Holger Petri, Bad Wildungen*
Konventionelle Basistherapeutika
Antimalariamittel
Chloroquin und Hydroxychloroquin sind primär zur Behandlung von Plasmodien-Infektionen entwickelt worden. Zum Metabolismus von Chloroquin liegen nur Ergebnisse aus In-vitro-Untersuchungen vor. Demzufolge wird Chloroquin primär über CYP2C8 und 3A4 verstoffwechselt [10]. Chloroquin kann potenziell lebensbedrohliche Torsade-de-pointes(TdP)-Arrhythmien auslösen [18]. Leflunomid hemmt über seinen Metaboliten Teriflunomid CYP2C8 (Abb. 1), wodurch die Chloroquin-Plasmaspiegel steigen können. Antiinfektiva wie Clarithromycin, Erythromycin und Fluconazol sind als CYP3A4-Inhibitoren selbst mit einem hohen TdP-Risiko behaftet [18]. Zum Abbau von Hydroxychloroquin über CYP-Isoenzyme existieren keine evidenten Daten. Hydroxychloroquin hemmt den Metabolismus des CYP2D6-Testsubstrats Metoprolol. In einer Untersuchung mit Probanden erhöhte sich die AUC (Fläche unter der Konzentrations-Zeit-Kurve) des Betablockers um 65% [15]. Somit ist Vorsicht geboten bei Kombinationstherapien mit Substanzen, deren Abbau klinisch relevant von der CYP2D6- Funktionalität abhängt.
Sulfasalazin
Sulfasalazin wird CYP-unabhängig metabolisiert. Es ist Substrat des Effluxtransporters BCRP (Breast Cancer Resistance Protein), neben P-gp (P-Glykoprotein) der wichtigste ABC(ATP-binding-cassette)-Transporter. Durch Blockade von BCRP können die Plasmaspiegel von Sulfasalazin steigen. Ceritinib, Gefitinib und Lapatinib sind beispielsweise Arzneimittel mit BCRP-hemmenden Eigenschaften [14].
Immunsuppressiva/ antineoplastische Mittel Leflunomid
Leflunomid ist ein Prodrug. Die therapeutische Wirksamkeit beruht auf der Bildung des aktiven Metaboliten Teriflunomid. Für dessen Bildung sind die CYP-Isoenzyme 1A2, 2C19 und 3A4 verantwortlich. Es entstehen zudem toxische Metabolite, die maßgeblich für die Therapieabbruchrate von bis zu 40% verantwortlich sind [9]. Für die Detoxifikation von Leflunomid ist die Funktionalität von CYP1A2 und 2C19 von Bedeutung. Patienten mit einer verminderten Stoffwechselleistung eines der beiden polymorphen CYP-Enzyme haben ein höheres Risiko, die Therapie aufgrund von Nebenwirkungen abzubrechen. Homo- und heterozygote C-Allel-Träger des CYP1A2*1F-Genotyps exprimieren Enzyme mit verminderter Aktivität. Das Absetzrisiko ist hierdurch 2,29- bis 9,7-fach erhöht [9]. Langsame und intermediäre Metabolisierer von CYP2C19 beenden eine Leflunomid-Therapie aufgrund nicht tolerabler Beschwerden im Vergleich zu ultraschnellen Metabolisierern mit 53,3% gegenüber 23,8% deutlich häufiger [17]. Leflunomid ist eine gleichwertig wirksame Methotrexat- Alternative und preisgünstiger als Biologika. Da die Risikofaktoren für eine Unverträglichkeit bekannt sind, kann vor Therapiebeginn eine Genotypisierung/Phänotypisierung angebracht sein. Inhibitoren von CYP1A2 und CYP2C19 erhöhen das Risiko für einen nebenwirkungsbedingten Therapieabbruch und sollten gemieden werden.
Cylophosphamid
Cyclophosphamid ist ein Prodrug. Das Zytostatikum wird über verschiedene CYP-Enzyme zu aktiven und toxischen Metaboliten verstoffwechselt. CYP2B6 hat den größten Anteil bei der Bioaktivierung, CYP3A4 an der Detoxifikation. Modulatoren dieser beiden Isoenzyme können somit Einfluss haben auf Wirksamkeit und Verträglichkeit [2].
Ciclosporin
Das Immunsuppressivum Ciclosporin wird primär über CYP3A4 abgebaut. Induktoren wie Rifampicin beschleunigen den intestinalen und hepatischen Metabolismus von Ciclosporin. Als Folge müssen die Ciclosporin-Dosen bis auf das 3- bis 5-Fache erhöht werden. CYP3A4-hemmende Antiinfektiva wie Clarithromycin, Itraconazol und Voriconazol können die Ciclosporin-Exposition mehr als verdoppeln, Telaprevir erhöht die AUC um das 4,6-Fache [6].
Zu weiteren pharmakokinetischen Interaktionen kommt es durch Inhibition des Effluxtransporters P-gp und des Influxtransporters OATP1B1 (Organic anion-transporting polypeptide 1B1). Vorsicht ist geboten bei Kombination mit Arzneimitteln, die Substrate dieser Arzneimitteltransporter sind und eine geringe therapeutische Breite haben. Beispielsweise ist Ciclosporin unter Anwendung des Nicht-Vitamin-K-Antagonisten Dabigatranetexilat, einem P-gp-Substrat, kontraindiziert [4]. OATP1B1 ist ein hepatischer Influxtransporter, über den HMG-CoA-Reduktase-Inhibitoren in die Leberzelle aufgenommen werden, woran sich die metabolischen Stoffwechselprozesse anschließen. Ciclosporin ist unter einer Simvastatin-Therapie kontraindiziert, Atorvastatin soll nicht höher als 10 mg pro Tag dosiert werden [7, 8].
Azathioprin
Azathioprin ist ein Prodrug von 6-Mercaptopurin (6-MP) [3]. Das polymorphe Enzym Thiopurin-S-Methyltransferase (TPMT) ist mitverantwortlich für die Entgiftung von Thiopurinwirkstoffen. 10% der kaukasischen Population sind heterozygote Träger eines funktionsdefizienten Enzyms. Aminosalicylsäurederivate wie Sulfasalazin blockieren TPMT. Dadurch kann das Risiko für myelotoxische Nebenwirkungen besonders bei diesen Patienten aufgrund einer zusätzlichen Abbauhemmung erhöht sein [11, 16].
Methotrexat
Methotrexat unterliegt keiner CYP-abhängigen Metabolisierung. Die Pharmakokinetik von Methotrexat im Niedrigdosisbereich ist primär abhängig von Efflux- und Influxtransportern [12]. Hierdurch sind zahlreiche Interaktionen möglich.
Biologika
Bei Patienten mit chronisch entzündlichen Erkrankungen kann sich die Pharmakokinetik von Arzneistoffen verändern [1, 13]. Grund hierfür ist unter anderem eine Zytokin-vermittelte Unterdrückung der CYP-Bildung. In-vitro-Studien mit kultivierten menschlichen Hepatozyten zeigten, dass Interleukin(IL)-6 einen Rückgang der CYP1A2, 2C9-, 2C19- und 3A4-Enzymexpression bewirkte. Der IL-6-Hemmer Tocilizumab normalisiert die Bildung dieser Enzyme [5]. So beeinflussen Biologika indirekt die Pharmakokinetik der Komedikamente. Bei Einleitung einer Therapie mit Biologika sollte dies berücksichtigt werden besonders bei Patienten, die Arzneimittel mit enger therapeutischer Breite einnehmen. Die gleichen Vorsichtsmaßnahmen sollten generell bei konventionellen und auch bei der neuen Generation von Basisantirheumatika, den Januskinase-Inhibitoren, gelten.

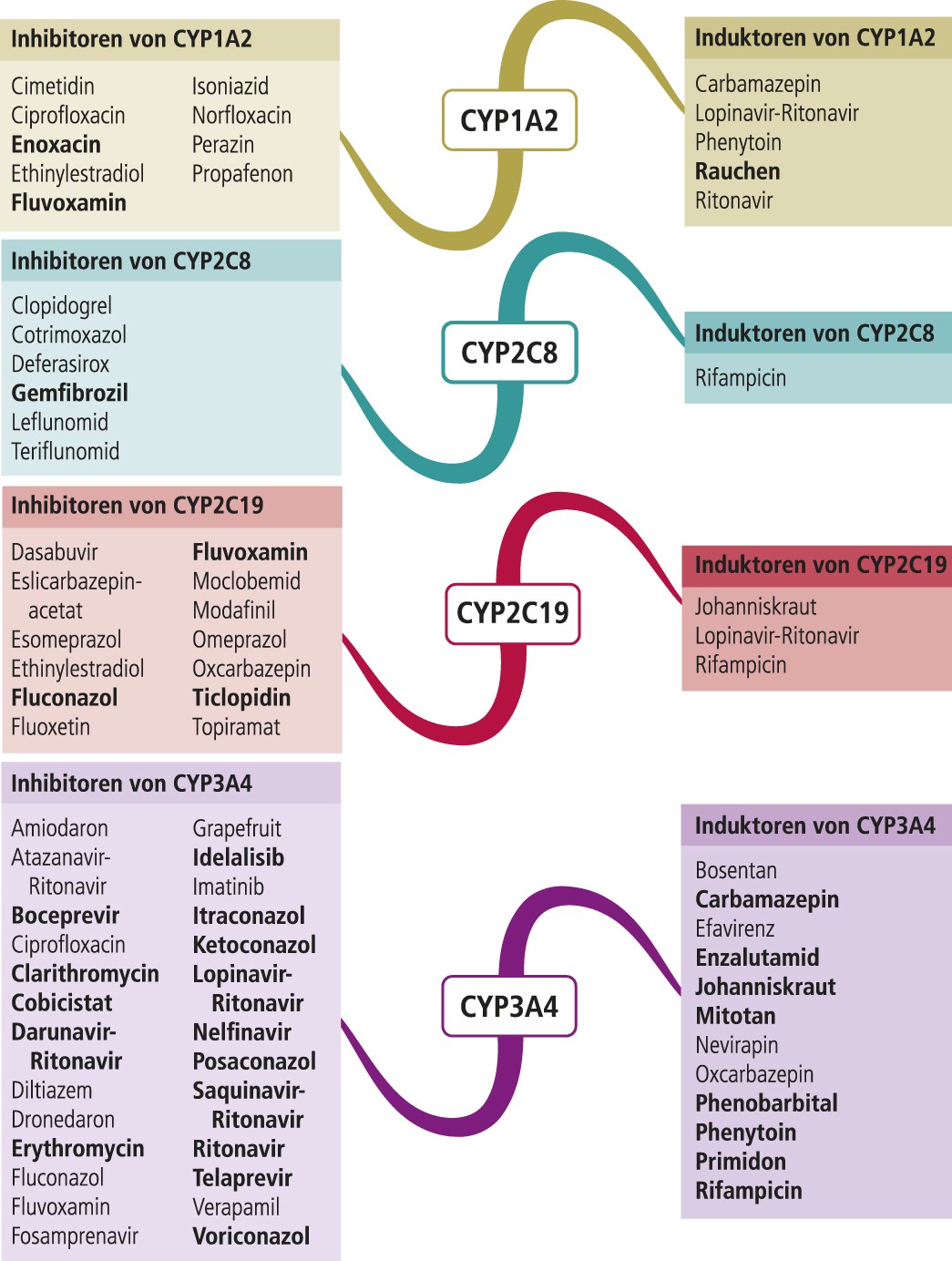
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 1A2, 2C8, 2C19 und 3A4 (Stand: 6/2017) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Christensen H, Hermann, M. Immunological responses as a source to variability in drug metabolism and transport. Front Pharmacol 2012;3:8. doi: 10.3389/fphar.2012.00008.
2. De Jonge ME, Huitema AD, Rodenhuis S, Beijnen JH. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 2005;44:1135-64.
3. Fachinformation Imurek®. Stand: Dezember 2013.
4. Fachinformation Pradaxa®. Stand: Februar 2017.
5. Fachinformation Roactemra®. Stand: Juli 2016
6. Fachinformation Sandimmun®. Stand: Juli 2015.
7. Fachinformation Sortis®. Stand: November 2016.
8. Fachinformation Zocor®. Stand: Juni 2015.
9. Hopkins AM, Wiese MD, Proudman SM, et al. Genetic polymorphism of CYP1A2 but not total or free teriflunomide concentrations is associated with leflunomide cessation in rheumatoid arthritis. Br J Clin Pharmacol 2016;81:113–23.
10. Kim KA; Park JY, Lee JS, et al. Cytochrome P4502C8 und CYP3A4/5 are involved in chloroquine metabolism in human liver microsomes. Arch Pharm Res 2003;26:631–7.
11. Lennard L. Clinical implications of thiopurine methyltransferase-optimization of drug dosage and potential drug interactions. Ther Drug Moni 1998;20:527–31.
12. Mikkelsen TS, Thorn CF, Yang JJ, et al. PharmGKB summary:methotrexate pathway. Pharmacogenet Genomics 2011;21:679–86.
13. Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther 2009;85:434–8.
14. Petri, H. Pharmakokinetik von Onkologika. Dtsch Arztebl 2017;114:26–8 www.aerzteblatt.de/pdf.asp?id=186148 (Zugriff am 30.05.2017).
15. Somer M, Kallio J, Pesonen U, et al. Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br J Clin Pharmacol 2000;49:549–54.
16. Szumlanski CL, Weinshilboum RM. Sulphasalazine inhibition of thiopurine methyltransferase: possible mechanism for interaction with 6-mercaptopurine and azathioprine. Br J Clin Pharmacol 1995;39:456–9.
17. Wiese MD, Schabl M, O‘Doherty C, et al. Polymorphisms in cytochrome P4502C19 enzyme and cessation of leflunomide in patients with rheumatoid arthritis. Arthritis Res Ther 2012;14:R163.
18. www.crediblemeds.org (Zugriff am 03.05.2017).
*Nachdruck aus Krankenhauspharmazie 2017;38:350–3.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2017; 24(04)