Karl Messlinger, Erlangen, Dagny Holle-Lee, Essen, und Hans-Christoph Diener, Essen
Historische Theorien und die Entdeckung von CGRP als Schlüsselmediator
Die Entwicklung der Pharmakotherapie der Migräne ist eng mit den historischen Migränetheorien verbunden. Bereits in den 30er-Jahren des letzten Jahrhunderts hatte man entdeckt, dass Ergotamintartrat, ein unspezifischer 5-HT1-Agonist, bei Migräneanfällen nicht nur den Schmerz verminderte, sondern gleichzeitig auch die Dilatation und Pulsation der Arteria temporalis reduzierte [92]. Daraus schloss man, dass der Schmerz bei „vaskulären Kopfschmerzen“ durch Erweiterung intra- und extrakranialer Gefäße zustande kommt, indem die umgebenden trigeminalen Nervenfasern gereizt werden. Von dieser vaskulären Theorie als alleinige Erklärung für den Migräneschmerz rückte man in der Folgezeit langsam ab, da es immer mehr Widersprüche gab, zum Beispiel die fehlende Korrelation zwischen der durch Bildgebung gemessenen Arterienweite und dem Kopfschmerz [84]. Die vaskuläre Hypothese regte aber wesentlich die Entwicklung spezifischer 5-HT1B/D-Agonisten (Triptane) als Migränetherapeutika an, darunter als erste klinisch verwendete Substanz das Sumatriptan, das nach wie vor in verschiedenen Formulierungen eingesetzt wird [54]. In den 80er-Jahren wurde bei tierexperimentellen Untersuchungen entdeckt, dass Ergotamine und Sumatriptan die Plasmaextravasation aus der Dura mater encephali hemmen. Dies begründete die nächste Theorie zur Migräneentstehung, die Theorie der neurogenen Entzündung [14, 82]. Neurogene Entzündungsvorgänge werden durch die Freisetzung von proinflammatorischen Neuropeptiden aus aktivierten Nervenfasern ausgelöst, wenn man zum Beispiel das Ganglion trigeminale elektrisch stimuliert. Die Plasmaextravasation als wichtigstes Element der neurogenen Entzündung kommt hauptsächlich durch die Wirkung von Substanz P aus afferenten Fasern zustande. Man setzte zunächst große Hoffnungen darauf, dass sich Migräneschmerz durch Blockade der Substanz-P-(NK1-)Rezeptoren und Hemmung der Plasmaextravasation behandeln lässt [3]. Als sich aber neu entwickelte Hemmsubstanzen der Plasmaextravasation bei Migräne als unwirksam erwiesen [71, 81], konnte auch die neurogene Entzündungstheorie der Migränentstehung in ihrer ursprünglichen Form nicht weiter aufrechterhalten werden. Bei den Untersuchungen zu neurogenen Entzündungsvorgängen wurde man auf das mit Substanz P kolokalisierte Calcitonin gene-related peptide (CGRP) aufmerksam. Es wurde durch Stimulation des Ganglion trigeminale in den Sinus sagittalis superior freigesetzt. Dieser Vorgang ließ sich durch Dihydroergotamin und Sumatriptan vermindern [13]. Die damals praktizierte Behandlung einer ganz anderen Schmerzerkrankung, der Trigeminusneuralgie, führte auch beim Menschen auf die Spur des CGRP: Die Thermokoagulation des Ganglion trigeminale löste eine ipsilaterale Rötung des Gesichts aus, und im Jugularvenenblut der Patienten fand man gleichzeitig hohe Plasmaspiegel von CGRP [36]. So lag der Gedanke nahe, dass auch die spontane Aktivierung trigeminaler Afferenzen mit einer erhöhten CGRP-Freisetzung einhergehen könnte. Tatsächlich wurden daraufhin auch bei Migräne- und Clusterattacken erhöhte Plasmaspiegel von CGRP im venösen Abstromgebiet des Kopfes gefunden, die sich nach erfolgreicher Therapie durch Sumatriptan normalisierten [37]. Seither konzentrierte man sich auf die neuronalen und vaskulären Wirkungen von CGRP als Schlüsselmediator der Migräne. Da meningeale Blutgefäße mit ihrer trigeminalen Innervation an der Kopfschmerzentstehung beteiligt sind, spricht man häufig vom „trigeminovaskulären System“ und bezeichnet Migräne und Clusterkopfschmerz – unabhängig von der internationalen Kopfschmerzklassifikation – als vaskuläre Kopfschmerzen.
Bedeutung von CGRP bei Migräne und Clusterkopfschmerz
Dass CGRP als Schlüsselmediator beim Migräneschmerz und anderen primären Kopfschmerzen angesehen wird, hat weitere gute Gründe. Migränepatienten reagierten auf die Infusion geringer Mengen von CGRP mit verzögert auftretenden Kopfschmerzen, die häufig die diagnostischen Kriterien für einen Migräneanfall erfüllten, während bei gesunden Vergleichspersonen nur vorübergehend leichte Kopfschmerzen auftraten [41]. Aus Messungen des regionalen zerebralen Blutflusses und der Blutflussgeschwindigkeit in der Arteria cerebri media während der CGRP-Infusion konnte dabei allerdings keine signifikante Vasodilatation berechnet werden [61].
Das wichtigste Argument für die Schlüsselrolle von CGRP bei Migräne und Clusterkopfschmerz sind die klaren therapeutischen Effekte bei Hemmung der CGRP-Freisetzung oder der CGRP-Rezeptoren [28]. Man hatte zunächst erkannt, dass Dihydroergotamin und Sumatriptan durch Aktivierung hemmender 5-HT1B/D-Rezeptoren nicht nur Vasokonstriktion bewirken, sondern auch die Neuropeptidfreisetzung und dadurch die neurogene Entzündung hemmen [14]. Da Substanz P offensichtlich nicht die entscheidende Rolle spielte, konzentrierte man sich auf die Wirkungen des CGRP. CGRP-Rezeptorantagonisten wurden für die klinische Anwendung entwickelt und erreichten klinische Prüfungen bis zur Phase II (s.u.).
Vorkommen von CGRP und CGRP-Rezeptoren im trigeminovaskulären System
Das Neuropeptid CGRP mit den zwei Isoformen Alpha-CGRP und Beta-CGRP besteht aus 37 Aminosäuren und wird in Nervenzellen durch alternatives Spleißen bei der Expression des Calcitonin-Gens gebildet [1]. Neben CGRP gibt es weitere ähnliche Mitglieder der Calcitonin-Familie: Adrenomedullin (AM) und Amylin (AMY). CGRP wird im peripheren und im zentralen Nervensystem einschließlich des Gehirns exprimiert [101]. Im afferenten Nervensystem wird Alpha-CGRP von einem großen Teil der nozizeptiven Neuronen gebildet, die höchsten Konzentrationen findet man im Ganglion trigeminale [105]. In menschlichen Trigeminalganglien hat man bis zu 48% der Neurone als CGRP-positiv befunden [31]. Dies sind wahrscheinlich vor allem Afferenzen, welche die intrakranialen Blutgefäße innervieren [76]. Große intrazerebrale Arterien zeigen eine dichte CGRP-immunreaktive Innervation [26]. In der Dura mater wurde die Mehrzahl der CGRP-positiven Fasern in der Nähe von meningealen Arterien und den Sinus gefunden [57, 73, 89]. CGRP wird nach Aktivierung primärer Afferenzen aus peripheren und zentralen Fortsätzen durch Ca2+-abhängige Mechanismen freigesetzt [68] und kann auch im Liquor cerebrospinalis nachgewiesen werden [102].
Die Rezeptoren, an die CGRP bindet, sind Heteromere aus drei Proteinen: dem Calcitonin receptor-like Rezeptorprotein (CLR), dem kleinen Receptor activity modifying protein 1 (RAMP1) und dem intrazellulären Receptor component Protein (RCP). CLR und RAMP1 sind für die Bindung des CGRP-Moleküls notwendig, während RCP die Verbindung zur intrazellulären Signaltransduktionskaskade herstellt. Durch die Aktivierung einer Adenylylcyclase über ein Gs-Protein kommt es zum Anstieg der intrazellulären cAMP-Konzentration [33]. RAMP-Proteine sind für die Rezeptorspezifität der Calcitonin-Rezeptorfamilie bestimmend [72]. CLR zusammen mit RAMP1 bilden CGRP-Rezeptoren, zusammen mit RAMP2 oder RAMP3 Rezeptoren mit hoher Affinität für Adrenomedullin (AM), während die Kombination aus dem Calcitonin-Rezeptor (CTR) und einem der RAMP-Proteine Rezeptoren für Amylin (AMY) bildet. Der AMY1-Rezeptor, eine Kombination von CTR und RAMP1, bindet aber auch CGRP mit ähnlich großer Affinität wie CLR/RAMP1, sodass heute sogar von einem zweiten CGRP-Rezeptor gesprochen wird, dessen Aktivierung die gleiche intrazelluläre Signalkette auslöst [98].
CGRP-Rezeptoren werden vor allem in der glatten Gefäßmuskulatur von arteriellen Gefäßen, aber auch in trigeminalen Afferenzen und ihren Gliazellen exprimiert, soweit immunhistochemische Untersuchungen darüber Aufschluss geben. Immunreaktivität für die CGRP-Rezeptorkomponenten CLR und RAMP1 wurde in einem Teil der trigeminalen Neuronen gefunden, wobei eine Co-Lokalisation mit CGRP nur sehr selten zu beobachten war [31, 62]. Interessanterweise werden die CGRP-Rezeptoren nach diesen Befunden nur in die zentralen, aber nicht die peripheren Fortsätze der trigeminalen Afferenzen transportiert und eingebaut, was Schlussfolgerungen auf die Wirkungsmechanismen von CGRP-Rezeporantagonisten zulässt (s.u.). Außerdem wurde CLR- und RAMP1-Immunreaktivität in peripherer Glia (Schwannzellen) und Satellitenzellen im Ganglion trigeminale gefunden. Dies könnte eine von CGRP vermittelte Wechselwirkung zwischen diesen neuronalen Elementen nahelegen. Untersuchungen an trigeminalen Zellkulturen zeigen zum Beispiel, dass CGRP aus Ganglienzellen die umliegenden Satellitenzellen zur Genexpression und zur Freisetzung von Stickstoffmonoxid (NO) stimuliert [65, 96]. Umgekehrt kann NO die CGRP-Expression in trigeminalen Ganglienzellen stimulieren [5], CGRP-Rezeptorproteine hochregulieren [86] und so möglicherweise zu einer verstärkten CGRP-Freisetzung führen.
Wirkungen von CGRP im trigeminovaskulären System
CGRP bewirkt eine Relaxation glatter Gefäßmuskelzellen. CGRP ist der stärkste bekannte Vasodilatator humaner zerebraler Arterien [56]. Die arterielle Vasodilatation und verstärkte Durchblutung durch CGRP wurde in vielen tierexperimentellen Untersuchungen bestätigt [25, 38, 59, 100]. Diese Gefäßwirkung erinnert an die vaskuläre Theorie des Migräneschmerzes, da nach den klassischen intraoperativen Experimenten am Menschen (durch die Arbeitsgruppen um Harold Wolff und Wilder Penfield) die noxische Stimulation ausschließlich der arteriellen Blutgefäße der Dura mater und der großen intrazerebralen Arterien schmerzhaft war, während vom Gehirn selbst keine Empfindungen ausgelöst werden konnten [79]. Allerdings konnten im Tierexperiment trigeminale Ganglienzellen oder sekundäre Neurone im spinalen Trigeminuskern durch die Applikation von CGRP auf die Dura mater weder aktiviert noch sensibilisiert werden [35, 64], was auch nicht erstaunlich ist, da die peripheren Fortsätze der trigeminalen Afferenzen höchstwahrscheinlich keine CGRP-Rezeptoren tragen [62]. Kürzlich wurde vermutet, dass das Ganglion trigeminale ein Ort der CGRP-Wirkungen sein könnte [29], aber auch die Injektion von CGRP direkt in das Ganglion bewirkte keine akute Aktivitätsänderung bei spinalen trigeminalen Neuronen mit afferentem Zustrom aus der Dura mater [20].
Weitere Möglichkeiten für periphere CGRP-Wirkungen ergeben sich durch Expression von CGRP-Rezeptoren in nicht-neuronalen Zellen. Immunreaktivität für CGRP-Rezeptorproteine wurde bei der Ratte auch auf mononukleären Zellen und Mastzellen der Dura mater gefunden [32, 62]. CGRP bewirkt in hohen Konzentrationen Histamin-Freisetzung aus der Dura mater, was für die Degranulation von Mastzellen spricht [85]. Mastzelldegranulation aktiviert im Tierversuch nach einer Verzögerung von mehreren Minuten einen Teil der meningealen Nozizeptoren [63]. Leider gibt es keine tierexperimentellen Kopfschmerzmodelle, mit denen man Langzeitveränderungen durch CGRP-Wirkung zuverlässig untersuchen kann. In den Zellkulturexperimenten benötigen die durch CGRP induzierten Veränderungen wie verstärkte NO-Synthese mehrere Stunden, was dafür spricht, dass dabei Genexpressionsvorgänge zwischengeschaltet sind [5, 65, 96]. Dasselbe gilt auch für die vermehrte Synthese purinerger Rezeptoren, wodurch Neuronen für ATP und andere Purine sensibilisiert werden könnten [34]. Diese verzögerten Wirkungen erinnern an die verzögert einsetzenden Kopfschmerzen von Migränepatienten bei Infusion von CGRP [41, 60].
Wie oben bereits erwähnt wurde, werden CGRP-Rezeptoren trigeminaler Afferenzen in ihre zentralen Fortsätze eingebaut, die in den spinalen Trigeminuskern projizieren. Dort werden aus den zentralen Terminalen neben Glutamat auch Neuropeptide als Co-Transmitter freigesetzt, wie man experimentell leicht zeigen kann [83]. Durch iontophoretische Applikation von CGRP konnte beim Rattenmodell die Aktivität sekundärer Neurone im trigemino-zervikalen Komplex gesteigert werden [88]. Auch andere Strukturen im Zentralnervensystem werden wahrscheinlich durch CGRP beeinflusst. Bindungsstellen für CGRP wurden im Nucleus accumbens, der Amygdala, den Basalganglien und besonders dicht im Cerebellum gefunden [95]; im Thalamus und im Hypothalamus gab es davon weniger, obwohl diese Gebiete durch CGRP-immunreaktive (CGRP-ir) Fasern relativ dicht innerviert sind. Neurone der thalamischen A11-Kerngruppe, die inhibitorisch auf den trigemino-zervikalen Kernkomplex wirken, sind teilweise CGRP-immunpositiv [15]. Auch in den zirkumventrikulären Organen wie der Area postrema, die keine dichte Blut-Hirn-Schranke aufweisen, wurde mRNA für RAMP1 gefunden [94]. Bei Primaten wurden CGRP-Bindungsstellen in den pontinen und anderen Kerngebieten einschließlich Raphé-Kernen und dem Locus coeruleus lokalisiert, also in Strukturen der absteigenden Hemmung [30, 69]. Zu der Idee, dass CGRP vielleicht auch in diesen Strukturen wirkt, passen Befunde der funktionellen Bildgebung, nach denen Aktivitätsänderungen, die mit Schmerzen im Trigeminusgebiet korrelieren, nicht nur im spinalen Trigeminuskern, sondern auch in der dorsalen Pons zu sehen sind [87]. Vor kurzem wurde gezeigt, dass durch iontophoretische Injektion von CGRP-Rezeptorantagonisten in das periaquaeduktale Grau der Ratte die Antworten von Neuronen im trigemino-zervikalen Komplex auf meningeale Stimulation gehemmt werden [80].
Es gibt also verschiedene zentrale Angriffspunkte von CGRP, die bei der Schmerzverarbeitung eine Rolle spielen können, doch sie liegen fast alle innerhalb der Blut-Hirn-Schranke. Eine der wesentlichen Fragen ist somit, ob und zu welchem Anteil Therapeutika, die gegen CGRP-Wirkungen gerichtet sind, die Blut-Hirn-Schranke überwinden. In einer neuen Untersuchung beim Rhesusaffen und beim Menschen mittels Positronen-Emissions-Tomographie wurde ein radioaktiver Tracer ([11C]MK-4232) gegeben, der an CGRP-Rezeptoren bindet, und dann der Rezeptorbesatz mit und ohne vorherige Gabe des CGRP-Rezeptorantagonisten Telcagepant berechnet [52]. Das Ergebnis war, dass 4–10% der zentralen CGRP-Rezeptoren von Telcagepant besetzt sind, wenn dieses in klinischer Dosierung von 140 mg verabreicht wurde. Die Autoren befanden diesen Prozentsatz als zu gering, um daraus auf eine effektive zentrale Wirksamkeit von Telcagepant bei Migräne schließen zu können.
Vorkommen und Bedeutung von CGRP-Rezeptoren außerhalb des trigeminalen Systems
Ob eine andauernde Hemmung der CGRP-Wirkungen auch prophylaktisch bei Migräne sein kann, ist bislang nicht hinreichend bekannt, aber von großem klinischen Interesse, um vor allem chronische Migräne behandeln zu können. Allerdings können unter solchen Bedingungen schwerwiegende Nebenwirkungen nicht von vornherein ausgeschlossen werden, denn es gibt auch außerhalb des trigeminovaskulären Systems viele Wirkungen von CGRP. Fast alle arteriellen Blutgefäße einschließlich der Koronararterien sind von CGRP freisetzenden afferenten Fasern innerviert, was auf eine bedeutende Rolle bei der kardiovaskulären Regulation hinweist [4, 9, 51]. Obwohl CGRP vorwiegend von Neuronen gebildet wird, kann es auch von anderen Zellen wie Immunzellen, Endothelzellen, Adipozyten und Keratinozyten produziert werden [39, 53, 67, 99]. CGRP-Rezeptorproteine werden neben der glatten Gefäßmuskulatur in vielen anderen peripheren Geweben exprimiert. So sind Endothelia, Herz, Gastrointestinalsystem, der juxtaglomeruläre Apparat der Niere und die Langerhans-Inseln des Pankreas mögliche Ziele von CGRP-Wirkungen [19, 40].
Die systemische Gabe von CGRP senkt unter experimentellen Bedingungen bei der Ratte den Blutdruck [55], es scheint jedoch beim Menschen keinen entscheidenden Einfluss auf die physiologische Blutdruckregulation zu haben, denn weder Herzfrequenz noch systemischer Blutdruck zeigten signifikante Veränderungen bei Gabe von CGRP-Rezeptorantagonisten [2]. CGRP-blockierende Antikörper (s.u.) wirkten hemmend auf die neurogene Vasodilatation in der Haut und der Meningen, ohne dass sie kardiovaskuläre Messwerte beeinflussten [106]. Dabei gibt es allerdings noch keine ausreichenden Erfahrungen bei einer dauerhaften Hemmung der CGRP-Wirkungen oder bei Risikopatienten. Bei ischämischen Herzattacken ist CGRP als endogener Schutzfaktor beteiligt [66]. Bei diabetischen Mäusen mit defizientem CGRP-System ist die ischämische Präkonditionierung zur Kardioprotektion deutlich vermindert [108]. CGRP wirkt auch in anderen Organen wie dem Intestinalsystem, der Niere und dem Gehirn protektiv gegenüber ischämischen Verletzungen [75, 91, 107]. Außerdem ist CGRP heilungsfördernd: CGRP fördert die Proliferation von Fibroblasten und Keratinozyten und verstärkt die Aktivierung von Matrix-Metalloproteasen in menschlicher Haut [16]. Bei CGRP-defizienten Mäusen wird der vaskuläre endotheliale Wachstumsfaktor (VEGF) vermindert exprimiert. Die Angiogenese und Wundheilung sind damit erheblich beeinträchtigt [93].
CGRP-immunreaktive Afferenzen wurden auch im Knochenmark und anderen immunmodulatorischen Organen wie Thymus, Milz und Lymphknoten gefunden [58, 104]. Einige Immunzellen produzieren selbst CGRP [103], während CGRP-Rezeptorkomponenten von vielen hämatopoetischen Zellen wie Lymphozyten, dendritischen Zellen, Mastzellen und Makrophagen exprimiert werden [74]. Die Inaktivierung von CGRP-gesteuerten Regulationsvorgängen könnte somit zu einer verminderten Produktion hämatopoetischer Zellen und damit zu einer Defizienz der Abwehrfunktionen führen, wenn das Immunsystem besonders gefordert ist [11]. Dazu passt die Beobachtung aus den Studien zum Einsatz monoklonaler Antikörper gegen CGRP (s.u.). In den meisten Studien wurde unter Verum ein gehäuftes Auftreten von Infekten der oberen Atemwege beobachtet.
CGRP-Antagonisten zur Behandlung akuter Migräneattacken
Nachdem der mögliche pathophysiologische Zusammenhang zwischen der Ausschüttung von CGRP während akuter Migräneattacken beim Menschen als ein wichtiger Mechanismus erkannt war, wurden direkte Antagonisten für den CGRP-Rezeptor entwickelt und zur Behandlung akuter Migräneattacken untersucht (Tab. 1).
Tab. 1. CGRP-Antagonisten zur Behandlung akuter Migräneattacken (Ergebnisse für die jeweils höchste Dosis)
|
Substanz |
Studie |
Vergleich |
Schmerzfrei nach ... (Dosis) |
Verum |
Placebo |
Komparator |
Kommentar |
Referenz |
|
BIBN 4096 |
Dosisfindung, i.v. 0,25–10 mg |
Placebo |
30 Minuten |
17% |
2% |
– |
Entwicklung eingestellt |
[77] |
|
BI 44370 TA |
Oral 50, 200, 400 mg |
Placebo, Eletriptan 40 mg |
2 Stunden |
27,4% |
34,8% |
8,6% |
Entwicklung eingestellt |
[22] |
|
Telcagepant |
Oral 50,150, 300 mg |
Placebo |
2 Stunden |
23,8% |
10,7% |
– |
Dosisfindung |
[18] |
|
MK-0974 |
Oral 150, 300 mg |
Placebo Zolmitriptan 5 mg |
2 Stunden |
27% |
10% |
31% |
[47] |
|
|
MK-0974 |
Oral 25, 50, 100, 200, 300, 400, 600 mg |
Placebo |
2 Stunden |
32,1% |
14,3% |
33,4% |
Dosisfindung |
[50] |
|
Telcagepant |
Oral 2,5, 5, 10, 20, 50, 100, 200 mg |
Placebo |
2 Stunden |
36,2% |
9,8% |
– |
Dosisfindung |
[43] |
|
Telcagepant |
Oral 140, 280 mg |
Placebo |
2 Stunden |
25,2% |
10,2% |
– |
[45] |
|
|
Telcagepant |
Oral 19820 Attacken 300 mg |
Rizatriptan 10 mg 10981 Attacken |
– |
Langzeitsicherheitsstudie |
[17] |
|||
|
BMS-927711 |
Oral 10, 25, 75, 150, 300, 600 mg |
Placebo Sumatriptan 100 mg |
2 Stunden |
23% |
15,3% |
35% |
Dosisfindung |
[70] |
|
Ubrogepant |
Oral 1, 10, 25, 50, 100 mg |
Placebo |
2 Stunden |
25,5% |
8,9% |
– |
Dosisfindung |
[97] |
BIBN 4096 (Olcagepant)
Die erste Substanz war der CGRP-Rezeptor-Blocker BIBN 4096 (Olcagepant). In einer Dosisfindungsstudie wurde diese Substanz während einer akuten Migräneattacke intravenös gegeben. Eine Besserung der Kopfschmerzen innerhalb von zwei Stunden wurde bei 66% der Patienten beobachtet, die mit BIBN 4096 behandelt wurden, verglichen mit 27% der Patienten, die mit Placebo behandelt wurden [77]. Damit war der wissenschaftliche Beweis erbracht, dass eine Blockierung des CGRP-Rezeptors eine effektive Therapie von akuten Migräneattacken darstellt. BIBN 4096 hatte keine relevanten Nebenwirkungen, insbesondere keine vasokonstriktiven Eigenschaften. Die Weiterentwicklung der Substanz wurde eingestellt, da es nicht möglich war, das Molekül so zu modifizieren, dass eine wirksame orale Absorption stattfand.
BI 44370 TA
Die Folgesubstanz war der CGRP-Rezeptor-Antagonist BI 44370 TA. In einer Dosisfindungsstudie wurden 50, 200 und 400 mg BI 44370 TA mit Eletriptan 40 mg und Placebo verglichen. Den primären Endpunkt, nämlich Schmerzfreiheit nach zwei Stunden, erreichten 27,4% der Patienten in der 400 mg Dosis von BI 44370 TA, 34,8% mit Eletriptan und 8,6% in der Placebo-Gruppe [22]. Die niedrigeren Dosierungen von BI 44370 TA waren einer Behandlung mit Placebo nicht überlegen. Die Substanz wurde nicht weiterentwickelt, da sie zu viele Interaktionen mit anderen Medikamenten hatte, die zur Prophylaxe der Migräne oder bei einer Behandlung von vaskulären Erkrankungen eingesetzt werden.
Telcagepant
MSD entwickelte eine Reihe von CGRP-Rezeptor-Antagonisten, darunter Telcagepant und MK3207. Telcagepant wurde in insgesamt acht randomisierten Studien in der Behandlung von akuten Migräneattacken untersucht. Über alle Studien hinweg lag der Prozentsatz der Patienten, die nach zwei Stunden schmerzfrei waren, zwischen 22% und 35% und die entsprechenden Placebo-Raten zwischen 9,6% und 19% [21]. Metaanalytisch ergab sich daraus ein Odds-Ratio von 2,70, das mit einem p-Wert <0,01 signifikant war. In zwei der Studien [17, 47] wurde Telcagepant mit Zolmitriptan und Rizatriptan verglichen. In beiden Studien waren die Triptane signifikant besser wirksam als Telcagepant. Über alle Studien hinweg hatte Telcagepant ein sehr gutes Verträglichkeits- und Sicherheitsprofil. Numerisch lagen die Nebenwirkungsraten im Placebo-Bereich. Zolmitriptan und Rizatriptan hatten signifikant mehr Nebenwirkungen als Telcagepant. Telcagepant war auch reproduzierbar gut wirksam, was in einer Studie belegt wurde, in der vier konsekutive Migräneattacken behandelt wurden [45]. Im Rahmen einer Sicherheitsstudie wurden auch Patienten, die unter einer Migräne litten und eine koronare Herzerkrankung hatten, mit Telcagepant behandelt [48]. Bei den 114 Patienten, die Telcagepant einnahmen, traten keine kardiovaskulären Nebenwirkungen auf. In einer weiteren Studie wurde untersucht, ob die Kombination von Telcagepant plus Ibuprofen oder Paracetamol einer Monotherapie überlegen war [44]. Dies war nicht der Fall.
In der Folgezeit wurde dann Telcagepant zur Migräneprophylaxe untersucht (Tab. 2). In der ersten Studie [46] wurden Patienten eingeschlossen, die zwischen 3 und 14 Migränetage pro Monat hatten. Die Patienten wurden in drei Gruppen randomisiert und erhielten entweder 2×140 mg, 2×280 mg Telcagepant täglich oder Placebo über einen Zeitraum von zwölf Wochen. Die Studie wurde vom Sicherheitskomitee abgebrochen, nachdem 660 Patienten randomisiert worden waren. Bei 13 Patienten kam es zu einer signifikanten Erhöhung der Leberwerte und bei zwei Patienten zu einem massiven Anstieg der Leberwerte. In der zweiten Studie [49] untersuchte man Telcagepant zur Prävention der menstruellen Migräne. Es handelte sich um eine doppelblinde Placebo-kontrollierte Studie bei Frauen mit perimenstruellen Kopfschmerzen. Die Frauen erhielten entweder 140 mg Telcagepant oder Placebo in einem randomisierten Verhältnis von 2:1 über sieben konsekutive Tage während der Menstruation. In die Verum-Gruppe wurden 2659 und in die Placebo-Gruppe 1327 Frauen eingeschlossen. Bezüglich des primären Endpunktes, Migränetage pro Monat zu reduzieren, war die Studie negativ. Die Zahl der Migränetage in den sieben Tagen der Behandlung war allerdings unter Telcagepant signifikant geringer. Während der Studie kam es bei drei Frauen zu einer massiven Erhöhung der Leberwerte.
Als Konsequenz aus den zuletzt zitierten Studien wurde die Entwicklung von Telcagepant beendet. Grund dafür war, dass nicht ausgeschlossen werden konnte, dass selbst bei einer Einschränkung zur Behandlung akuter Migräneattacken die Substanz, wie alle Medikamente zur Attackentherapie, missbräuchlich häufig verwendet werden würde.
Tab. 2. CGRP-Antagonisten zur Migräneprophylaxe
|
Substanz |
Studie |
Vergleich |
Zielgröße |
Reduktion unter |
Komparator |
Kommentar |
Referenz |
|
|
Verum |
Placebo |
|||||||
|
Telcagepant |
Oral; 140, 280 mg |
Placebo |
Kopfschmerztage/Monat |
–3,1 |
–1,7 |
– |
Vom DSMB abgebrochen |
[46] |
|
Telcagepant |
Oral; 140 mg |
Placebo |
Kopfschmerztage/Monat |
–8,8 |
–9,3 |
– |
Menstruelle Migräne |
[49] |
DSMB: Daten- und Sicherheitsüberwachungskomitee
BMS-927711
Ein weiterer CGRP-Antagonist war BMS-927711. Die Substanz wurde in einer randomisierten doppelblinden Placebo-kontrollierten Dosisfindungsstudie an 885 Patienten untersucht [70]. Die eingesetzten Dosierungen waren 10, 25, 75, 150, 300 oder 600 mg BMS-927711. Als Vergleichssubstanz dienten 100 mg Sumatriptan und Placebo. Der primäre Endpunkt war Schmerzfreiheit nach zwei Stunden. Ab einer Dosis von 25 mg war die Substanz signifikant wirksamer als Placebo. Die beiden höchsten Dosierungen hatten eine Wirksamkeit, die mit der von Sumatriptan vergleichbar war. Die Rate an Nebenwirkungen entsprach der von Placebo. Die Entwicklung der Substanz wurde in der Folgezeit eingestellt, wobei nicht bekannt ist, was die Gründe dafür waren.
MK-162 (Ubrogepant)
Ein anderer neuer CGRP-Antagonist ist MK-162 (Ubrogepant). Das Molekül unterscheidet sich von Telcagepant und MK3207. In einer Dosisfindungsstudie wurden 834 Patienten eingeschlossen [97]. Die Dosierung von Ubrogepant betrug 1, 10, 25 oder 100 mg. Der primäre Endpunkt war Schmerzfreiheit nach zwei Stunden. Ab der Dosis von 25 mg war die Wirksamkeit verglichen mit Placebo signifikant besser. Die Besserung der Kopfschmerzen von mittelschwer oder schwer auf leicht bis keine Kopfschmerzen war jedoch nicht signifikant unterschiedlich gegenüber Placebo. Die berichteten Nebenwirkungen waren Mundtrockenheit, Übelkeit, Müdigkeit, Benommenheit und Somnolenz. Eine Erhöhung der Leberwerte wurde nicht beobachtet. Diese Studie zeigt eine dosisabhängige Wirkung von Ubrogepant in der Akuttherapie von Migräneattacken. Insgesamt ist der Therapieeffekt aber nicht sehr beeindruckend. Auffällig ist, dass die Ergebnisse der Studie erst vier Jahre nach Durchführung der Studie publiziert wurden. Bisher liegen auch keine Sicherheitsdaten zu Ubrogepant vor, wenn die Substanz täglich oder häufiger eingenommen wird.
CGRP- oder CGRP-Rezeptor-bindende Antikörper
Die Wirkungen CGRP-bindender Antikörper wurden in präklinischen Experimenten an Ratten getestet. Ein humanisierter CGRP-bindender monoklonaler Antikörper von Rinat Neuroscience hemmte in myographischen Experimenten den muskelrelaxierenden Effekt von Alpha-CGRP auf isolierte zerebrale Arterien [27]. Zwei Antikörper von Rinat aus der Maus (MuMab 4901 and MuMab 7E9) hemmten den durch elektrische Stimulation ausgelösten Blutflussanstieg und die neurogene Entzündung von meningealen Arterien im geschlossenen kranialen Fenster in einer Dosierung von 10 mg/kg i.v. in gleicher Größenordnung wie der CGRP-Antagonist Olcegepant (300 µg/kg), zwar mit langsam einsetzender Wirkung, aber langer Wirkdauer von mindestens einer Woche [106]. MuMab 4901 (30 mg/kg) hatte keinen signifikanten Effekt auf Herzfrequenz oder Blutdruck bei wachen Tieren.
Nachdem sich herausgestellt hatte, dass kleine Moleküle, die direkt den CGRP-Rezeptor antagonisieren, aufgrund ihrer Lebertoxizität oder schwierigen Wechselwirkungen wahrscheinlich für die Behandlung von akuter Migräne ungeeignet sind, wurden monoklonale humanisierte Antikörper gegen den CGRP-Rezeptor (AMG334) oder das CGRP-Molekül selbst entwickelt (LY2951742, TEV-48125, ALD-403). Letztere Substanzen wurden bisher in Phase-II-Studien zur Prophylaxe der episodischen Migräne untersucht, TEV-48125 auch zur Prophylaxe der chronischen Migräne (Tab. 3).
ALD403
ALD403 von Alder Biopharmaceuticals ist ein humanisierter selektiver Anti-CGRP-Immunglobulin-G(IgG)-Antikörper mit einer Größe von etwa 150 kDa, der in B-Zellen von Kaninchen durch Genetic Engineering hergestellt wurde. Er bindet mit hoher Potenz (Kd <20 pM) an Alpha- und Beta-CGRP und hat eine Plasma-Halbwertszeit von 31 Tagen. Die Substanz wird alle drei Monate in einer einstündigen Infusion intravenös appliziert. Die Phase-II-Studie war eine randomisierte doppelblinde Placebo-kontrollierte Untersuchung, in die Patienten im Alter zwischen 18 und 55 Jahren mit 5 bis 14 Migränetagen im Monat eingeschlossen wurden [23]. In der Verum-Gruppe erhielten die Patienten eine einmalige Infusion von 1000 mg ALD403. Es wurden 82 Patienten in die Placebo-Gruppe und 81 in die ALD403-Gruppe randomisiert. 80% der Patienten waren Frauen. Die durchschnittliche Zahl der Migränetage im Monat betrug 8,5 und die Zahl der Migräneattacken lag zwischen 6,0 und 6,7. Die mittlere Reduktion der Migränetage in den Wochen 5 bis 8 verglichen mit der Baseline betrug –5,6 Tage für ALD403 und –4,6 Tage für die Placebo-Gruppe. Der geringe Unterschied war statistisch signifikant. Die unter ALD403 berichteten häufigsten Nebenwirkungen waren Husten, Schnupfen, Heiserkeit sowie Harnwegsinfektionen. Alle anderen Nebenwirkungen waren genauso häufig wie in der Placebo-Gruppe. Insgesamt zeigt die Phase-II-Studie für ALD403 eine gute Verträglichkeit mit einer 50%-Responderrate (Anteil der Patienten mit einer mindestens 50%igen Reduktion der Migränetage) unter Verum von rund 60% (Tab. 3). Bemerkenswert war, dass 16% der mit Verum behandelten Patienten während der zwölfwöchigen Studiendauer keine einzige Migräneattacke hatten. Die Ergebnisse müssen jetzt in einer laufenden Phase-III-Studie bestätigt werden.
AMG 334
AMG 334 von Amgen ist ein vollständig humanisierter monoklonaler Antikörper, der mit hoher Affinität (Kd =25 pM) an CGRP-Rezeptoren bindet, ohne dass er agonistische Aktivität aufweist. Die Substanz wird einmal monatlich subkutan appliziert. In der Phase-II-Studie, die multizentrisch randomisiert und doppelblind durchgeführt wurde, wurden Migränepatienten im Alter zwischen 18 und 60 Jahren mit 4 bis 14 Migränetagen pro Monat eingeschlossen [90]. In der Dosisfindungsstudie wurden 7, 21 und 70 mg AMG 334 mit Placebo verglichen. Der primäre Endpunkt war die Reduktion der Migränetage pro Monat zwischen der Baselinephase von vier Wochen und der zwölfwöchigen doppelblinden Behandlungsdauer. In die Studie wurden insgesamt 483 Patienten eingeschlossen, 160 Patienten erhielten Placebo. 81% der Patienten waren Frauen. Die durchschnittliche Zahl der Migränetage in der Baseline betrug 8,7, die Zahl der Migräneattacken 5,4. Unter Placebo betrug die Reduktion der Migränetage 2,3 pro Monat. Die Reduktion unter Verum betrug 2,2 Tage bei 7 mg AMG-334, 2,4 Tage für 21 mg AMG 334 und 3,4 Tage bei der 70-mg-Dosis. Nur für die hohe Dosis war der Unterschied statistisch signifikant. Die 50%-Responderrate betrug für die hohe Dosis von AMG 334 46%. AMG 334 wurde gut vertragen. Es gab keine schwerwiegenden oder unerwünschten Arzneimittelwirkungen. Alle übrigen Nebenwirkungen waren in ihrer Häufigkeit für beide Dosierungen mit der in der Placebo-Gruppe vergleichbar. Auch hier läuft derzeit eine große Phase-III-Studie.
LY2951742
LY2951742 von Eli Lilly ist ein weiterer voll humanisierter monoklonaler Antikörper, der selektiv und mit hoher Potenz (Kd =30 pM) an CGRP bindet und eine Plasma-Halbwertszeit von etwa 28 Tagen hat. Die Substanz wird alle zwei Wochen subkutan verabreicht. In der Phase-II-Studie wurden Patienten im Alter zwischen 18 und 60 Jahren mit 4 bis 14 Migränetagen pro Monat eingeschlossen [24]. Der primäre Endpunkt war die Änderung in der Zahl der Migränetage in den Wochen 9 bis 12 nach Randomisierung. Insgesamt wurden 218 Patienten in die Studie eingeschlossen. Die mittlere Reduktion in der Zahl der Migränetage betrug 4,2 unter Verum und 3,0 unter Placebo. Dieser Unterschied war statistisch signifikant. Unter Verum kam es häufiger zu Atemwegsinfektionen und abdominellen Schmerzen als unter Placebo. Die übrigen Nebenwirkungen waren gleich häufig. Die 50%-Responderrate betrug 70% und bei 32% der Patienten kam es unter Verum in der zwölfwöchigen Behandlungsphase zu keiner weiteren Migräneattacke (Tab. 3).
Tab. 3. CGRP- oder CGRP-Rezeptor-Antikörper zur Prophylaxe der Migräne (Ergebnisse für die jeweils höchste Dosis)
|
Substanz Anzahl |
Studie |
Vergleich |
Zielgröße |
Reduktion unter |
50%-Responderrate |
Kommentar |
Referenz |
||
|
Verum |
Placebo |
Verum |
Placebo |
||||||
|
ALD403 n=163 |
Phase II, |
Placebo |
Migränetage |
–5,6 |
–4,6 |
61% |
33% |
[23] |
|
|
AMG 334 n=483 |
Phase II 4–14 Migränetage 7, 21, 70 mg alle 4 Wochen s.c. |
Placebo |
Migränetage |
–3,4 |
–2,3 |
46% |
30% |
[90] |
|
|
LY2951742 n=218 |
Phase II |
Placebo |
Migränetage |
–4,2 |
–3,0 |
70% |
45% |
[24] |
|
|
TEV 48125 n=297 |
Phase II |
Placebo |
Migränetage |
–6,1 |
–3,5 |
59% |
28% |
[7] |
|
|
TEV 48125 n=264 |
Phase II, >15 Migränetage |
Placebo |
Kopfschmerz- |
–67,5 |
–37,1 |
55% |
31% |
Chronische Migräne |
[6] |
TEV-48125
TEV-48125 wurde ursprünglich LBR-101 genannt. Der Antikörper wurde in Rattenzellen entwickelt und durch Gentechnik so modifiziert, dass mögliche Immuneffekte mit humanen Zellen minimiert wurden. Der Antikörper bindet selektiv an beide Isoformen von CGRP und hat eine Plasma-Halbwertszeit von 45 Tagen. Vorklinische Experimente umfassten Blutflussmessungen an der Dura mater der Ratte und Sicherheitsprüfungen in Ratten und Affen von mehr als drei Monaten. Dann wurden Sicherheit und Verträglichkeit von LBR-101 bei i.v. Dosen von 0,2 bis 2000 mg an 94 gesunden Personen getestet und mit 45 Personen verglichen, die Placebo erhielten [8]. Leichte Nebenwirkungen traten dabei bei etwa 20% der Personen in beiden Gruppen auf. In einer doppelblinden Placebo-kontrollierten Folgestudie mit 31 Frauen traten keine wesentlichen Änderungen des Blutdrucks, der Herzfrequenz oder anderer Parameter im Elektrokardiogramm nach LBR-101 in einer Dosis bis zu 2000 mg auf [9].
TEV-48125 wird einmal im Monat subkutan appliziert. In einer ersten Studie wurde die Substanz bei der hochfrequenten episodischen Migräne untersucht [7]. Es handelte sich um eine multizentrische randomisierte doppelblinde Placebo-kontrollierte Studie, in die Patienten mit 8 bis 14 Migränetagen pro Monat eingeschlossen wurden. Die Patienten erhielten in der Verum-Gruppe alle vier Wochen entweder 225 oder 675 mg TEV-48125 oder Placebo. Der primäre Endpunkt war die Reduktion der Migränetage zwischen Baseline und den Wochen 9 bis 12. In die Studie wurden 297 Teilnehmer eingeschlossen, 90% waren Frauen. Die mittlere Zahl der Migränetage in der Baseline betrug 11, die mittlere Zahl der Kopfschmerztage pro Monat 12,5, die Abnahme der Migränetage zwischen Baseline und den Wochen 9 bis 12 betrug 3,5 Tage für Placebo, 6,7 Tage für die niedrigere Dosis und 6,1 Tage für die hohe Dosis TEV-48125. Diese Unterschiede waren statistisch signifikant. Die 50%-Responderrate betrug rund 60%. Die Zahl der berichteten Nebenwirkungen unterschied sich nicht zwischen aktiver Therapie und Placebo.
TEV-48125 wurde auch in der Prophylaxe der chronischen Migräne untersucht [6]. In die multizentrische randomisierte doppelblinde Placebo-kontrollierte Parallelgruppen-Studie wurden Migränepatienten im Alter zwischen 18 und 65 Jahren mit chronischer Migräne eingeschlossen. Sie erhielten im Abstand von 28 Tagen drei subkutane Injektionen von TEV-48125, die Dosis betrug entweder initial 675 mg gefolgt von 225 mg oder über die gesamte Studienperiode 900 mg. Als Vergleich diente eine Placebo-Injektion. Der primäre Endpunkt war die Reduktion der Kopfschmerzstunden zwischen Baseline und der Wochen 9 bis 12. In die Studie wurden 264 Teilnehmer eingeschlossen, 85% waren Frauen. Die mittlere Zahl der Kopfschmerz-Stunden pro Monat betrug zwischen 160 und 170 Stunden. Die mittlere Reduktion der Kopfschmerzstunden in den Wochen 9 bis 12 verglichen mit der Baseline waren 60 Stunden für die niedrigere Dosis von TEV-48125, 67 Stunden in der hohen Dosis von TEV-48125 und 37 Stunden in der Placebo-Gruppe. Die Unterschiede waren signifikant. Nebenwirkungen waren mit denen von Placebo vergleichbar. In den beiden Verum-Gruppen traten etwas häufiger Atemwegsinfekte auf. Die 50%-Responderrate lag bei 54%.
Phase-III-Studien mit Antikörpern gegen den CGRP-Rezeptor
Die einzige neue Substanz, bei der die Phase III abgeschlossen ist, die AMG-334 (Erenumab). Die Studien waren im Januar 2017 noch nicht publiziert. In der STRIVE-Studie erhielten die Patienten entweder Placebo oder 70 beziehungsweise 140 mg Erenumab subkutan einmal pro Monat über sechs Monate (Tab. 4). In der Baseline hatten die Patienten im Mittel 8,3 Migränetage pro Monat. Die Reduktion der Migränetage pro Monat betrug unter niedrigen Dosen Erenumab 3,2 Tage und in der hohen Dosis 3,7 Tage. Verglichen mit der Placebo-Gruppe, in der die Reduktion 1,8 Tage betrug, war der Unterschied signifikant. Bezüglich Nebenwirkungen ergaben sich keine relevanten Unterschiede zwischen Erenumab und Placebo. Lediglich Husten, Schnupfen und Nebenhöhlenentzündungen waren unter Erenumab häufiger.
Tab. 4. CGRP- oder CGRP-Rezeptor-Antikörper zur Prophylaxe der Migräne (Ergebnisse für die jeweils höchste Dosis) – Ergebnisse bisher nur in Abstract-Form
|
Substanz Anzahl |
Studie |
Vergleich |
Primärer |
Verum |
Placebo |
75%-Responderrate |
Kommentar |
Referenz |
|
|
Verum |
Placebo |
||||||||
|
ALD403 n=616 |
Phase II 15–28 Migränetage 10, 20, 100, 300 mg 1 × i.v. |
Placebo |
Migränetage; 75% Responder |
–9,0 |
–5,2 |
33% |
21% |
Chronische |
– |
|
AMG 334 n=577 |
Phase III 4–14 Migränetage |
Placebo |
Migränetage |
–2,9 |
–1,8 |
– |
– |
ARISE-Studie |
– |
|
AMG 334 n=955 |
Phase III 70, 140 mg alle 4 Wochen s.c. für 6 Monate |
Placebo |
Migränetage |
–3,7 |
–1,8 |
– |
– |
STRIVE-Studie |
– |
i.v.: intravenös; n: Anzahl; s.c.: subkutan
ARISE war eine weitere Phase-III-Studie für AMG 334. In die Studie wurden 577 Migränepatienten eingeschlossen, die entweder AMG 334 70 mg subkutan einmal monatlich oder Placebo erhielten. Die Patienten hatten zwischen 4 und 14 Migränetage pro Monat, und im Mittel 8 Migränetage in der Baseline. Unter AMG 334 kam es zu einer signifikanten Reduktion der Migränetage pro Monat um 2,9 Tage verglichen mit Placebo um 1,8 Tage. Auch hier waren die häufigsten Nebenwirkungen unter AMG 334 obere Atemwegsinfektionen und Schnupfen.
Beurteilung der monoklonalen Antikörper gegen CGRP oder den CGRP-Rezeptor und ungeklärte Fragen
Die Ergebnisse der bisher publizierten Phase-II- und Phase-III-Studien zur Prophylaxe der episodischen und chronischen Migräne mit den neuen Substanzen sind ermutigend (Abb. 1). Die Antikörper zeigen eine Wirksamkeit, die mit der anderer Migräneprophylaktika wie Betablocker, Topiramat, Valproinsäure und Amitriptylin bei der episodischen Migräne sowie Topiramat und Botulinumtoxin bei der chronischen Migräne vergleichbar sind. Bemerkenswert ist die geringe Rate an Nebenwirkungen. Insbesondere wurden keine zentralen Nebenwirkungen beobachtet, was dafür sprechen würde, dass die Antikörper die intakte Blut-Hirn-Schranke nicht überwinden können. Ein weiterer Vorteil der subkutanen oder intravenösen Gabe ist die Gewährleistung der Compliance. Dies ist bei einer medikamentösen Therapie mit Tabletten ein sehr großes Problem, da die Compliance mit der medikamentösen Migräneprophylaxe nach drei Monaten nur noch 30–40% beträgt [42]. Die meisten Patienten brechen wegen Nebenwirkungen ab.
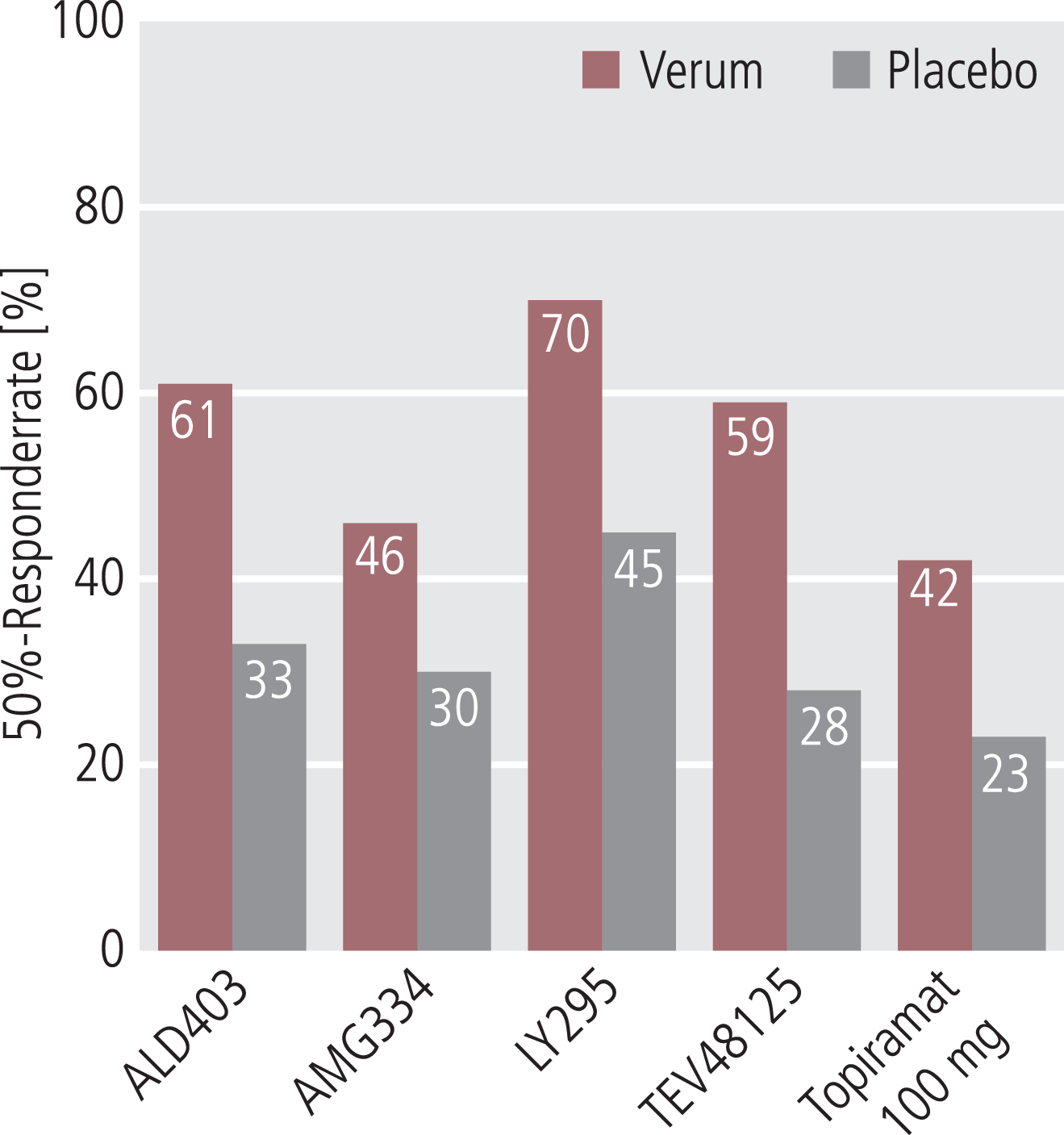
Abb. 1. 50%-Responderraten in den Phase-II-Studien der Antikörper gegen CGRP oder den CGRP-Rezeptor zur Migräneprophylaxe. Als Vergleich sind die Ergebnisse von Topiramat 100 mg dargestellt [12]
Die CGRP-Antikörper haben darüber hinaus den Vorteil, dass sie auch bei Patienten gegeben werden können, die die derzeitigen Migräneprophylaktika entweder nicht vertragen oder bei denen sie kontraindiziert sind. Ungelöst ist die Frage, ob Antikörper gegen CGRP oder den CGRP-Rezeptor angesichts der ubiquitären Verteilung von CGRP-Rezeptoren im Körper und im Gehirn langfristige Nebenwirkungen haben können. Was passiert beispielsweise, wenn im Rahmen einer Meningoenzephalitis die Blut-Hirn-Schranke undicht wird? Ungeklärt ist ebenfalls, wie sich die Antikörper auswirken, wenn bei einem Patienten mit Migräne unter Therapie ein akutes Koronarsyndrom oder ein Schlaganfall auftritt. Diese Fragen werden erst dann beantwortet werden können, wenn nach Zulassung der Substanzen Langzeitsicherheitsdaten vorliegen.
Eine Herausforderung in der Zukunft wäre, herauszufinden, welche der Patienten auf die Behandlung am ehesten ansprechen, was vielleicht von einem bestimmten genetischen Profil abhängt, also eine Art personalisierte Medizin.
Eine andere, genauso wichtige Frage, die mit der Langzeitbehandlung dieser Art zusammenhängen mag, ist der immer noch ungeklärte Mechanismus und Wirkungsort von CGRP und damit der Inaktivierung von CGRP oder von CGRP-Rezeptoren. Wie bereits diskutiert, wurde auch eine direkte antinozizeptive Wirkung bei Hemmung der CGRP-Rezeptoren im spinalen Trigeminuskern vermutet. Im Tierversuch verstärkt dort die lokale Gabe von CGRP die synaptische Übertragung und Hemmung von CGRP-Rezeptoren schwächt diese ab. Es ist aber unwahrscheinlich, dass die großen Antikörper ohne weiteres durch die intakte Blut-Hirn-Schranke treten können, wobei diese Annahme allerdings nur für die akute Wirkung gilt und bei Langzeitgabe durchaus anders sein kann. Die Transzytose von IgG-Antikörpern durch die Blut-Hirn-Schranke ist nämlich prinzipiell möglich [78]. Manchmal funktioniert dieser Transport unidirektional in Richtung Gehirn [109]. Eine sorgfältige Analyse der Vorgänge Wochen und Monate nach der Gabe von monoklonalen Antikörpern wäre somit sehr sinnvoll, um diese Frage zu beantworten.
Wenn die Antikörper keine Veränderungen im Zentralnervensystem (ZNS) bewirken, muss man den peripheren Mechanismus genauer untersuchen. Dies ist umso wichtiger, als viele Organe und Gewebe unter der Kontrolle von CGRP stehen, wie bereits diskutiert wurde. Es könnte zum Beispiel sein, dass CGRP-Antikörper oder CGRP-Rezeptor-Antikörper die Bindung von CGRP an den Rezeptor verhindern. Möglicherweise werden aber auch CGRP-Rezeptoren vermehrt abgebaut oder das „Trafficking“ von CGRP-Rezeptoren wird gehemmt, sodass Rezeptormoleküle nicht mehr in die Membran eingebaut werden können und funktionelle CGRP-Rezeptoren bilden.
Zusammengefasst weisen die vorliegenden Daten auf eine Wirksamkeit der CGRP-Antagonisten als auch CGRP-Antikörper beziehungsweise CGRP-Rezeptor-Antikörper in der akuten beziehungsweise prophylaktischen Therapie der Migräne hin. Zudem haben grundlagenwissenschaftliche Untersuchungen die Wichtigkeit von CGRP bei Migräne und Clusterkopfschmerz gezeigt und unser Verständnis für die zugrundeliegenden pathophysiologischen Mechanismen erweitert. Größere Phase-III-Studien, die derzeit durchgeführt werden, müssen nun zeigen, ob sich die Wirksamkeit auch in einem größeren Patientenkollektiv zeigen lässt und inwieweit sich das bislang weitgehend unauffällige Sicherheitsprofil ändert. Die aktuellen Studien schließen dabei Patienten mit hochfrequenter und chronischer Migräne sowie episodischen und chronischen Clusterkopfschmerz ein. Möglichweise stehen in Zukunft dann gute Alternativen zu den bislang verwendeten Medikamenten zur Verfügung, die nicht immer gut wirksam sind und häufig mit zahlreichen Nebenwirkungen einhergehen.
Interessenkonflikterklärung
KM war 2016 honoriertes Mitglied des AMG 334 Scientific Advisory Board von Novartis. Forschungsprojekte wurden durch Allergan und NOXXON Pharma sowie durch die Universität Erlangen-Nürnberg (Marohn-Stiftung, Emerging Fields Project Medicinal Redox Inorganic Chemistry), die Humboldt-Stiftung, die Pfleger-Stiftung und die Europäische Union (FP7 EU-ROHEADPAIN, Grant Agreement 602633) unterstützt. KM besitzt keine Aktien oder Anteile von Pharmafirmen.
DH hat Honorare für Vorträge oder Forschungsprojekte erhalten von Allergan Pharma, Lilly, Novartis Pharma, Desitin Arzneimittel, Hormosan Pharma, ElectroCore und Grünenthal erhalten. DH besitzt keine Aktien oder Anteile von Pharmafirmen.
HCD hat Honorare für die Planung, Ausführung oder Teilnahme an Klinischen Studien, Teilnahme an Advisory Boards oder Vorträge erhalten von: Addex Pharma, Alder, Allergan, Almirall, Amgen, AstraZeneca, Autonomic Technology, Bayer Vital, Berlin Chemie, Boehringer Ingelheim, Bristol-Myers Squibb, Chordate, CoLucid, Coherex, Electrocore, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Labrys Biologicals, Lilly, La Roche, 3M Medica, Menarini, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper und Brümmer, Sanofi, St. Jude Medical, TEVA und Weber & Weber. Finanzielle Unterstützung für Forschungsprojekte wurde gewährt von: Allergan, Almirall, AstraZeneca, Bayer, Electrocore, GSK, Janssen-Cilag, MSD und Pfizer. Kopfschmerzforschung an der Universitätsklinik für Neurologie und dem Westdeutschen Kopfschmerzzentrum Essen erfolgt durch: Deutsche Forschungsgemeinschaft (DFG), Bundesministerium für Bildung und Forschung (BMBF), und die Europäische Union (EU). HCD besitzt keine Aktien oder Anteile von Pharmafirmen.
Literatur
1. Amara SG, Jonas V, Rosenfeld MG, Ong ES, et al. Alternative RNA processing in calcitonin gene expression generates mRNAs encoding different polypeptide products. Nature 1982;298:240–4.
2. Arulmani U, Schuijt MP, Heiligers JP, Willems EW, et al. Effects of the calcitonin gene-related peptide (CGRP) receptor antagonist BIBN4096BS on alpha-CGRP-induced regional haemodynamic changes in anaesthetised rats. Basic Clin Pharmacol Toxicol 2004;94:291–7.
3. Beattie DT, Connor HE, Hagan RM. Recent developments in tachykinin NK1 receptor antagonists: prospects for the treatment of migraine. Can J Physiol Pharmacol 1995;73:871–7.
4. Bell D, McDermott BJ. Calcitonin gene-related peptide in the cardiovascular system: characterization of receptor populations and their (patho)physiological significance. Pharmacol Rev 1996;48:253–88.
5. Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci 2006;23:2057–66.
6. Bigal ME, Dodick DW, Krymchantowski AV, VanderPluym JH, et al. TEV-48125 for the preventive treatment of chronic migraine: Efficacy at early time points. Neurology 2016;87:41–8.
7. Bigal ME, Dodick DW, Rapoport AM, Silberstein SD, et al. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol 2015;14:1081–90.
8. Bigal ME, Escandon R, Bronson M, Walter S, et al. Safety and tolerability of LBR-101, a humanized monoclonal antibody that blocks the binding of CGRP to its receptor: Results of the Phase 1 program. Cephalalgia 2014;34:483–92.
9. Bigal ME, Walter S, Bronson M, Alibhoy A, et al. Cardiovascular and hemodynamic parameters in women following prolonged CGRP inhibition using LBR-101, a monoclonal antibody against CGRP. Cephalalgia 2014;34:968–76.
10. Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev 2004;84:903–34.
11. Broome CS, Miyan JA. Neuropeptide control of bone marrow neutrophil production. A key axis for neuroimmunomodulation. Ann N Y Acad Sci 2000;917:424–34.
12. Bussone G, Diener H, Pfeil J, Schwalen S. Topiramate 100 mg/day in migraine prevention: a pooled analysis of double-blind randomised controlled trials. Int J Clin Pract 2005;59:961–8.
13. Buzzi MG, Carter WB, Shimizu THH, Moskowitz MA. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology 1991;30:1193–200.
14. Buzzi MG, Moskowitz MA. Evidence for 5-HT1B/1D receptors mediating the antimigraine effect of sumatriptan and dihydroergotamine. Cephalalgia 1991;11:165–8.
15. Charbit AR, Akerman S, Holland PR, Goadsby PJ. Neurons of the dopaminergic/calcitonin gene-related peptide A11 cell group modulate neuronal firing in the trigeminocervical complex: an electrophysiological and immunohistochemical study. J Neurosci 2009;29:12532–41.
16. Cheret J, Lebonvallet N, Buhe V, Carre JL, et al. Influence of sensory neuropeptides on human cutaneous wound healing process. J Dermatol Sci 2014;74:193–203.
17. Connor KM, Aurora SK, Loeys T, Ashina M, et al. Long-term tolerability of telcagepant for acute treatment of migraine in a randomized trial. Headache 2010;51:73–84.
18. Connor KM, Shapiro RE, Diener HC, Lucas S, et al. Randomized, controlled trial of telcagepant for the acute treatment of migraine. Neurology 2009;73:970–7.
19. Cottrell GS, Alemi F, Kirkland JG, Grady EF, et al. Localization of calcitonin receptor-like receptor (CLR) and receptor activity-modifying protein 1 (RAMP1) in human gastrointestinal tract. Peptides 2012;35:202–11.
20. Covasala O, Stirn SL, Albrecht S, De Col R, et al. Calcitonin gene-related peptide receptors in rat trigeminal ganglion do not control spinal trigeminal activity. J Neurophysiol 2012;108:431–40.
21. Cui XP, Ye JX, Lin H, Mu JS, et al. Efficacy, safety, and tolerability of telcagepant in the treatment of acute migraine: a meta-analysis. Pain pract 2015;15:124–31.
22. Diener HC, Barbanti P, Dahlof C, Reuter U, et al. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: results from a phase II study. Cephalalgia 2010;31:573–84.
23. Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 2014;13:1100–7.
24. Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, et al. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol 2014;13:885–92.
25. Dux M, Rosta J, Santha P, Jancso G. Involvement of capsaicin-sensitive afferent nerves in the proteinase-activated receptor 2-mediated vasodilatation in the rat dura mater. Neuroscience 2009;161:887–94.
26. Edvinsson L, Ekman R, Jansen I, McCulloch J, et al. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 1987;7:720–8.
27. Edvinsson L, Nilsson E, Jansen-Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8–37), a CGRP antibody and an RNA-Spiegelmer on CGRP induced vasodilatation in the perfused and non-perfused rat middle cerebral artery. Br J Pharmacol 2007;150:633–40.
28. Edvinsson L, Villalon CM, MaassenVanDenBrink A. Basic mechanisms of migraine and its acute treatment. Pharmacol Ther 2012;136:319–33.
29. Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol 2015;80:193–9.
30. Eftekhari S, Gaspar RC, Roberts R, Chen TB, et al. Localization of CGRP receptor components and receptor binding sites in rhesus monkey brainstem: A detailed study using in situ hybridization, immunofluorescence, and autoradiography. J Comp Neurol 2016;524:90–118.
31. Eftekhari S, Salvatore CA, Calamari A, Kane SA, et al. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 2010;169:683–96.
32. Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 2013;14:1289–303.
33. Evans BN, Rosenblatt MI, Mnayer LO, Oliver KR, et al. CGRP-RCP, a novel protein required for signal transduction at calcitonin gene-related peptide and adrenomedullin receptors. J Biol Chem 2000;275:31438–43.
34. Fabbretti E, D‘Arco M, Fabbro A, Simonetti M, et al. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci 2006;26:6163–71.
35. Fischer MJ, Koulchitsky S, Messlinger K. The nonpeptide calcitonin gene-related peptide receptor antagonist BIBN4096BS lowers the activity of neurons with meningeal input in the rat spinal trigeminal nucleus. J Neurosci 2005;25:5877–83.
36. Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of man and the cat during activation of the trigeminovascular system. Ann Neurol 1988;23:193–6.
37. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993;33:48–56.
38. Gupta S, Akerman S, van den Maagdenberg AM, Saxena PR, et al. Intravital microscopy on a closed cranial window in mice: a model to study trigeminovascular mechanisms involved in migraine. Cephalalgia 2006;26:1294–303.
39. Gupta S, Mehrotra S, Villalon CM, Perusquia M, et al. Potential role of female sex hormones in the pathophysiology of migraine. Pharmacol Ther 2007;113:321–40.
40. Hagner S, Stahl U, Knoblauch B, McGregor GP, et al. Calcitonin receptor-like receptor: identification and distribution in human peripheral tissues. Cell Tissue Res 2002;310:41–50.
41. Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 2010;30:1179–86.
42. Hepp Z, Dodick DW, Varon SF, Gillard P, et al. Adherence to oral migraine-preventive medications among patients with chronic migraine. Cephalalgia 2015;35:478–88.
43. Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 2011;31:712–22.
44. Hewitt DJ, Martin V, Lipton RB, Brandes J, et al. Randomized controlled study of telcagepant plus ibuprofen or acetaminophen in migraine. Headache 2011;51:533–43.
45. Ho AP, Dahlof CG, Silberstein SD, Saper JR, et al. Randomized, controlled trial of telcagepant over four migraine attacks. Cephalalgia 2010;30:1443–57.
46. Ho TW, Connor KM, Zhang Y, Pearlman E, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology 2014;83:958–66.
47. Ho TW, Ferrari MD, Dodick DW, Galet V, et al. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet 2008;372:2115–23.
48. Ho TW, Ho AP, Chaitman BR, Johnson C, et al. Randomized, controlled study of telcagepant in patients with migraine and coronary artery disease. Headache 2012;52:224–35.
49. Ho TW, Ho AP, Ge YJ, Assaid C, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for prevention of headache in women with perimenstrual migraine. Cephalalgia 2016;36:148–61.
50. Ho TW, Mannix LK, Fan X, Assaid C, et al. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurology 2008;70:1304–12.
51. Holzer P. Peptidergic sensory neurons in the control of vascular functions: mechanisms and significance in the cutaneous and splanchnic vascular beds. Rev Physiol Biochem Pharmacol 1992;121:49–146.
52. Hostetler ED, Joshi AD, Sanabria-Bohorquez S, Fan H, et al. In vivo quantification of calcitonin gene-related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK-4232. J Pharmacol Exp Ther 2013;347:478–86.
53. Hou Q, Barr T, Gee L, Vickers J, et al. Keratinocyte expression of calcitonin gene-related peptide beta: implications for neuropathic and inflammatory pain mechanisms. Pain 2011;152:2036–51.
54. Humphrey PPA, Feniuk W, Perren M, Oxford AW, et al. Sumatriptan succinate. Drugs Future 1989;14:35–9.
55. Itabashi A, Kashiwabara H, Shibuya M, Tanaka K, et al. The interaction of calcitonin gene-related peptide with angiotensin II on blood pressure and renin release. J Hypertens Suppl 1988;6:S418–20.
56. Jansen-Olesen I, Mortensen A, Edvinsson L. Calcitonin gene-related peptide is released from capsaicin-sensitive nerve fibres and induces vasodilatation of human cerebral arteries concomitant with activation of adenylyl cyclase. Cephalalgia 1996;16:310–6.
57. Keller JT, Marfurt CF. Peptidergic and serotoninergic innervation of the rat dura mater. J Comp Neurol 1991;309:515–34.
58. Kendall MD, al-Shawaf AA. Innervation of the rat thymus gland. Brain Behav Immun 1991;5:9–28.
59. Kurosawa M, Messlinger K, Pawlak M, Schmidt RF. Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene-related peptide. Br J Pharmacol 1995;114:1397–402.
60. Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, et al. CGRP may play a causative role in migraine. Cephalalgia 2002;22:54–61.
61. Lassen LH, Jacobsen VB, Haderslev PA, Sperling B, et al. Involvement of calcitonin gene-related peptide in migraine: regional cerebral blood flow and blood flow velocity in migraine patients. J Headache Pain 2008;9:151–7.
62. Lennerz JK, Ruhle V, Ceppa EP, Neuhuber WL, et al. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol 2008;507:1277–99.
63. Levy D, Burstein R, Kainz V, Jakubowski M, et al. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 2007;130:166–76.
64. Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 2005;58:698–705.
65. Li J, Vause CV, Durham PL. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res 2008;1196:22–32.
66. Li YJ, Song QJ, Xiao J. Calcitonin gene-related peptide: an endogenous mediator of preconditioning. Acta Pharmacol Sin 2000;21:865–9.
67. Linscheid P, Seboek D, Schaer DJ, Zulewski H, et al. Expression and secretion of procalcitonin and calcitonin gene-related peptide by adherent monocytes and by macrophage-activated adipocytes. Crit Care Med 2004;32:1715–21.
68. Lundberg JM, Franco-Cereceda A, Alving K, Delay-Goyet P, et al. Release of calcitonin gene-related peptide from sensory neurons. Ann N Y Acad Sci 1992;657:187–93.
69. Ma W, Chabot JG, Powell KJ, Jhamandas K, et al. Localization and modulation of calcitonin gene-related peptide-receptor component protein-immunoreactive cells in the rat central and peripheral nervous systems. Neuroscience 2003;120:677–94.
70. Marcus R, Goadsby PJ, Dodick D, Stock D, et al. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia 2014;34:114–25.
71. May A, Gijsman HJ, Wallnöfer A, Jones R, et al. Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain 1996;67:375–8.
72. McLatchie LM, Fraser NJ, Main MJ, Wise A, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998;393:333–9.
73. Messlinger K, Hanesch U, Baumgartel M, Trost B, et al. Innervation of the dura mater encephali of cat and rat: ultrastructure and calcitonin gene-related peptide-like and substance P-like immunoreactivity. Anat Embryol (Berl) 1993;188:219–37.
74. Mikami N, Matsushita H, Kato T, Kawasaki R, et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J Immunol 2011;186:6886–93.
75. Mizutani A, Okajima K, Murakami K, Mizutani S, et al. Activation of sensory neurons reduces ischemia/reperfusion-induced acute renal injury in rats. Anesthesiology 2009;110:361–9.
76. O‘Connor TP, van der Kooy D. Pattern of intracranial and extracranial projections of trigeminal ganglion cells. J Neurosci 1986;6:2200–7.
77. Olesen J, Diener HC, Husstedt IW, Goadsby PJ, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 2004;350:1104–10.
78. Pardridge WM, Kang YS, Buciak JL, Yang J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm Res 1995;12:807–16.
79. Penfield W, McNaughton FL. Dural headache and the innervation of the dura mater. Arch Neurol Psychiatry 1940;44:43–75.
80. Pozo-Rosich P, Storer RJ, Charbit AR, Goadsby PJ. Periaqueductal gray calcitonin gene-related peptide modulates trigeminovascular neurons. Cephalalgia 2015;35:1298–307.
81. Roon KI, Olesen J, Diener HC, Ellis P, et al. No acute antimigraine efficacy of CP-122,288, a highly potent inhibitor of neurogenic inflammation: results of two randomized, double-blind, placebo-controlled clinical trials. Ann Neurol 2000;47:238–41.
82. Saito K, Markowitz S, Moskowitz MA. Ergot alkaloids block neurogenic extravasation in dura mater: proposed action in vascular headaches. Ann Neurol 1988;24:732–7.
83. Schaible HG, Ebersberger A, Peppel P, Beck U, et al. Release of immunoreactive substance P in the trigeminal brain stem nuclear complex evoked by chemical stimulation of the nasal mucosa and the dura mater encephali – a study with antibody microprobes. Neuroscience 1997;76:273–84.
84. Schoonman GG, van der Grond J, Kortmann C, van der Geest RJ, Terwindt GM, Ferrari MD. Migraine headache is not associated with cerebral or meningeal vasodilatation–a 3T magnetic resonance angiography study. Brain 2008;131:2192–200.
85. Schwenger N, Dux M, de Col R, Carr R, et al. Interaction of calcitonin gene-related peptide, nitric oxide and histamine release in neurogenic blood flow and afferent activation in the rat cranial dura mater. Cephalalgia 2007;27:481–91.
86. Seiler K, Nusser JI, Lennerz JK, Neuhuber WL, et al. Changes in calcitonin gene-related peptide (CGRP) receptor component and nitric oxide receptor (sGC) immunoreactivity in rat trigeminal ganglion following glyceroltrinitrate pretreatment. J Headache Pain 2013;14:74.
87. Stankewitz A, Aderjan D, Eippert F, May A. Trigeminal nociceptive transmission in migraineurs predicts migraine attacks. J Neurosci 2011;31:1937–43.
88. Storer RJ, Akerman S, Goadsby PJ. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol 2004;142:1171–81.
89. Strassman AM, Weissner W, Williams M, Ali S, et al. Axon diameters and intradural trajectories of the dural innervation in the rat. J Comp Neurol 2004;473:364–76.
90. Sun H, Dodick DW, Silberstein S, Goadsby PJ, et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15:382–90.
91. Tache Y. Brainstem neuropeptides and vagal protection of the gastric mucosal against injury: role of prostaglandins, nitric oxide and calcitonin-gene related peptide in capsaicin afferents. Curr Med Chem 2012;19:35–42.
92. Tfelt-Hansen PC, Koehler PJ. One hundred years of migraine research: major clinical and scientific observations from 1910 to 2010. Headache 2011;51:752–78.
93. Toda M, Suzuki T, Hosono K, Kurihara Y, et al. Roles of calcitonin gene-related peptide in facilitation of wound healing and angiogenesis. Biomed Pharmacother 2008;62:352–9.
94. Ueda T, Ugawa S, Saishin Y, Shimada S. Expression of receptor-activity modifying protein (RAMP) mRNAs in the mouse brain. Brain Res Mol Brain Res 2001;93:36–45.
95. van Rossum D, Hanisch UK, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev 1997;21:649–78.
96. Vause CV, Durham PL. Calcitonin gene-related peptide differentially regulates gene and protein expression in trigeminal glia cells: findings from array analysis. Neurosci Lett 2010;473:163–7.
97. Voss T, Lipton RB, Dodick DW, Dupre N, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia 2016;36:887–98.
98. Walker CS, Eftekhari S, Bower RL, Wilderman A, et al. A second trigeminal CGRP receptor: function and expression of the AMY1 receptor. Ann Clin Transl Neurol 2015;2:595–608.
99. Wang H, Xing L, Li W, Hou L, et al. Production and secretion of calcitonin gene-related peptide from human lymphocytes. J Neuroimmunol 2002;130:155–62.
100. Williamson DJ, Hargreaves RJ, Hill RG, Shepheard SL. Intravital microscope studies on the effects of neurokinin agonists and calcitonin gene-related peptide on dural vessel diameter in the anaesthetized rat. Cephalalgia 1997;17:518–24.
101. Wimalawansa SJ, Emson PC, MacIntyre I. Regional distribution of calcitonin gene-related peptide and its specific binding sites in rats with particular reference to the nervous system. Neuroendocrinology 1987;46:131–6.
102. Wimalawansa SJ, MacIntyre I. The presence of calcitonin gene-related peptide in human cerebrospinal fluid. Brain 1987;110:1647–55.
103. Xing LY, Xing YT, Tang YM, Guo JX, et al. Expression of calcitonin gene-related peptide (CGRP) mRNA in rat lymphocytes. Sheng Li Xue Bao 1998;50:423–30.
104. Xu G, Jiang D. The role and mechanism of exogenous calcitonin gene-related peptide on mesenchymal stem cell proliferation and osteogenetic formation. Cell Biochem Biophys 2014;69:369–78.
105. Zaidi M, Breimer LH, MacIntyre I. Biology of peptides from the calcitonin genes. Q J Exp Physiol 1987;72:371–408.
106. Zeller J, Poulsen KT, Sutton JE, Abdiche YN, et al. CGRP function-blocking antibodies inhibit neurogenic vasodilatation without affecting heart rate or arterial blood pressure in the rat. Br J Pharmacol 2008;155:1093–103.
107. Zhang JY, Yan GT, Liao J, Deng ZH, et al. Leptin attenuates cerebral ischemia/reperfusion injury partially by CGRP expression. Eur J Pharmacol 2011;671:61–9.
108. Zheng LR, Han J, Yao L, Sun YL, et al. Up-regulation of calcitonin gene-related peptide protects streptozotocin-induced diabetic hearts from ischemia/reperfusion injury. Int J Cardiol 2012;156:192–8.
109. Zlokovic BV, Skundric DS, Segal MB, Lipovac MN, et al. A saturable mechanism for transport of immunoglobulin G across the blood-brain barrier of the guinea pig. Exp Neurol 1990;107:263–70.
Prof. Dr.med. Karl Messlinger, Institut für Physiologie und Pathophysiologie, Friedrich-Alexander Universität Erlangen-Nürnberg, Universitätsstr. 17, 91054 Erlangen
Priv.-Doz. Dr.med. Dagny Holle-Lee, Prof. Dr.med. Hans-Christoph Diener, Westdeutsches Kopfschmerzzentrum und Neurologische Klinik der Universität Duisburg-Essen, Hufelandstr. 55, 45147 Essen, E-Mail: hans.diener@uk-essen.de
Significance of the neuropeptide CGRP in primary headache
Etiology and pathophysiology of primary headache disorders such as migraine and cluster headache is only partly understood. Calcitonin gene -related peptide (CGRP) seems to be involved as biological mediator in both headache entities. CGRP is a vasoactive neuropetide that is secreted by primary afferent nerve fibres. In most organs it has protective effects, but it might also cause headache attacks in migraineurs. The underlying mechanisms are largely unknown. Particularly, it is currently intensively discussed if CGRP has predominantly peripherial effects or central effects. Despite this uncertainty, actual and future pharmacotherapy of migraine and cluster headache targets at the release inhibition of CGRP through selective 5-HT1B/D-agonists (so-called triptans) or the blockade of CGRP receptors by antagonists (so-called gepants). The newest approach is the application of humanized monoclonal antibodies. These antibodies, which are right now in clinical development, inactivate CGRP molecules themselves or their receptors. Much hope is set on antibodies especially regarding therapy of high-frequent and chronic migraine and cluster headache, as they might offer a therapeutic approach without severe cardiovascular or hepatic side effects.
Key words: Migraine, cluster headache, triptans, CGRP, acute therapy, prophylactic therapy
Psychopharmakotherapie 2017; 24(02)