Holger Petri, Bad Wildungen*
Venlafaxin
Venlafaxin ist das in Deutschland meistverordnete SNRI-Antidepressivum [12]. Es wird in der Leber extensiv verstoffwechselt. Durch Demethylierung, primär über CYP2D6 vermittelt, entsteht mit O-Desmethylvenlafaxin (ODV) der Hauptmetabolit. ODV hat die gleiche pharmakologische Potenz wie die Muttersubstanz und ist in den USA als SNRI-Antidepressivum Desvenlafaxin zugelassen [9, 11]. Der Referenzbereich zur Anwendung des therapeutischen Drug-Monitorings (TDM) umfasst die Summenspiegel aus Venlafaxin plus ODV [4]. Beide Substanzen werden unter Beteiligung von CYP2C19 und 3A4 zu inaktiven Metaboliten abgebaut (Abb. 1) [11].
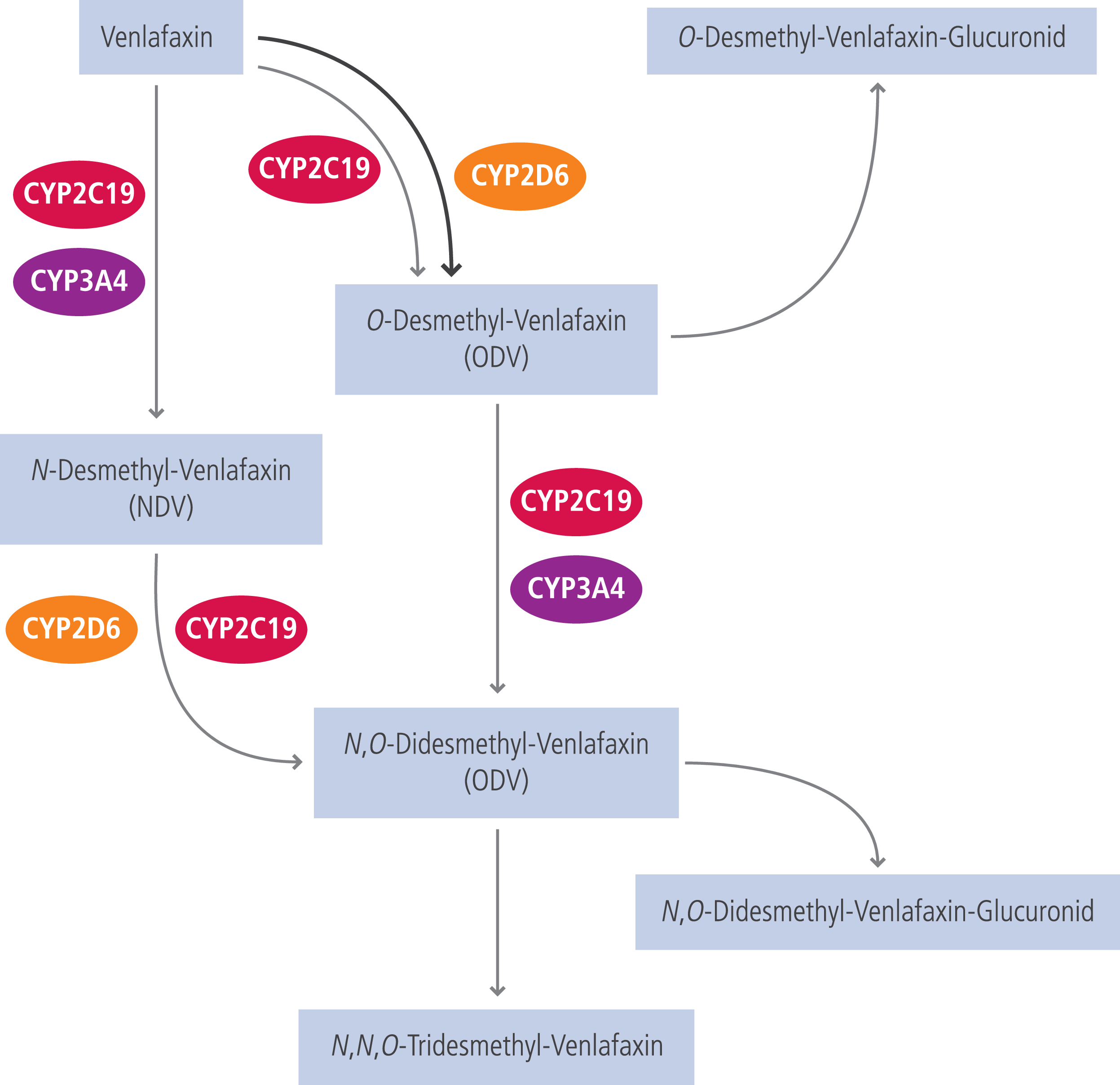
Abb. 1. Schematische Darstellung des Metabolismus von Venlafaxin (nach [11])
CYP2D6 wird polymorph exprimiert. Bei Patienten mit einem langsamen CYP2D6-Stoffwechsel (Poor Metabolizer [PM], und intermediärem Stoffwechel (Intermediate Metabolizer [IM]) ist das Verhältnis Metabolit/Muttersubstanz zugunsten des Venlafaxins verschoben. Ultrarapid Metabolizer (UM) bilden verstärkt den Metaboliten verglichen mit extensiven Metabolisierern (EM) [4].
Die Expertengruppe der Dutch Pharmacogenetics Working Group (DPWG) empfiehlt, sofern jeweils nicht auf einen alternativen Wirkstoff ausgewichen werden kann, bei Patienten mit verminderter CYP2D6-Aktivität eine Anpassung der Erhaltungsdosis an das klinische Ansprechen und an die durch TDM ermittelten Plasmaskonzentrationen. Die Datenlage ist jedoch nicht ausreichend, um eine Dosisempfehlung zu geben. Bei Patienten mit einem stark beschleunigten Metabolismus (UM) empfiehlt die DPWG eine Dosistitration bis zu einem Maximum von 150% der Normdosis [13]. Werden diese Empfehlungen auf mögliche Arzneimittelinteraktionen übertragen, so können durch potente CYP2D6-Hemmer (Abb. 2) Patienten mit einem extensiven Stoffwechsel in einen PM- bzw. IM-Phänotyp konvertiert werden. Beispielsweise erhöhte Terbinafin in einer pharmakokinetischen Studie mit Probanden die AUC (Fläche unter der Konzentrations-Zeit-Kurve) von Venlafaxin um 490%, während die AUC von ODV um 57% sank [5]. Die Neuansetzung von Bupropion, einem starken CYP2D6-Inhibitor, kann zu deutlich erhöhten Summenspiegeln von Venlafaxin und ODV führen, wodurch verstärkt Venlafaxin-assoziierte Arzneimittelnebenwirkungen auftreten können [10]. Dies sollte in der antidepressiven Kombinationstherapie bedacht werden. Venlafaxin und ODV sind Substrate des Effluxtransporters P-Glykoprotein (P-gp) [9]. Propafenon hemmt sowohl CYP2D6 als auch P-gp [9]. In einer Kasuistik löste das Antiarrhythmikum Halluzinationen und psychomotorische Überaktivität aus. Es wird neben höheren Plasmaspiegeln auch eine gesteigerte Exposition des Antidepressivums im Zentralnervensystem durch die Blockade des Effluxtransporters in der Blut-Hirn-Schranke angenommen [3].
Klinisch relevante CYP2D6-Induktoren sind nicht bekannt (Abb. 2).
Welchen Einfluss CYP2C19, das wie CYP2D6 auch polymorph exprimiert wird, auf die klinische Wirksamkeit von Venlafaxin hat, bedarf zur Klärung weiterer Untersuchungen [11]. Die Komedikation mit CYP2C19-Hemmern wie Omeprazol erhöht signifikant die Exposition von Venlafaxin und ODV, wenn auch geringer als durch CYP2D6-Inhibitoren [7]. Sind CYP2D6 und CYP2C19 durch genetische Ursachen oder durch Arzneimittelinteraktionen in ihrer Aktivität verändert, kann dies Einfluss auf die Wirksamkeit und Verträglichkeit unter einer Venlafaxin-Normdosis haben. Unter diesen Voraussetzungen können auch starke CYP3A4-Modulatoren wie Ketoconazol möglichweise klinisch relevante Interaktionen auslösen [8].
Duloxetin
Duloxetin wird in vitro über CYP1A2 und CYP2D6 zu pharmakologisch inaktiven Metaboliten verstoffwechselt. Gemäß pharmakokinetischen Interaktionsstudien wird Duloxetin bevorzugt über CYP1A2 abgebaut [6]. In Kombination mit dem starken CYP1A2-Inhibitor Fluvoxamin erhöht sich der AUC-Wert von Duloxetin um das 5,6-Fache. Der starke CYP2D6-Hemmer Paroxetin hingegen erhöht die Duloxetin-Exposition um nur 60% [6]. Starke CYP1A2-Hemmer (Abb. 2) sind folglich unter einer Duloxetin-Therapie kontraindiziert, wobei die Fachinformation explizit auch Ciprofloxacin mit aufführt [2]. Rauchen induziert die Bildung von CYP1A2. Dies führt zu einer Senkung der Duloxetin-Plasmaspiegel um 30% im Vergleich mit Werten nichtrauchender Patienten [6].
Duloxetin erhöht die AUC des CYP2D6-Testsubstrats Desipramin um das 3-Fache. Vorsicht ist daher geboten bei Anwendung von Duloxetin mit Arzneimitteln, deren Metabolismus hauptsächlich von CYP2D6 abhängt (z.B. Flecainid, Propafenon und Metoprolol) [2].
Milnacipran
Milnacipran wird CYP-unabhängig abgebaut [1]. Somit sind keine metabolisch bedingten pharmakokinetischen Wechselwirkungen zu beachten.
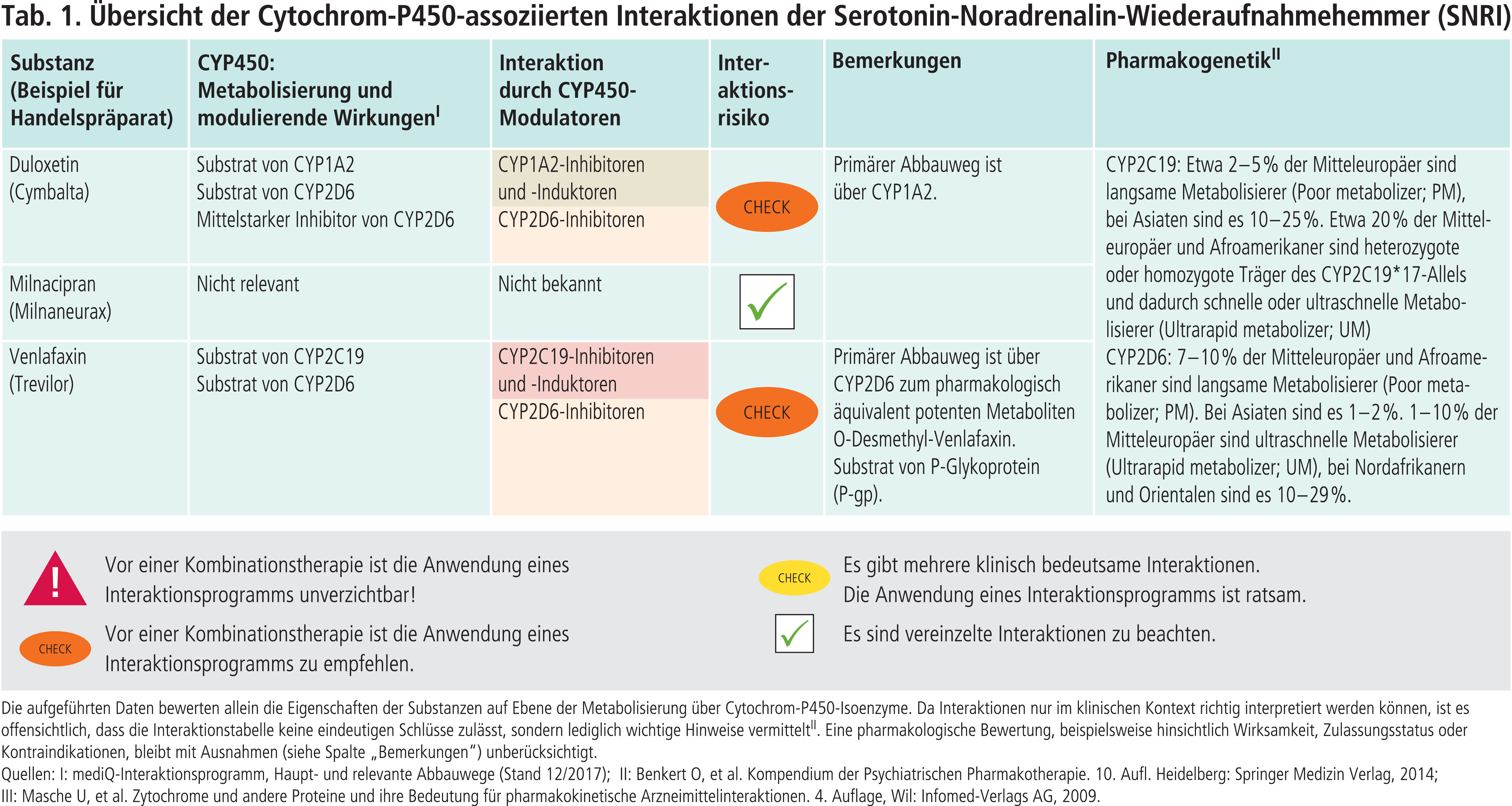
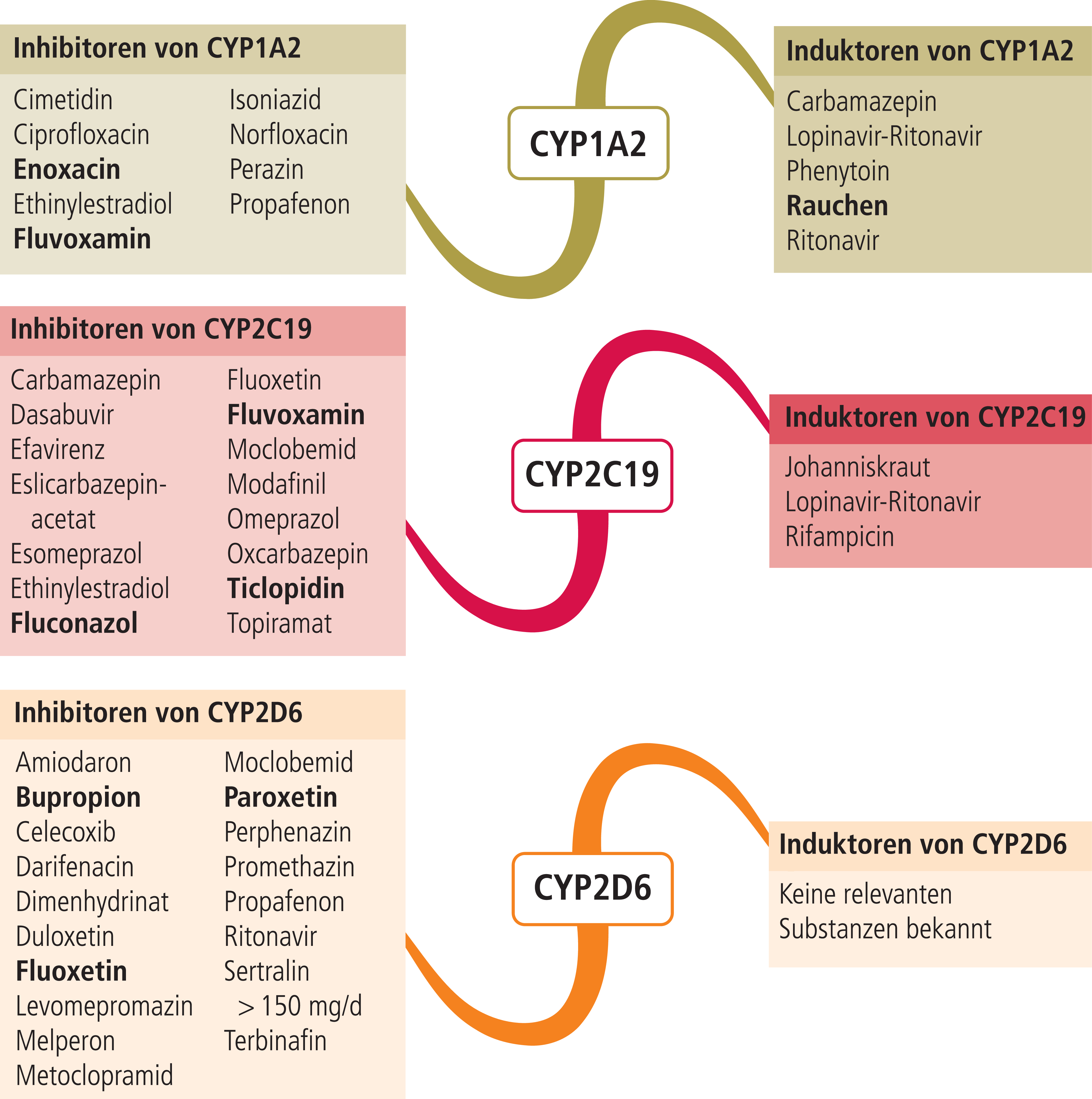
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2C19 und 2D6 (Stand: 12/2017) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Briley M. Milnacipran, a well-tolerated specific serotonin and norepinephrine reuptake inhibiting antidepressant. CNS Drugs 1998;4:137–48.
2. Fachinformation Cymbalta®. Stand: Januar 2016.
3. Gareri P, De Fazio P, Galleli, L. Venlafaxine-propafenone interaction resulting in hallucinations and psychomotor agitation. Ann Pharmacother 2008;42:434–8.
4. Hiemke C, Baumann P, Bergemann N, et al. AGNP-Konsensus-Leitlinien für therapeutisches Drug Monitoring in der Psychiatrie: Update 2011. Psychopharmakotherapie 2012;19:91–122.
5. Hynninen VV, Olkkola KT, Bertilsson L. et al. Effect of terbinafine and voriconazole on the pharmacokinetics of the antidepressant venlafaxine. Clin Pharmacol Ther 2008;83:342–8.
6. Knadler MP, Lobo E, Chappell J, et al. Duloxetine. Clin Pharmacokinet 2011;50:281–94.
7. Kuzin M, Schoretsanitis G, Haen E, et al. Effects of the proton pump inhibitors omeprazole and pantoprazole on the cytochrome P450-mediated metabolism of venlafaxine. Clin Pharmacokinet 2017. DOI 10.1007/s40262-017-0591-8.
8. Lindh JD, Annas A, Meurling L, et al. Effect of ketoconazole on venlafaxine plasma concentrations in extensive and poor metabolisers of debrisoquine. Eur J Clin Pharmacol 2003;59:401–6.
9. Magalhães P, Alves G, LLerena A, et al. Clinical drug-drug interactions: focus on venlafaxine. Drug Metab Pers Ther 2015;30:3–17.
10. Paslakis G, Gilles M, Deuschle M. Clinically relevant pharmacokinetic interaction between venlafaxine and bupropion. J Clin Psychopharmacol 2010;30:473–4.
11. Sangkuhl K, Stingl JC, Turpeinen M, et al. PharmGKB summary: venlafaxine pathway. Pharmacogenet Genomics 2014;24:62–72.
12. Schwabe U, Paffrath D, Ludwig WD, et al. Arzneiverordnungs-Report 2017. Berlin, Heidelberg: Springer Verlag GmbH, 2017.
13. Swen JJ, Nijenhuis M., de Boer A, et al. Pharmacogenetics: from bench to byte – an update of guidelines. Clinical pharmacology and therapeutics 2011;89:662–73.
*Nachdruck aus Krankenhauspharmazie 2018;39:21–4.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2018; 25(01):40-43