Holger Petri, Bad Wildungen*
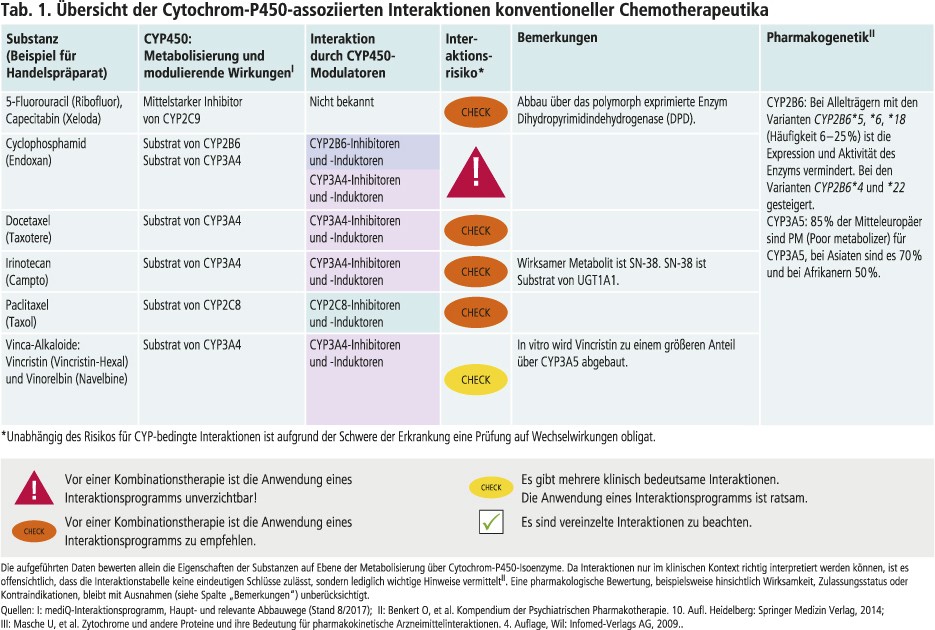
Cyclophosphamid
Cyclophosphamid ist ein Prodrug. An der Bioaktivierung und an der Detoxifikation sind Cytochrom-P450(CYP)-Isoenzyme maßgeblich beteiligt. Das polymorph exprimierte Enzym CYP2B6 hat den größten Anteil an der Bildung des aktiven Metaboliten 4-Hydroxycyclophosphamid, das weiter zum letztlich zytotoxischen Phosphoramidlost abgebaut wird. Über CYP3A4 findet primär die Detoxifikation statt [4]. Modulatoren dieser beiden Isoenzyme können somit Einfluss auf die Wirksamkeit und Verträglichkeit einer Cyclophosphamid-Therapie haben. Carbamazepin und Phenytoin sind Induktoren von CYP2B6 und 3A4 (Abb. 1). Bei einem Patienten unter Hochdosischemotherapie verursachte Carbamazepin an Tag 1 einen Anstieg der AUC (Fläche unter der Konzentrations-Zeit-Kurve) von 4-Hydroxycyclophosphamid um 58% [7]. In einem anderen Fallbericht steigerte Phenytoin die Bildung von 4-Hydroxycyclophosphamid um 51% und die maximale Plasmakonzentration war um 600% erhöht. In der Konsequenz wurde die Absolutdosis von Cyclophosphamid von 2835 mg auf 1500 mg gesenkt, ohne dass sich dies negativ auf den Behandlungserfolg ausgewirkt hätte [5].
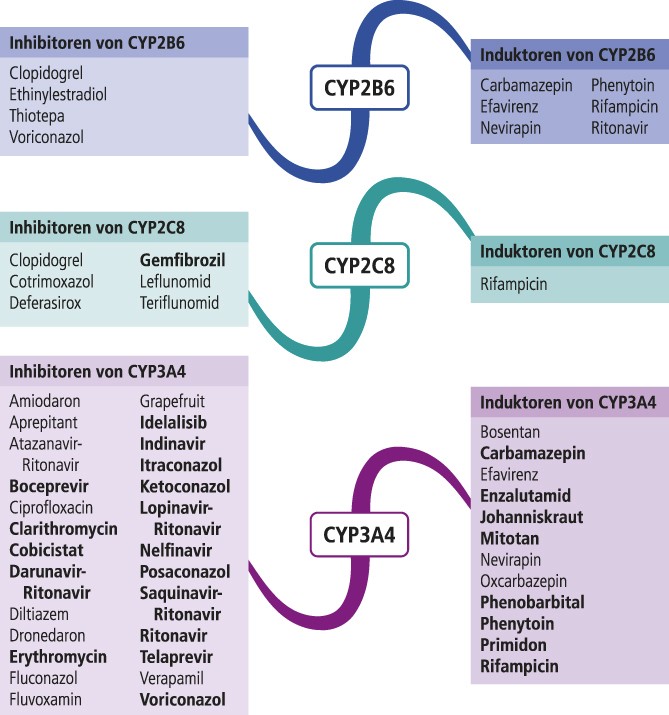
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2B6, 2C8 und 3A4 (Stand: 8/2017) [Quelle: mediQ-Interaktionsprogramm]
5-Fluorouracil und Capecitabin
Der Antimetabolit 5-Fluorouracil und sein Prodrug Capecitabin sind potente Hemmer von CYP2C9. Das Antikonvulsivum Phenytoin wird primär über CYP2C9 abgebaut. Die Kombination mit den beiden Antimetaboliten führte in Fallberichten bis zu einem vierfachen Anstieg der Phenytoin-Exposition mit hieraus folgenden Intoxikationen [2]. Auch Vitamin-K-Antagonisten wie Warfarin sind Substrate von CYP2C9. Die beiden Fluoropyrimidine können die Antikoagulanzien-Plasmaspiegel und somit das Blutungsrisiko erhöhen [11]. Andere Substrate von CYP2C9 sind die Sulfonylharnstoff-Antidiabetika Glibenclamid und Glimepirid [17]. Es besteht die Gefahr von Hypoglykämien. 5-Fluorouracil wird CYP-unabhängig über das polymorphe Enzym Dihydropyrimidindehydrogenase (DPD) abgebaut [14].
Irinotecan
Der Topoisomerase-I-Inhibitor Irinotecan wird zu etwa 3% über Carboxylesterasen zum Metaboliten SN-38 verstoffwechselt, dessen antineoplastische Aktivität 100- bis 1000-mal höher ist als die seiner Muttersubstanz. SN-38 wird weiter über die Uridin-5’-diphosphat-Glucuronosyltransferase (UGT) 1A1 abgebaut. Andere inaktive Metaboliten werden über CYP3A4 gebildet [20].
Ketoconazol ist ein starker Inhibitor von CYP3A4 (Abb. 1) und hemmt UGT1A1 [23]. Ketoconazol verminderte in einer Studie die AUC des über CYP3A4 gebildeten oxidativen Hauptmetaboliten APC um 87%, während die AUC von SN-38 um 109% erhöht war [12]. Johanniskraut induziert CYP3A4 und UGT1A1. In einer Interaktionsstudie mit fünf Patienten senkte Johanniskraut die Plasmakonzentration von SN-38 um 42% und sollte daher in Kombination mit Irinotecan vermieden werden [13].
Eine auf dem CYP3A4-Phänotyp basierende Dosisadjustierung von Irinotecan führte in einer Untersuchung zu einem selteneren Auftreten schwerer Neutropenien und Blutbildstörungen [20].
Docetaxel und Paclitaxel
Docetaxel wird durch CYP3A4 metabolisiert. Der starke CYP3A4-Inhibitor Ketoconazol senkte in einer Studie mit sieben Patienten die Docetaxel-Clearance um 49% [8]. Die Hemmung des Abbaus von Docetaxel durch Ritonavir wird für schwerwiegende Nebenwirkungen des Taxans bei drei HIV-Patienten verantwortlich gemacht [15]. In einem Tierversuch erhöhte sich die AUC von Docetaxel durch Ritonavir und Ketoconazol um das 6,9- bzw. 3,1-Fache. CYP3A4-Induktoren wie Dexamethason und Efavirenz blieben ohne nennenswerten Effekt auf pharmakokinetische Parameter [19]. In einer Studie mit Johanniskraut sank die AUC in Kombination mit Docetaxel um 12% [9].
Paclitaxel wird primär über CYP2C8 und nachgeordnet über CYP3A4 abgebaut. Das Nebenwirkungsprofil ist bei gleichzeitiger Medikation von CYP2C8-Inhibitoren verschlechtert. Beispielsweise verdoppelt der Glucuronid-Metabolit von Clopidogrel als potenter CYP2C8-Inhibitor das Risiko für schwerwiegende periphere Neuropathien und Blutbildstörungen bei einer höheren Taxandosis [1]. CYP3A4-hemmende Proteasehemmer führten in einer Pilotstudie zu einer erhöhten Paclitaxel-Exposition. Hierdurch wurden aber weder Verträglichkeit noch Wirksamkeit beeinflusst [3].
Vinca-Alkaloide
Vinca-Alkaloide wie Vincristin und Vinorelbin werden über CYP3A4 verstoffwechselt [18]. Die Komedikation von Azol-Antimykotika, mit Ausnahme von Fluconazol, führt bei Patienten unter Vincristin-Therapie häufiger zu schweren neurotoxischen Nebenwirkungen. Die CYP3A4-hemmende Potenz von Fluconazol ist schwächer ausgeprägt als die anderer Azole [16]. Die CYP3A4-induzierenden Antikonvulsiva Carbamazepin und Phenytoin senken die AUC von Vincristin durchschnittlich um 43% [22].
CYP3A5 hat in vitro einen größeren Anteil am Abbau von Vincristin als CYP3A4 [18]. Es gibt Hinweise, dass Patienten, die CYP3A5 exprimieren, ein niedrigeres Risiko neurotoxischer Nebenwirkungen aufweisen [6]. In der mitteleuropäischen Bevölkerung sind zum Teil über 90% Träger von Genen, die für funktionsdefiziente Enzyme kodieren [21]. In einer Untersuchung mit pädiatrischen Patienten ließ sich kein Zusammenhang feststellen zwischen dem Polymorphismus von CYP3A5 und dem Vincristin-Plasmaspiegel [10]. Welchen Einfluss CYP3A5-bedingte Interaktionen auf den Vincristin-Plasmaspiegel haben, bleibt somit noch zu klären.
Fazit
Der Stoffwechsel über CYP-Enzyme ist für den Metabolismus zahlreicher konventioneller Zytostatika von Bedeutung. Der Toxizität der Substanzen ist es geschuldet, dass Daten aus klassischen Interaktionsstudien in der Regel mit gesunden, jungen Probanden fehlen. Die Übertragbarkeit der aus In-vitro- und Tierstudien gewonnenen Erkenntnisse auf den Menschen ist nicht ohne Weiteres möglich. Alternativ können In-vivo-Studien mit Patienten und Kasuistiken nützliche Hinweise zu potenziell klinisch relevanten Interaktionen bieten. Hierzu ist es notwendig, die Literatur auf diesbezügliche Signale fortlaufend zu beobachten. Für die in diesem Beitrag nicht beschriebenen konventionellen Zytostatika gilt bei der Bewertung CYP-bedingter Interaktionen in Analogie die Fragestellung, welche Bedeutung die Metaboliten für Wirkung und Verträglichkeit haben. Nicht zuletzt spielen zahlreiche weitere Vorgänge in der Biotransformation (Phase-II-Reaktionen, Verteilung durch Arzneimitteltransporter) eine Rolle, sodass zu jeder individuellen Therapie aufgrund der Schwere der Erkrankung eine Interaktionsprüfung obligat ist.
Literatur
1. Agergaard K, Mau-Sørensen M, Stage TB, et al. Clopidogrel-paclitaxel drug-drug interaction: a pharmacoepidemiologic study. Clin Pharmacol Ther 2017 Feb 22 doi: 10.1002/cpt.674.
2. Brickell K, Porter D, Thompson P. Phenytoin toxicity due to fluoropyrimidines (5FU/capecitabine): three case reports Br J Cancer 2003;89:615–6.
3. Cianfrocca M, Lee S, Von Roenn J, et al. Pilot study evaluating the interaction between paclitaxel and protease inhibitors in patients with human immunodeficiency virus-associated Kaposi’s sarcoma: an Eastern Cooperative Oncology Group (ECOG) and AIDS Malignancy Consortium (AMC) trial. Cancer Chemother Pharmacol 2011;68:827–33.
4. De Jonge ME, Huitema ADR, Rodenhuis S, et al. Clinical pharmacokinetics of cyclophosphamide, Clin Pharmacokinet 2005;44: 1135–64.
5. De Jonge ME, Huitema ADR, van Dam SM, et al. Significant induction of cyclophosphamide and thiotepa metabolism by phenytoin. Cancer Chemother Pharmacol 2005;55: 507–10.
6. Egbelakin A, Ferguson MJ, MacGill EA, et al. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011;56:361–7.
7. Ekhart C, Rodenhuis S, Beijnen JH, et al. Carbamazepine induces bioactivation of cyclophosphamide and thiotepa. Cancer Chemother Pharmacol 2009;63:543–7.
8. Fachinformation Taxotere®. Stand: Mai 2017.
9. Goey AK, Meijerman I, Rosing H, et al. The effect of St John‘s wort on the pharmacokinetics of docetaxel. Clin Pharmacokinet 2014;53:103–10.
10. Guilhaumou R, Simon N, Quaranta S, et al. Population pharmacokinetics and pharmacogenetics of vincristine in paediatric patients treated for solid tumour diseases. Cancer Chemother Pharmacol 2011;68:1191–8.
11. Gunes A, Coskun U, Boruban C, et al. Inhibitory effect of 5-fluorouracil on cytochrome P450 2C9 activity in cancer patients. Basic Clin Pharmacol Toxicol 2006;98:197–200.
12. Kehrer DF, Mathijssen RH, Verweij J, et al. Modulation of irinotecan metabolism by ketoconazole. J Clin Oncol 2002;20:3122–9.
13. Mathijssen RH, Verweij J, de Bruijn P, et al. Effects of St. John‘s wort on irinotecan metabolism. J Natl Cancer Inst 2002;94: 1247–9.
14. Mazzuca F, Borro M, Botticelli A, et al. Pre-treatment evaluation of 5-fluorouracil degradation rate: association of poor and ultra-rapid metabolism with severe toxicity in a colorectal cancer patients cohort. Oncotarget 2016;7:20612–20.
15. Mir O, Dessard-Diana B, Louet AL, et al. Severe toxicity related to a pharmacokinetic interaction between docetaxel and ritonavir in HIV-infected patients. Br J Clin Pharmacol 2010;69:99–101.
16. Moriyama B, Henning SA, Leung J, et al. Adverse interactions between antifungal azoles and vincristine: review and analysis of cases. Mycoses 2012;55:290–7.
17. Petri H. Das Interaktionspotenzial der oralen Antidiabetika. Krankenhauspharmazie 2017; 38:42–6.
18. PharmGKB. Vinka Alkaloid Pathyway, Pharmacokinetics. www.pharmgkb.org/pathway/PA150981002 (Zugriff 18.07.2017).
19. Rudek MA, Chang CY, Steadman K, et al. Combination antiretroviral therapy (cART) component ritonavir significantly alters docetaxel exposure. Cancer Chemother Pharmacol 2014;73:729–36.
20. Van der Bol JM, Mathijssen RH, Creemers GJ, et al. A CYP3A4 phenotype-based dosing algorithm for individualized treatment of irinotecan. Clin Cancer Res 2010;16:736–42.
21. Van Schaik RH, van der Heiden IP, van den Anker JN, et al. CYP3A5 variant allele frequencies in Dutch Caucasians. Clin Chem 2002;48:1068–71.
22. Villikka K, Kivistö KT, Mäenpää H, et al. Cytochrome P450-inducing antiepileptics increase the clearance of vincristine in patients with brain tumors. Clin Pharmacol Ther 1999;66:589–93.
23. Yong WP, Ramirez J, Innocenti F, et al. Effects of ketoconazole on glucuronidation by UDP-glucuronosyltransferase enzymes. Clin Cancer Res 2005;11:6699–704.
*Nachdruck aus Krankenhauspharmazie 2017;38:434–7.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2017; 24(05)