Holger Petri, Bad Wildungen*
Fluconazol
Fluconazol unterliegt minimalen metabolischen Reaktionen. Über 80% von oral eingenommenem Fluconazol werden unverändert renal ausgeschieden [1]. Als starker CYP2C9- und CYP2C19-Hemmer sind zahlreiche Wechselwirkungen zu beachten [14, 20]. Zu den CYP2C9-Substraten gehören die nichtsteroidalen Antirheumatika Celecoxib, Ibuprofen und Naproxen, die Antidiabetika Glibenclamid und Glimepirid, die Vitamin-K-Antagonisten Phenprocoumon und Warfarin sowie Fluvastatin und Phenytoin [13]. In der Fachinformation wird eine engmaschige Überwachung der Patienten auf dosisabhängige Nebenwirkungen empfohlen [5]. Unter einer Phenytoin-Therapie ist ein therapeutisches Drug-Monitoring (TDM) obligat [10]. Bei den CYP2C19-Substraten sind unter anderem Interaktionen mit dem Thrombozytenaggregationshemmer Clopidogrel und den Serotonin-Wiederaufnahmehemmern (SSRI) Citalopram/Escitalopram von klinischer Relevanz. Clopidogrel ist ein Prodrug. Fluconazol kann dessen Bioaktivierungsschritt mindern und folglich die Wirksamkeit verringern [6]. Das Risiko für eine Serotonin-Intoxikation ist bei Komedikation mit Citalopram erhöht [22].
Die CYP3A4-hemmende Potenz ist, verglichen mit anderen Azol-Antimykotika, geringer [37]. Die Anwendung von Fluconazol ist mit CYP3A4-Substraten, die QTc-Zeit-verlängernde Eigenschaften besitzen wie Amiodaron und Erythromycin, kontraindiziert [5]. Fluconazol hat selbst ein hohes Potenzial, Torsade de pointes zu verursachen [35]. Die Kombination mit den beiden SSRI Citalopram und Escitalopram ist schon aus diesem Grund kontraindiziert [26].
Isavuconazol
Isavuconazol ist Substrat von CYP3A4. Ketoconazol erhöht die Exposition von Isavuconazol um 422%, Lopinavir/Ritonavir um das Doppelte [32, 36]. Mit Ausnahme von Ketoconazol dürfen andere starke CYP3A4-Inhibitoren ohne Dosisanpassung mit Isavuconazol kombiniert werden [4]. Der starke CYP3A4-Induktor Rifampicin senkt die Plasmaspiegel des Antimykotikums auf ein Zehntel [32]. Starke und mittelstarke CYP3A4-Induktoren (Abb. 1) sind unter Isavuconazol-Therapie kontraindiziert [4]. Es sollte beachtet werden, dass selbst nach deren Absetzen die induktiven Effekte noch bis zu zwei Wochen anhalten können (Deinduktionszeit) [28]. Schwache CYP3A4-Induktoren sollten gemieden werden, sofern der potenzielle Nutzen nicht die Risiken überwiegt [4]. Isavuconazol ist als mittelstarker CYP3A4-Hemmer einzustufen [4]. In einer Phase-I-Studie erhöhte das Azol-Antimykotikum die AUC (Fläche unter der Konzentrations-Zeit-Kurve) des oral eingenommen CYP3A4-Modellsubstrats Midazolam um das Doppelte [32].
Itraconazol
Itraconazol wird hauptsächlich über CYP3A4 metabolisiert. Es werden sequenziell die drei Itraconazol-Metabolite Hydroxy-Itraconazol (OH-ITZ), Keto-Itraconazol (keto-ITZ) und N-Desalkyl-Itraconazol (ND-ITZ) gebildet. Diese Metaboliten tragen signifikant zur starken CYP3A4-Hemmung unter Itraconazol-Anwendung in vivo bei [11, 31]. Die gleichzeitige Anwendung von Itraconazol mit CYP3A4-Substraten, die die QTc-Zeit verlängern können, ist kontraindiziert [7]. Tabelle 2 enthält eine Auswahl an CYP3A4-Substraten, die zusammen mit potenten CYP3A4-Inhibitoren nicht oder nur mit Vorsicht anwendbar sind. Selbst bei Inhalation von Budesonid wird dessen Abbau durch Itraconazol so stark gehemmt, dass trotz lokaler Anwendung mit systemischen Glucocorticoid-Wirkungen zu rechnen ist [27].
Tab. 2. CYP3A4-Substrate (Beispiele)
|
CYP3A4-Substrate |
|
|
Antiinfektiva |
Atazanavir, Clarithromycin, Clindamycin, Cobicistat, Darunavir, Erythromycin, Lopinavir, Ritonavir |
|
Antineoplastische Mittel |
Axitinib, Bortezomib, Bosutinib, Cabazitaxel, Ceritinib, Crizotinib, Cyclophosphamid, Dabrafenib, Erlotinib, Idelalisib, Ifosfamid, Imatinib, Irinotecan, Lapatinib, Olaparib, Pazopanib, Regorafenib, Temsirolimus, Vincristin, Vinorelbin |
|
Blut-, Herz- und Kreislaufmittel |
Amiodaron, Atorvastatin, Dronedaron, Eplerenon, Felodipin, Ivabradin, Lercanidipin, Nitrendipin, Ranolazin, Simvastatin, Ticagrelor, Verapamil |
|
Immunsuppressiva |
Ciclosporin, Everolimus, Sirolimus, Tacrolimus |
|
Urologika |
Avanafil, Alfuzosin, Darifenacin, Dutasterid, Finasterid, Sildenafil, Solifenacin, Tadalafil, Vardenafil |
|
ZNS-Mittel |
Carbamazepin, Donepezil, Eletriptan, Fentanyl, Galantamin, Midazolam oral, |
|
Weitere |
Colchicin, Dexamethason, Domperidon, Ergotamin, Etoricoxib, Tolvaptan |
Quelle: mediQ-Interaktionsprogramm (Stand 10/2016)
Die Plasmaspiegel von Itraconazol werden durch CYP3A4-Induktoren erniedrigt. Bei gesunden Probanden sanken die Itraconazol-AUC-Werte unter Phenytoin- und Rifampicin-Therapie um etwa 90% [2, 12]. Somit sollte Itraconazol nicht früher als zwei Wochen nach Absetzen einer Behandlung mit CYP3A4-induzierenden Wirkstoffen angewendet werden. Die gemeinsame Anwendung von Itraconazol mit diesen Wirkstoffen kann zu subtherapeutischen Plasmakonzentrationen von Itraconazol und damit zum Therapieversagen führen [7].
Posaconazol
Posaconazol wird in nur geringem Umfang über CYP-Enzyme metabolisiert. Das Potenzial für CYP-bedingte Veränderungen der Plasmaspiegel ist gering [15]. Posaconazol unterliegt primär Phase-II-Reaktionen, wobei der Hauptmetabolit über die Uridin-5’-diphosphat-Glucuronosyltransferase (UGT) 1A4 gebildet wird [8]. Die UGT-Induktion durch Efavirenz, Phenytoin und Rifabutin beschleunigt die Clearance von Posaconazol, die zu einer Abnahme der AUC auf die Hälfte führt [17–19].
Posaconazol ist ein starker Inhibitor von CYP3A4 [16].
Voriconazol
Voriconazol wird primär über das polymorphe Enzym CYP2C19 und nachgeordnet über CYP2C9 und CYP3A4 abgebaut. Personen mit nicht oder eingeschränkt funktionsfähigen CYP2C19-Enzymen (poor und intermediate metabolizer, PM und IM) haben gegenüber normalen Verstoffwechslern (extensive metabolizer, EM) drei- (PM-Status) bzw. zweifach (IM-Status) erhöhte AUC-Werte [21, 24]. Personen mit gesteigerter Enzymaktivität (ultrarapid metabolizer, UM) zeigen eine erhöhte Voriconazol-Clearance und es drohen subtherapeutische Wirkstoffspiegel [9, 25, 33]. Der CYP2C19-Genotyp ist auch von Relevanz für potenzielle Interaktionen. Die Pharmakokinetik des CYP2C19-Modellsubstrats S-Mephenytoin blieb unverändert bei Probanden mit einem PM-Status, die über einen Zeitraum von 14 Tagen den CYP2C19-Induktor Johanniskraut (Abb. 1) einnahmen [34]. Daher gewinnt die Interaktion mit CYP3A4-Modulatoren bei Patienten mit dem PM-Phänotyp an Bedeutung, weil ein größerer Anteil von Voriconazol über diesen Nebenweg abgebaut wird. Beispielsweise sinkt die Clearance durch die CYP3A4-Inhibitoren Erythromycin und Ritonavir bei CYP2C19-Poor-metabolizern stärker als bei extensive metabolizern [23, 30]. Die Interaktionstabelle enthält Angaben zur Häufigkeit von CYP2C19-Polymorphismen, auch in Abhängigkeit von der ethnischen Herkunft.
Voriconazol ist ein starker CYP3A4-Inhibitor [29].
Fazit
Azol-Antimykotika bergen ein hohes pharmakokinetisches Interaktionspotenzial. Die Überprüfung der klinischen Relevanz von Wechselwirkungen mithilfe einer Interaktionsdatenbank ist obligat. Bei oberflächlichen Mykosen ist Terbinafin eine Alternative. Terbinafin hemmt als CYP2D6-Inhibitor den Metabolismus von CYP2D6-Substraten wie Metoprolol, Fluoxetin, Tamoxifen u.a. [3]. Bei systemischen Mykosen kommen Echinocandine infrage, die jedoch für eine orale Einnahme nicht zur Verfügung stehen.
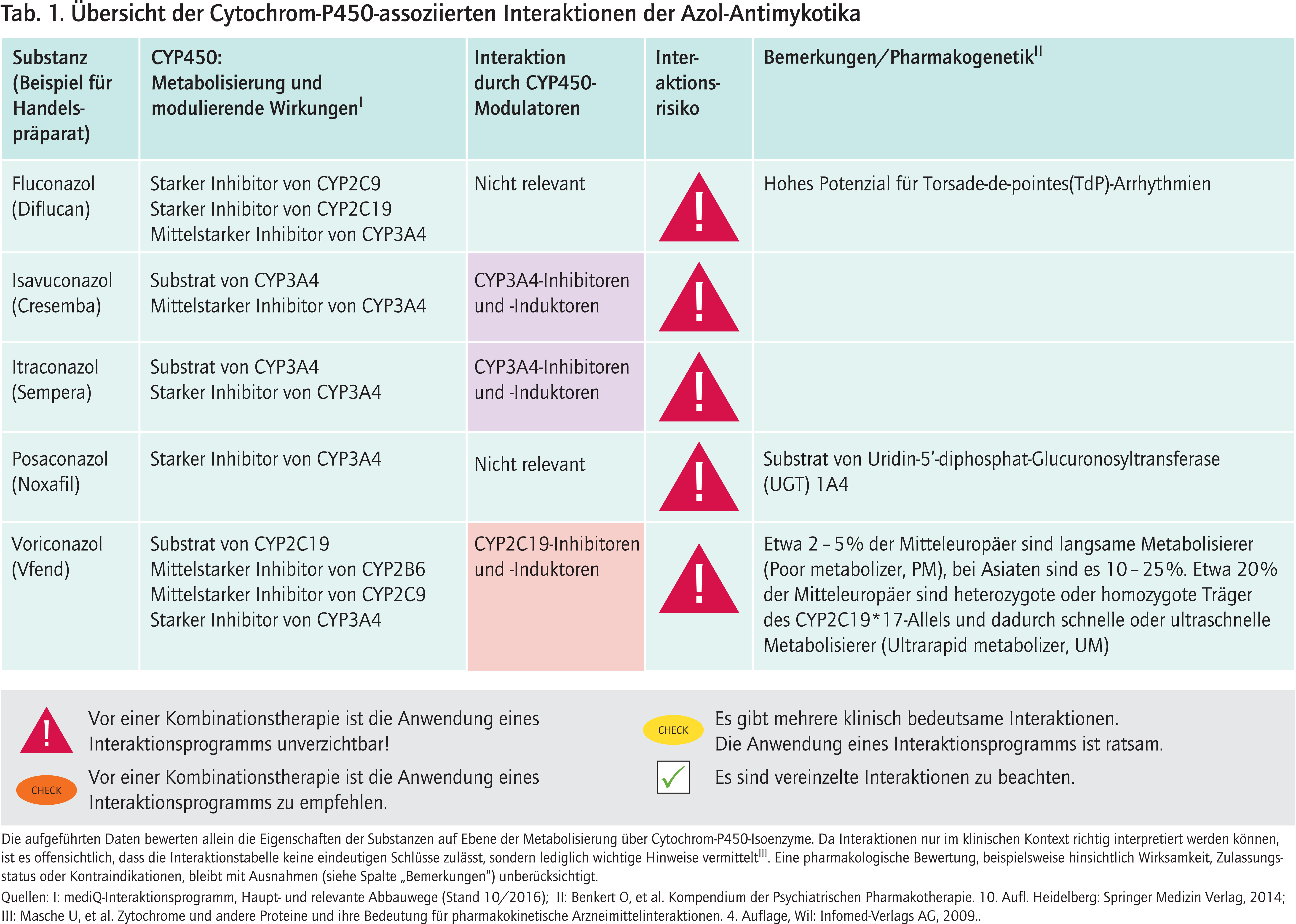
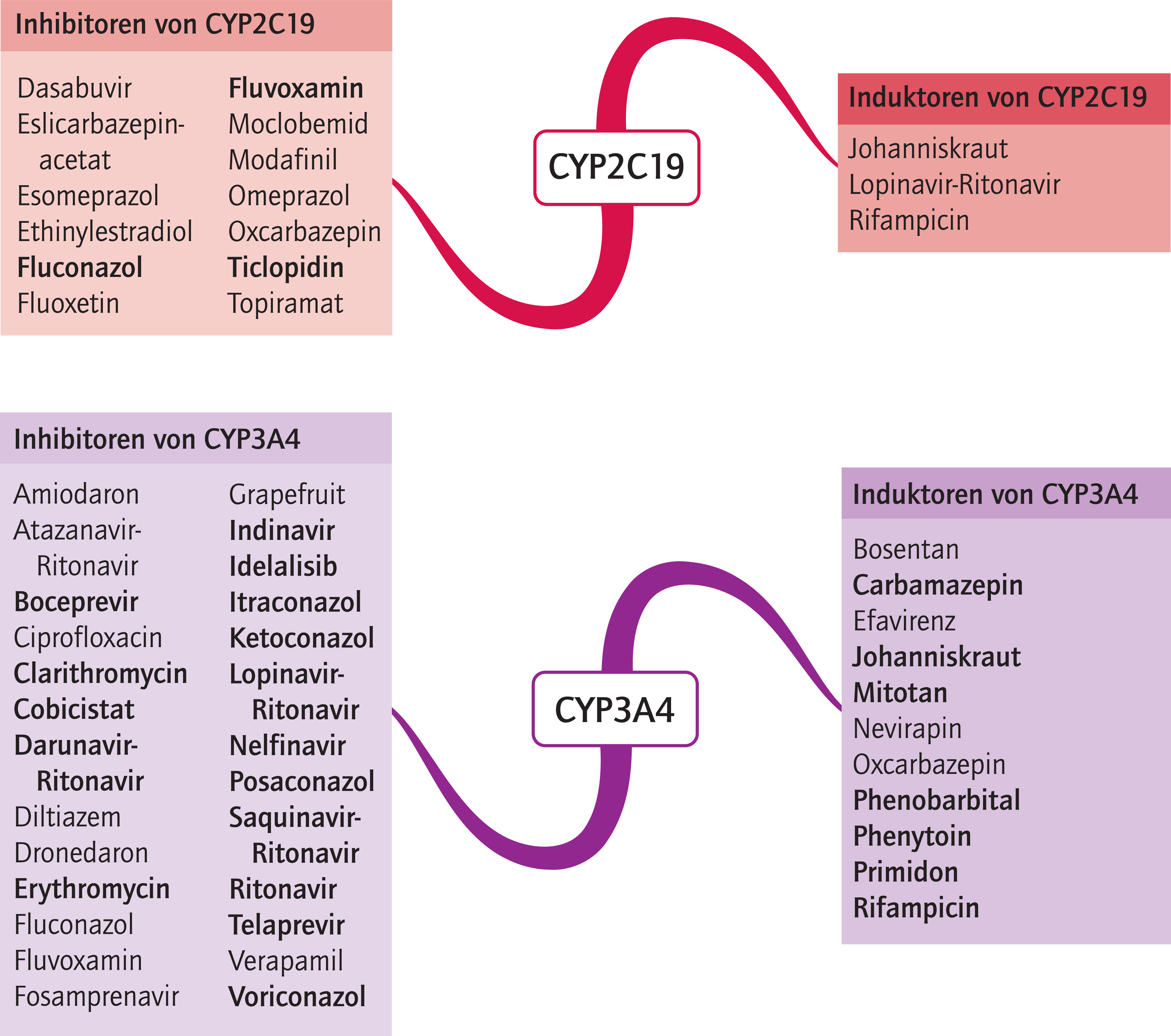
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 (CYP) 2C19 und 3A4 (Stand: 10/2016) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Brammer KW, Coakley AJ, Jezequed SG, et al. The disposition and metabolism of [14C]fluconazole in humans. Drug Metab Dispos 1991;19:764–7.
2. Ducharme MP, Slaughter RL, Warbasse LH, et al. Itraconazole and hydroxyitraconazole serum concentrations are reduced more than tenfold by phenytoin. Clin Pharmacol Ther 1995;58:617–24.
3. Dürrbeck A, Nenoff P. Terbinafin-relevante Arzneimittelinteraktionen und deren Management. Hautarzt 2016;67:718–23.
4. Fachinformation Cresemba. Stand: Oktober 2015.
5. Fachinformation Diflucan®. Stand: Juli 2015.
6. Fachinformation Plavix®. Stand: September 2015.
7. Fachinformation Sempera®. Stand: Dezember 2014.
8. Ghosal A, Hapangama N, Yuan Y, et al. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of posaconazole (Noxafil). Drug Metab Dispos 2004;32:267–71.
9. Hicks JK, Crews KR, Flynn Pet al. Voriconazole plasma concentrations in immunocompromised pediatric patients vary by CYP2C19 diplotypes. Pharmacogenomics 2014;15:1065–78.
10. Hiemke C, Baumann P, Bergemann N, et al. AGNP-Konsensus-Leitlinien für therapeutisches Drug-Monitoring in der Psychiatrie: Update 2011. Psychopharmakotherapie 2012;19:91–122.
11. Isoherranen N, Kunze KL, Allen KE, et al. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos 2004;10:1121–31.
12. Jaruratanasirikul S, Sriwiriyajan S. Effect of rifampicin on the pharmacokine- tics of itraconazole in normal volunteers and AIDS patients. Eur J Clin Pharmacol 1998;54:155–8.
13. Kämmerer W. Porträt eines Enzyms – CYP2C9. Arzneimitteltherapie 2012;30:123–5.
14. Kang BC, Yang CQ, Cho HK, et al. Influence of fluconazole on the pharmacokinetics of omeprazole in healthy volunteers. Biopharm Drug Dispos 2002;23:77–81.
15. Krieter P, Flannery B, Musick T, et al. Disposition of posaconazole following single-dose oral administration in healthy subjects. Antimicrob Agents Chemoter 2004;48:3543–51.
16. Krishna G, Ma L, Prasad P, et al. Effect of posaconazole on the pharmacokinetics of simvastatin and midazolam in healthy volunteers. Expert Opin Drug Metab Toxicol 2012;8:1–10.
17. Krishna G, Moton A, Ma L, et al. Effects of oral posaconazole on the pharmacokinetics of atazanavir alone and with ritonavir or with efavirenz in healthy adult volunteers. J Acquir Immune Defic Syndr 2009;51:437–44.
18. Krishna G, Parsons A, Kantesaria B. Evaluation of the pharmacokinetics of posaconazole and rifabutin following co-administration to healthy men. Curr Med Res Opin 2007;23:545–52.
19. Krishna G, Sansone-Parsons A, Kantesaria B. Drug interaction assessment following concomitant administration of posaconazole and phenytoin in healthy men. Curr Med Res Opin 2007;23:1415–22.
20. Kumar V, Brundage RC, Oetting WS, et al. Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab Dispos 2008;36:1242–8.
21. Lee S, Kim BH, Nam WS. Effect of CYP2C19 polymorphism on the pharmacokinetics of voriconazole after single and multiple doses in healthy volunteers. J Clin Pharmacol 2012;52:195–203.
22. Levin TT, Cortes-Ladino A, Weiss M, et al. Life-threatening serotonin toxicity due to a citalopram-fluconazole drug interaction: case reports and discussion. Gen Hosp Psychiatry 2008;30:372–7.
23. Mikus G, Schöwel V, Drzewinska M, et al. Potent cytochrome P4502C19 genotype related interaction between voriconazole and the cytochrome P4503A4 inhibitor ritonavir. Clin Pharmacol Ther 2006;80:126–35.
24. Mikus G, Scholz IM, Weiss J. Pharmacogenomics of the triazole antifungal agent voriconazole. Pharmacogenomics 2011;12:861–72.
25. Pascual A, Calandra T, Bolay S et al. Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin Infect Dis 2008;46:201–11.
26. Petri H. Das Interaktionspotenzial der selektiven Serotonin-Wiederaufnahmehemmer. Krankenhauspharmazie 2013;34:85–7.
27. Raaska K, Niemi M, Neuvonen M. Plasma concentrations of inhaled budesonide and its effects on plasma cortisol are increased by the cytochrome P4503A4 inhibitor itraconazole. Clin Pharmacol Ther 2002;72:362–9.
28. Reitman ML, Chu X, Cai X, et al. Rifampin᾽s acute inhibitory and chronic inductive drug interactions: experimental and model-based approaches to drug-drug interaction trial design. Clin Pharmacol Ther 2011;89:234–42.
29. Saabri TI, Laine K, Leino K, et al. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin Pharmacol Ther 2006;79:362–70.
30. Shi HY, Yan j, Zhu WH, et al. Effects of erythromycin on voriconazole pharmacokinetics and association with CYP2C19 polymorphism. Eur J Clin Pharmacol 2010;66:1131–6.
31. Templeton I, Peng CC, Thummel KE. Accurate prediction of dose-dependent CYP3A4 inhibition by itraconazole and its metabolites from in vitro inhibition data. Clin Pharmacol Ther 2010;88:499–505.
32. Townsend R, Dietz A, Hale C, Akhtar S, et al. Pharmacokinetic evaluation of CYP3A4-mediated drug-drug interactions of isavuconazole with rifampin, ketoconazole, midazolam, and ethinyl estradiol/norethindrone in healthy adults. Clin Pharmacol Drug Dec 2016 Jun 8. doi: 10.1002/cpdd.285.
33. Wang G, Lei HP, Li Z, et al. The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol 2009; 65:281–5.
34. Wang LS, Zhu B, Abd El-Aty AM, et al. The influence of St John᾽s Wort on CYP2C19 activity with respect to genotype. J Clin Pharmacol 2004;44:577–81.
35. Woosley RL and Romero KA. www.crediblemeds.org/index.php/login/dlcheck (letzter Zugriff am 14 Sep 2016).
36. Yamazaki T, Desai A, Han D, et al. Pharmacokinetic interaction between isavuconazole and a fixed-dose combination of lopinavir 400 mg/ritonavir 100 mg in healthy subjects. Clin Pharmacol Drug Dev 2016 Jun 8. doi: 10.1002/cpdd.282.
37. Yang J, Atkins WM, Isoherranen N, et al. Evidence of CYP3A allosterism in vivo: analysis of interaction between fluconazole and midazolam. Clin Pharmacol Ther 2012;91:442–9.
*Nachdruck aus Krankenhauspharmazie 2016;37:506–10.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2016; 23(06)