzentrationen außerhalb des therapeutischen oder dosisbezogenen Referenzbereichs vermieden werden, wenn dies aufgrund von Genpolymorphismen zu begründen gewesen wäre (UM, PM; pharmakokinetische Ebene). Situationen und Fälle, wo pharmakogenetische Tests mit TDM kombiniert werden könnten, sind in Abbildung 6 erklärt. In Übereinstimmung mit Empfehlungen der deutschen Gendiagnostik-Kommission (GeKO) und des Clinical Pharmacogenetics Implementation Consortium (CPIC) [46] sowie regulatorischer Behörden wie FDA und EMA sind die wichtigsten Indikationen für eine Genotypisierung von Arzneistoff-metabolisierenden Enzymen in Kombination mit TDM:
- A-priori-Genotypisierung, wenn ein Arzneistoff einen engen therapeutischen Bereich besitzt und ein erhöhtes Toxizitätsrisiko im Falle eines genetisch beeinträchtigten Metabolismus aufweist (z.B. HLA-B*1502 vor der Verordnung von Carbamazepin; siehe FDA-Warnung)
- A-priori-Genotypisierung, wenn ein Patient mit einem Substrat mit einer hohen interindividuellen Variabilität im Metabolismus behandelt wird und ein bedeutsames Toxizitätsrisiko im Falle einer Überdosierung besteht (z.B. trizyklische Antidepressiva)
- Post-hoc-Genotypisierung, wenn der Patient ungewöhnliche Arzneistoff- oder Metabolit-Konzentrationen im Blut entwickelt, um den metabolischen Status vor der Verabreichung anderer Arzneistoffe zu bestimmen (z.B. Codein im Falle eines ultraschnellen Metabolisiererstatus; siehe Warnung: Codein für ultraschnelle Metabolisierer)
Sofern ein Patient anhand einer Genotypisierung als PM oder UM identifiziert wurde, muss die Medikation nicht automatisch gewechselt werden, aber die Dosis könnte anhand TDM und klinischer Beurteilung angepasst werden.
Pharmakogenetische Tests für pharmakodynamische Parameter werden bisher in der klinischen Praxis nicht empfohlen.
Kommerziell erhältliche Testbatterien für die Detektion von pharmakokinetischen und pharmakodynamischen Genvarianten werden heutzutage schon vermarktet, aber die Evidenz erlaubt bisher keine Empfehlung bezüglich deren unkritischem Einsatz in der klinischen Praxis. Diesbezüglich besteht Bedarf weiterer Forschung in großen, multizentrischen Studien.
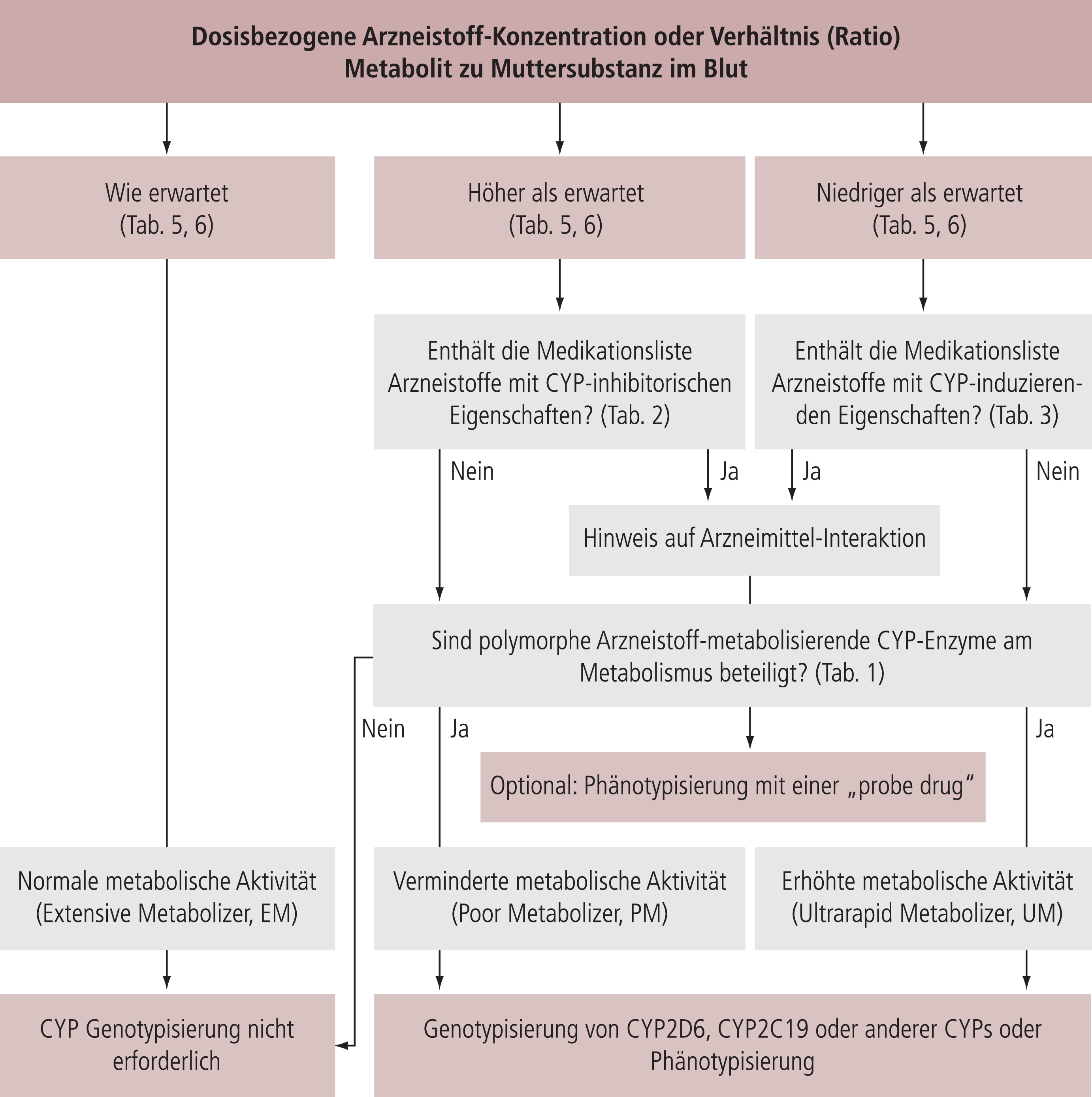
Abb. 6. Algorithmus für den Einsatz einer Genotypisierung von Cytochrom-P450-(CYP-)Enzymen und einer Phänotypisierung mit einem Testsubstrat („probe drug“), in Kombination mit TDM
Klinische Entscheidung
Ein TDM-Ergebnis führt zu einer optimalen Arzneistoff-Dosis für den individuellen Patienten (Abb. 7). Der Arzt sollte sich vergegenwärtigen, dass unter optimalen Bedingungen den Dosisempfehlungen und Kommentaren des Labors die beste verfügbare Evidenz zugrunde liegt. Das Labor besitzt jedoch nur ein begrenztes Wissen über die klinische Situation des Patienten. Auf der anderen Seite haben die meisten behandelnden Ärzte nur ein begrenztes pharmakokinetisches Wissen. Deshalb ist es essenziell anzuerkennen, dass ein optimales TDM eine interdisziplinäre, enge Kommunikation zwischen Labor und klinischen Experten erfordert.
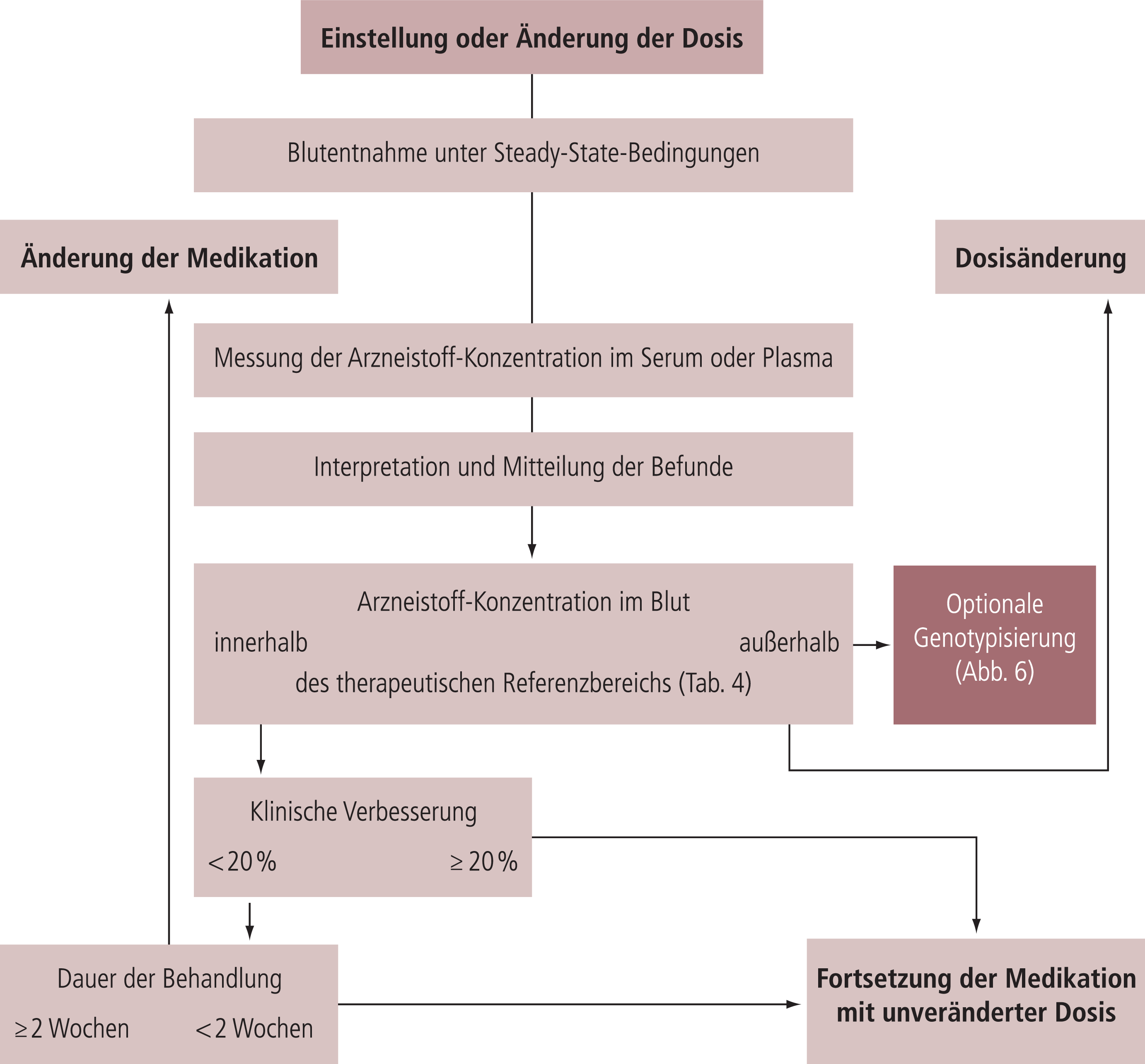
Abb. 7. TDM-geleitete Dosis-Titration für die Behandlung mit stimmungsstabilisierenden, antidepressiven, antipsychotischen oder antiepileptischen Arzneistoffen. Die klinische Entscheidung sollte die Steady-State-Konzentrationen des Arzneistoffs im Blut, die klinische Verbesserung und die Behandlungsdauer berücksichtigen. 94% des Steady-State sind nach vier Eliminationshalbwertszeiten des Arzneistoffs oder aktiven Metaboliten erreicht (siehe Tab. 4).
Wenn die gemessene Arzneistoff-Konzentration innerhalb des therapeutischen Referenzbereichs liegt, wird eine Dosisänderung natürlich nur empfohlen, wenn klinische Gründe, beispielsweise unerwünschte Arzneimittelwirkungen oder unzureichendes Therapieansprechen, solch eine Entscheidung klar rechtfertigen. Der behandelnde Arzt hat zu entscheiden, ob die Behandlungsstrategie geändert werden sollte oder nicht. Wenn der TDM-Empfehlung nicht gefolgt wird, sollte andererseits der Grund hierfür dokumentiert werden, um die Entscheidung des Arztes nachvollziehen zu können, falls der Patient zu Schaden kommt. Empfehlungen für solch eine Evaluation vor Gericht wurden von der TDM-AGNP-Gruppe publiziert [48].
Bei Patienten mit bekannter beschleunigter Elimination kann es sinnvoll sein, Dosen oberhalb der maximal empfohlenen Dosis zu verschreiben, da diese Patienten unter Standarddosen Arzneistoff-Konzentrationen unterhalb der Referenzbereiche entwickeln können. Die Medikation sollte erst dann geändert werden, wenn der Patient ausreichend hohe Arzneistoff-Konzentrationen im Blut für einen ausreichend langen Behandlungszeitraum (mindestens zwei Wochen) entwickelt und sich die Symptomatik nicht um wenigstens 20% verbessert hat. Eine weitere Option wäre die Verschreibung eines Arzneistoffs, der nicht über CYP metabolisiert wird, wie das Antidepressivum Milnacipran oder das Antipsychotikum Amisulprid.
Wenn unerwünschte Arzneimittelwirkungen mit der klinischen Verbesserung unter empfohlenen Dosen einhergehen, kann die Messung der Arzneistoff-Konzentration im Blut klären, ob die unerwünschten Arzneimittelwirkungen mit zu hohen Arzneistoff-Konzentrationen im Blut zu begründen sind. In dieser Situation kann die Dosis reduziert werden, normalerweise ohne Risiko eines Wirkverlusts.
Für die Behandlung mit Antidepressiva, Antipsychotika oder stimmungsstabilisierenden Arzneistoffen besteht eine gute Evidenz, dass ein Nichtansprechen in Woche 2 hoch prädiktiv für ein späteres Therapieversagen ist [26, 41]. Für die Dosistitration von Antidepressiva oder Antipsychotika empfehlen wir deshalb ein zusätzliches ärztliches Rating der Symptomatik eines Patienten zu Beginn der Therapie und an Woche 2, neben der Bestimmung der Arzneistoff-Konzentration im Blut.
Abbildung 7 fasst die genannten Empfehlungen in einem Flussdiagramm zusammen.
Wenn nach einer Modifikation der Dosis oder nach Verschreibung einer Komedikation, die den Metabolismus des gemessenen Arzneistoffs inhibiert oder induziert, weitere Messungen der Arzneistoff-Konzentration im Blut empfohlen werden, sollte das nächste TDM erst ausgeführt werden, wenn wieder Steady-State-Konzentrationen erreicht sind. Dafür ist die terminale Eliminationshalbwertszeit (t1/2) des Arzneistoffs zu berücksichtigen (Tab. 4).
Falls sich die Symptomatik des Patienten bei Arzneistoff-Konzentrationen unterhalb des Referenzbereichs gebessert hat, sollte ein (schrittweises) Absetzen der Medikation in Betracht gezogen werden, da die Medikation nur als Placebo eingesetzt wird, aber weiterhin das Risiko für unerwünschte Arzneimittelwirkungen gegeben und die medikamentöse Therapie kostspielig ist.
Kosteneffektivität von TDM
Eine Kosteneffektivität von TDM wurde in verschiedenen Studien gezeigt (Review siehe [42]). Für trizyklische Antidepressiva wurde dies anhand einer Reduktion von Intoxikationen beschrieben. Wenn Patienten nach Gabe einer Testdosis von Amitriptylin oder Nortriptylin einem pharmakokinetischen Monitoring unterzogen wurden, um die Eliminationsrate und die Eliminationshalbwertszeit zu ermitteln und um die erforderliche Dosis für das Erreichen von therapeutisch effektiven Steady-State-Konzentrationen des Arzneistoffs im Blut zu berechnen, senkte die pharmakokinetische Dosierung die Kosten deutlich. Die Patienten wurden sechs Tage früher aus der stationären Behandlung entlassen und kehrten 55 Tage früher zur Arbeit zurück als die empirisch dosierten Patienten. Für SSRI ermittelten Lundmark und Kollegen in einer Gruppe von 127 älteren ambulanten Patienten, dass die Anwendung von TDM in 38 Fällen zu einer Dosisreduktion und somit zu einer Kostenreduktion der Arzneimittel um 16% führte. Für Citalopram verkürzte TDM die stationäre Aufenthaltsdauer. Die TDM-geleitete Pharmakotherapie bei stationären Patienten, bei denen ausreichend hohe Citalopram-Konzentrationen (>50 ng/ml) eingestellt waren, führte zu einer Reduktion der stationären Aufenthaltsdauer um 23 Tage im Vergleich zu einer Patientengruppe mit subtherapeutischen Citalopram-Konzentrationen. Arzneistoff-Konzentrationen unterhalb von 50 ng/ml an Behandlungstag 7 waren hochprädiktiv für ein späteres Therapieversagen. Ähnliche Ergebnisse wurden für mit Venlafaxin behandelte depressive Patienten berichtet [38]. Darüber hinaus kann angenommen werden, dass TDM das Potenzial besitzt, die Rückfallraten zu reduzieren. Falls TDM Non-Adhärenz zur Medikation vor einer Rehospitalisierung aufdeckt, ist dies ebenfalls hoch kosteneffektiv. Ein stationärer Tag ist 4- bis 16-mal teurer als eine Bestimmung der Arzneistoff-Konzentration im Blut.
Zusammengefasst kann TDM die Gesundheitskosten deutlich reduzieren, unter anderem aufgrund der Verbesserung der Adhärenz, Beschleunigung der klinischen Verbesserung oder Verkürzung der Aufenthalts- oder Erkrankungsdauer. Mehr Studien werden jedoch bezüglich der Kosteneffektivität von TDM benötigt.
Schlussfolgerung und Perspektiven
Dieses zweite Update der AGNP-Leitlinien beschreibt die Praxis von TDM für Neuropsychopharmaka, um die angemessene Anwendung von TDM voranzutreiben. Wenn TDM adäquat angewendet wird, ist dies ein exzellentes Instrument der Präzisionsmedizin, um die Pharmakotherapie des individuellen Patienten zu optimieren. Während der letzten Jahrzehnte hat das Wissen über metabolische Abbauwege und Arzneimittelwirkungen im menschlichen Organismus stark zugenommen. Dennoch besteht eine Lücke zwischen dem vorhandenen Wissen in der Pharmakologie und der tatsächlichen Anwendung im Gesundheitsbereich. TDM überbrückt diese Lücke. Für dieses Update wurde der Berechnung von Faktoren zur Analyse von dosisbezogenen Referenzbereichen spezielle Beachtung geschenkt mit dem Ziel einer verbesserten pharmakokinetischen Charakterisierung des Patienten vom behandelnden Arzt. Die Kombination von Informationen über therapeutische Referenzbereiche, dosisbezogene Referenzbereiche, Metabolit-zu-Muttersubstanz-Quotienten sowie Eigenschaften von eingenommenen Arzneistoffen wie CYP-Substrat, Inhibitor- und Induktoreigenschaften und schließlich Genotypen von CYP450-Enzymen und Arzneistoff-Transportern ermöglicht es, individuelle Charakteristika in der Pharmakodynamik und Pharmakokinetik von Neuropsychopharmaka zu erkennen und zu dokumentieren. Die Information kann für rationale Dosiskorrekturen angewendet werden, um die Wirksamkeit und Verträglichkeit dieser Arzneistoffe sowie die Therapiekosten zu optimieren. Ungeachtet objektiver Fortschritte bezüglich der Anwendung von TDM in der klinischen Praxis muss die Qualität von TDM weiterhin verbessert werden. Des Weiteren besteht der Bedarf, pharmakokinetische Messungen in klinische Studien zur Arzneistoffentwicklung einzubeziehen. Es ist ein wesentliches Defizit bei der Arzneimittelzulassung, dass Daten zu Arzneistoff-Konzentrationen im Blut, die eine hohe Wahrscheinlichkeit eines optimalen Therapieansprechens ermöglichen, gesetzlich nicht vorgeschrieben sind. Die Produktinformation sollte um TDM-bezogene Daten erweitert werden. Schließlich ist die Schulung von diesen Themen auf einem postgraduellen Level notwendig.
Abkürzungen
|
BHS |
Blut-Hirn-Schranke |
|
C/D |
Konzentration-zu-Dosis-Verhältnis |
|
CL |
Totale Clearance |
|
CL/F |
Apparente totale Clearance |
|
Cmax |
Maximale Konzentration |
|
Cmin |
Talspiegel, minimale Konzentration |
|
CYP |
Cytochrom P450 |
|
EMA |
European Medicines Agency |
|
F |
Bioverfügbarkeit |
|
FDA |
Food and Drug Administration |
|
GeKO |
Gendiagnostik-Kommission |
|
MPR |
Metabolit-zu-Muttersubstanz-Verhältnis |
|
P-gp |
P-Glykoprotein |
|
t1/2 |
Eliminationshalbwertszeit |
|
TDM |
Therapeutisches Drug-Monitoring |
|
tmax |
Zeit von maximalen Arzneistoff-Konzentrationen |
|
tmin |
Zeit von minimalen Arzneistoff-Konzentrationen |
Interessenkonflikterklärung
Christoph Hiemke erhielt Honorare für Vorträge und Beratungen von den Firmen Janssen, Stada, LTS und Servier.
Pierre Baumann erhielt Honorare für Vorträge und Beratungen von nahezu allen pharmazeutischen Firmen, welche psychotrope Arzneistoffe in der Schweiz verkaufen.
Ekkehard Haen ist Vorsitzender und leitender Direktor der AGATE (www.amuep-agate.de), Arbeitsgemeinschaft Arzneimitteltherapie bei psychiatrischen Erkrankungen. Er ist Gesellschafter der psiac GmbH (www.psiac.de), einem internetbasierten Arzneistoff-Interaktionsprogramm.
Gerhard Gründer diente als Berater für Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson und Otsuka. Er war Sprecher für Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, Janssen Cilag, Otsuka, Pfizer, Servier und Wyeth. Er erhielt subventionierte Unterstützung von Alkermes, Bristol-Myers Squibb, Eli Lilly und Johnson & Johnson. Er ist Mitbegründer von Pharma-Image – Molecular Imaging Technologies GmbH.
Gerd Laux erhielt Honorare für Vorträge und Beratungen oder unbeschränkte Subventionen für Schulungen von Bayer, Janssen-Cilag, Lundbeck, Pfizer und Servier.
Gudrun Hefner wurde von der pharmazeutischen Industrie nicht unterstützt.
Literatur
1. Adli M, Baethge C, Heinz A, Langlitz N, et al. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci 2005;255:387–400.
2. Baumann P. Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors. Clin Pharmacokinet 1996;31:444–69.
3. Baumann P, Hiemke C, Ulrich S, Eckermann G, et al. The AGNP-TDM expert group consensus guidelines: Therapeutic drug monitoring in psychiatry. Pharmacopsychiatry 2004;37:243–65.
4. Baumann P, Rougemont M, Corruble E, Hiemke C, et al. Recommendations for therapeutic monitoring of antidepressants. Rev Med Suisse 2013;9:577–86.
5. Bergemann N, Rommel F, Conca A. Therapeutisches Drug-Monitoring von Psychopharmaka in der Schwangerschaft. J Neurol Neurochir Psychiatr 2009;10:38–40.
6. Brünen S, Vincent PD, Baumann P, Hiemke C, et al. Therapeutic drug monitoring for drugs used in the treatment of substance-related disorders: literature review using a therapeutic drug monitoring appropriateness rating scale. Ther Drug Monit 2011;33:561–72.
7. Crettol S, de Leon J, Hiemke C, Eap CB. Pharmacogenomics in psychiatry: From therapeutic drug monitoring to genomic medicine. Clin Pharmacol Ther 2014;95:254–57.
8. de Leon J. The crucial role of the therapeutic window in understanding the clinical relevance of the poor versus the ultrarapid metabolizer phenotypes in subjects taking drugs metabolized by CYP2D6 or CYP2C19. J Clin Psychopharmacol 2007;27:241–45.
9. Deligiannidis KM. Therapeutic drug monitoring in pregnant and postpartum women: Recommendations for SSRIs, lamotrigine, and lithium. J Clin Psychiatry 2010;71:649–50.
10. Egberts K, Karwautz A, Plener PL, Mehler-Wex C, et al. Pharmacovigilance in child and adolescent psychiatry. Z Kinder Jugendpsychiatr Psychother 2015;43:21–8.
11. Egberts KM, Mehler-Wex C, Gerlach M. Therapeutic drug monitoring in child and adolescent psychiatry. Pharmacopsychiatry 2011;44:249–53.
12. Gerlach M, Egberts K, Dang SY, Plener P, et al. Therapeutic drug monitoring as a measure of proactive pharmacovigilance in child and adolescent psychiatry. Expert Opin Drug Saf 2016;15:1477–82.
13. Greil W, Häberle A, Schuhmann T, Grohmann R, et al. Age and adverse drug reactions from psychopharmacological treatment: Data from the AMSP drug surveillance programme in Switzerland. Swiss Med Wkly 2013;143:w13772.
14. Grohmann R, Engel RR, Rüther E, Hippius H. The AMSP drug safety program: methods and global results. Pharmacopsychiatry 2004;37(Suppl 1):S4–11.
15. Gründer G, Hiemke C, Paulzen M, Veselinovic T, et al. Therapeutic plasma concentrations of antidepressants and antipsychotics: Lessons from PET imaging. Pharmacopsychiatry 2011;44:236–48.
16. Haen E. Der TDM-Befund. Psychopharmakotherapie 2012;19:129–34.
17. Haen E. Therapeutic drug monitoring in pharmacovigilance and pharmacotherapy safety. Pharmacopsychiatry 2011;44:254–58.
18. Haen E, Greiner C, Bader W, Wittmann M. [Expanding therapeutic reference ranges using dose-related reference ranges]. Nervenarzt 2008;79:558–66.
19. Hefner G, Laib AK, Sigurdsson H, Hohner M, et al. The value of drug and metabolite concentration in blood as a biomarker of psychopharmacological therapy. Int Rev Psychiatry 2013;25:494–508.
20. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry 2011;44:195–235.
21. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP-Konsensus-Leitlinie für therapeutisches Drug-Monitoring in der Psychiatrie. Update 2011. Psychopharmakotherapie 2012;19:91–122.
22. Hiemke C, Shams M. Phenotyping and genotyping of drug metabolism to guide pharmacotherapy in psychiatry. Curr Drug Deliv 2013;10:46–53.
23. Holt S, Schmiedl S, Thurmann PA. Potentially inappropriate medications in the elderly: The PRISCUS list. Dtsch Arztebl Int 2010;107:543–51.
24. Jang SH, Yan Z, Lazor JA. Therapeutic drug monitoring: A patient management tool for precision medicine. Clin Pharmacol Ther 2016;99:148–50.
25. Landmark CJ, Johannessen SI, Tomson T. Dosing strategies for antiepileptic drugs: From a standard dose for all to individualised treatment by implementation of therapeutic drug monitoring. Epileptic Disord 2016;18:367–83.
26. Leucht S, Busch R, Kissling W, Kane JM. Early prediction of antipsychotic nonresponse among patients with schizophrenia. J Clin Psychiatry 2007;68:352–60.
27. Mann K, Hiemke C, Schmidt LG, Bates DW. Appropriateness of therapeutic drug monitoring for antidepressants in routine psychiatric inpatient care. Ther Drug Monit 2006;28: 83–8.
28. Müller H, Eusterschulte B, Havemann-Reinecke U, Stetter R, et al. Forensische Aspekte des therapeutischen Drug-Monitorings (TDM) in der Psychiatrie. Psychopharmakotherapie 2009;16:52–6.
29. Ostad Haji E, Tadic A, Wagner S, Dragivevic A, et al. Early improvement and serum concentrations of citalopram to predict antidepressant drug response of patients with major depression. Pharmacopsychiatry 2013; 46:261–66.
30. Patsalos PN, Berry DJ, Bourgeois BF, Cloyd JC, et al. Antiepileptic drugs – best practice guidelines for therapeutic drug monitoring: A position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008;49:1239–76.
31. Patteet L, Maudens KE, Sabbe B, Morrens M, et al. High throughput identification and quantification of 16 antipsychotics and 8 major metabolites in serum using ultra-high performance liquid chromatography-tandem mass spectrometry. Clin Chim Acta 2014;429:51–8.
32. Patteet L, Morrens M, Maudens KE, Niemegeers P, et al. Therapeutic drug monitoring of common antipsychotics. Ther Drug Monit 2012;34:629–51.
33. Pozzi M, Cattaneo D, Baldelli S, Fucile S, et al. Therapeutic drug monitoring of second-generation antipsychotics in pediatric patients: An observational study in real-life settings. Eur J Clin Pharmacol 2016;72: 285–93.
34. Preskorn SH. Patients who do not respond to the „usual“ dose: Why Terry fell off the dose-response curve. J Psychiatr Pract 2009;15:460–66.
35. Preskorn SH. Therapeutic drug monitoring (TDM) in psychiatry (part I): Why studies attempting to correlate drug concentration and antidepressant response don’t work. J Psychiatr Pract 2014;20:133–7.
36. Reis M, Aberg-Wistedt A, Agren H, Akerblad AC, et al. Compliance with SSRI medication during 6 months of treatment for major depression: an evaluation by determination of repeated serum drug concentrations. J Affect Disord 2004;82:443–6.
37. Shin C, Han C, Pae CU, Patkar AA. Precision medicine for psychopharmacology: a general introduction. Expert Rev Neurother 2016;16:831–9.
38. Stamm TJ, Becker D, Sondergeld LM, Wiethoff K, et al. Prediction of antidepressant response to venlafaxine by a combination of early response assessment and therapeutic drug monitoring. Pharmacopsychiatry 2014;47:174–9.
39. Stingl J, Viviani R. Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs. J Intern Med 2015;277:167–77.
40. Stingl JC, Brockmoller J, Viviani R. Genetic variability of drug-metabolizing enzymes: The dual impact on psychiatric therapy and regulation of brain function. Mol Psychiatry 2013;18:273–87.
41. Szegedi A, Jansen WT, van Willigenburg AP, van der Meulen E, et al. Early improvement in the first 2 weeks as a predictor of treatment outcome in patients with major depressive disorder: A meta-analysis including 6562 patients. J Clin Psychiatry 2009;70:344–53.
42. Touw DJ, Neef C, Thomson AH, Vinks AA, et al. Cost-effectiveness of therapeutic drug monitoring: A systematic review. Ther Drug Monit 2005;27:10–7.
43. Uchida H, Mamo DC, Mulsant BH, Pollock BG, et al. Increased antipsychotic sensitivity in elderly patients: Evidence and mechanisms. J Clin Psychiatry 2009;70:397–405.
44. Uhr M, Tontsch A, Namendorf C, Ripke S, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 2008;57:203–20.
45. Unterecker S, Riederer P, Proft F, Maloney J, et al. Effects of gender and age on serum concentrations of antidepressants under naturalistic conditions. J Neural Transm (Vienna) 2013;120:1237–46.
46. Valdes R Jr, Payne DA, Linder MW (eds.). Laboratory medicine practice guidelines and recommendations for laboratory analysis and application of pharmacogenetics to clinical practice. Washington, DC: National Academy of Clinical Biochemistry, 2010.
47. Whitney Z, Boyda HN, Procyshyn RM, Elbe D, et al. Therapeutic drug levels of second generation antipsychotics in youth: a systematic review. J Child Adolesc Psychopharmacol 2015;25:234–45.
48. Zernig G, Hiemke C, Havemann-Reinecke U, Laux G, et al. Empfehlungen für die gutachterliche Bewertung von Medikamentenspiegeln in der Psychiatrie im gerichtsanhängigen Schadensfall. Psychopharmakotherapie 2009;16:57–64.
*Diese Arbeit ist eine leicht gekürzte Übersetzung/adaptierte Fassung von Hiemke C. et al. AGNP Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017. Pharmacopsychiatry 2018;51:9–62, ©Georg Thieme Verlag, Stuttgart 2018.
Dr. Gudrun Hefner für die TDM-Gruppe der AGNP, Vitos Klinik, Hochtaunus, Emil-Sioli-Weg 1–3, 61081 Friedrichsdorf, E-Mail: Gudrun.Hefner@vitos-rheingau.de
Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017
Therapeutic drug monitoring (TDM) is the quantification and interpretation of drug concentrations in blood to optimize pharmacotherapy. TDM is a valuable tool of precision medicine. It considers the high interindividual variability of pharmacokinetics and thus enables personalized pharmacotherapy. Non-response at therapeutic doses, uncertain drug adherence (compliance), suboptimal tolerability, or pharmacokinetic drug-drug interactions are typical indications for TDM. In psychiatry and neurology, patient populations that may particularly benefit from TDM are children and adolescents, pregnant women, elderly patients, individuals with intellectual disabilities, substance abuse disorders, forensic psychiatric patients or patients with known or suspected pharmacokinetic abnormalities or pharmacokinetically relevant comorbidities. The potential benefits of TDM for optimization of pharmacotherapy can only be obtained if the method is adequately integrated in the clinical treatment process. The interdisciplinary TDM task force of the working group on neuropsychopharmacology (Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie, AGNP) has updated the guidelines now: Therapeutic and dose related reference ranges, laboratory alert levels, indications and levels of recommendation to use TDM were reevaluated and updated for more than 154 neuropsychiatric compounds. Moreover, supportive information on cytochrome P450 substrate, inhibitor and inducer properties of drugs and normal ranges of metabolite-to-parent concentration ratios, required for the interpretation of results from the laboratory, were updated. Recommendations are given when and how TDM should be combined with pharmacogenetic tests for cytochrome P450 enzymes and drug transporters. Following these guidelines holds the potential to improve the outcome of neuropsychopharmacotherapy, accelerate the recovery of many patients, especially in case of pharmacokinetic problems, and reduce health care costs.
Key words: consensus guidelines, therapeutic drug monitoring, pharmacogenetics, plasma concentration, therapeutic window
Psychopharmakotherapie 2018; 25(03):92-140