Holger Petri, Bad Wildungen*
Dihydropyridin-Calciumkanalblocker: Amlodipin und Lercanidipin
Die Arzneimittel aus der Dihydropyridingruppe werden primär über das Cytochrom-P450(CYP)-Isoenzym 3A4 verstoffwechselt [1]. Das klinische Ansprechen auf die medikamentöse Therapie kann von einer Komedikation mit CYP3A4-Modulatoren abhängen. Die gleichzeitige Anwendung mit starken oder moderaten CYP3A4-Hemmern erhöht die Dihydropyridin-Exposition [1]. Folglich resultiert ein erhöhtes Hypotonierisiko. Der Blutdruck sollte daher überwacht werden und die Dosis bei Bedarf gesenkt werden [5]. Das Antibiotikum Clarithromycin ist ein starker Inhibitor von CYP3A4 (Abb. 1). In einer bevölkerungsbasierten Studie führte die Verordnung von Clarithromycin, verglichen mit Azithromycin, bei älteren Patienten, die Calciumkanalblocker vom Dihydropyridintyp einnahmen, zu häufigeren Hospitalisierungen aufgrund von akutem Nierenversagen [9]. Azithromycin ist kein CYP3A4-Inhibitor [1].
Lercanidipin ist nach Amlodipin der am zweithäufigsten verordnete Calciumkanalblocker [12]. In einer Interaktionsstudie mit dem starken CYP3A4-Inhibitor Ketoconazol (in Deutschland zugelassen zur Therapie des Cushing-Syndroms) stieg die AUC (area under the curve) von Lercanidipin um das 15-Fache [3]. Starke CYP3A4-Hemmer (Abb. 1) sind unter einer Lercanidipin-Therapie daher kontraindiziert.
Bei gleichzeitiger Verordnung von CYP3A4-Induktoren wie Carbamazepin oder Johanniskraut (Abb. 1) mit Dihydropyridinen kann es zu einem Wirkverlust kommen. Es wird empfohlen, den Blutdruck häufiger als üblich zu überwachen und bei Bedarf die Dosis zu erhöhen [5].
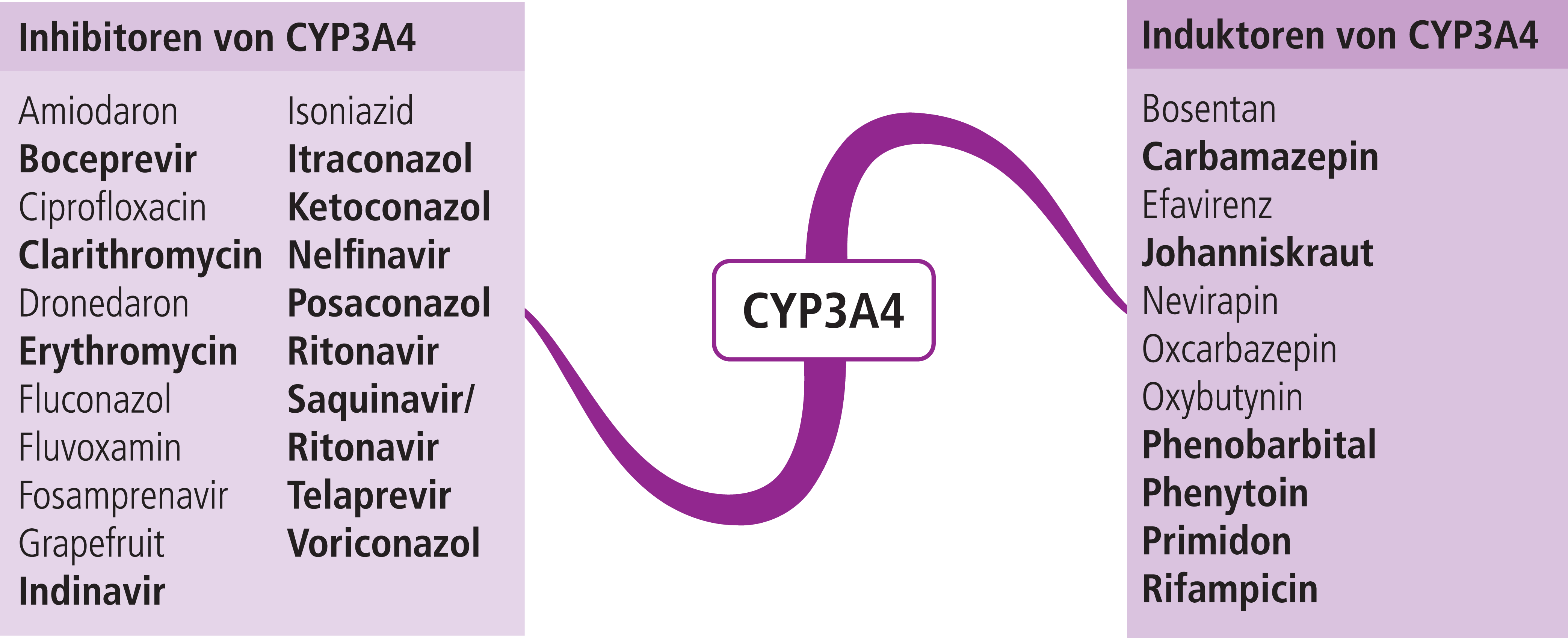
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf Cytochrom P450 3A4 (Stand: 10/2015) [Quelle: mediQ-Interaktionsprogramm]
Diltiazem und Verapamil
Die im Gegensatz zu den Dihydropyridinen auch antiarrhythmisch wirkenden Calciumkanalblocker Diltiazem und Verapamil sind ebenfalls Substrate von CYP3A4 [1]. Gleichzeitig sind sie moderate CYP3A4-Inhibitoren (Tab. 1). Die Plasmaspiegel von Substanzen mit einer engen therapeutischen Breite, die primär über CYP3A4 metabolisiert werden, können bei Komedikation mit Diltiazem oder Verapamil in einen kritischen Bereich steigen. Beispielsweise wird die Anwendung von Verapamil bei Patienten unter Fentanyl-Pflastertherapie nicht empfohlen, es sei denn, der Patient wird engmaschig überwacht [4].
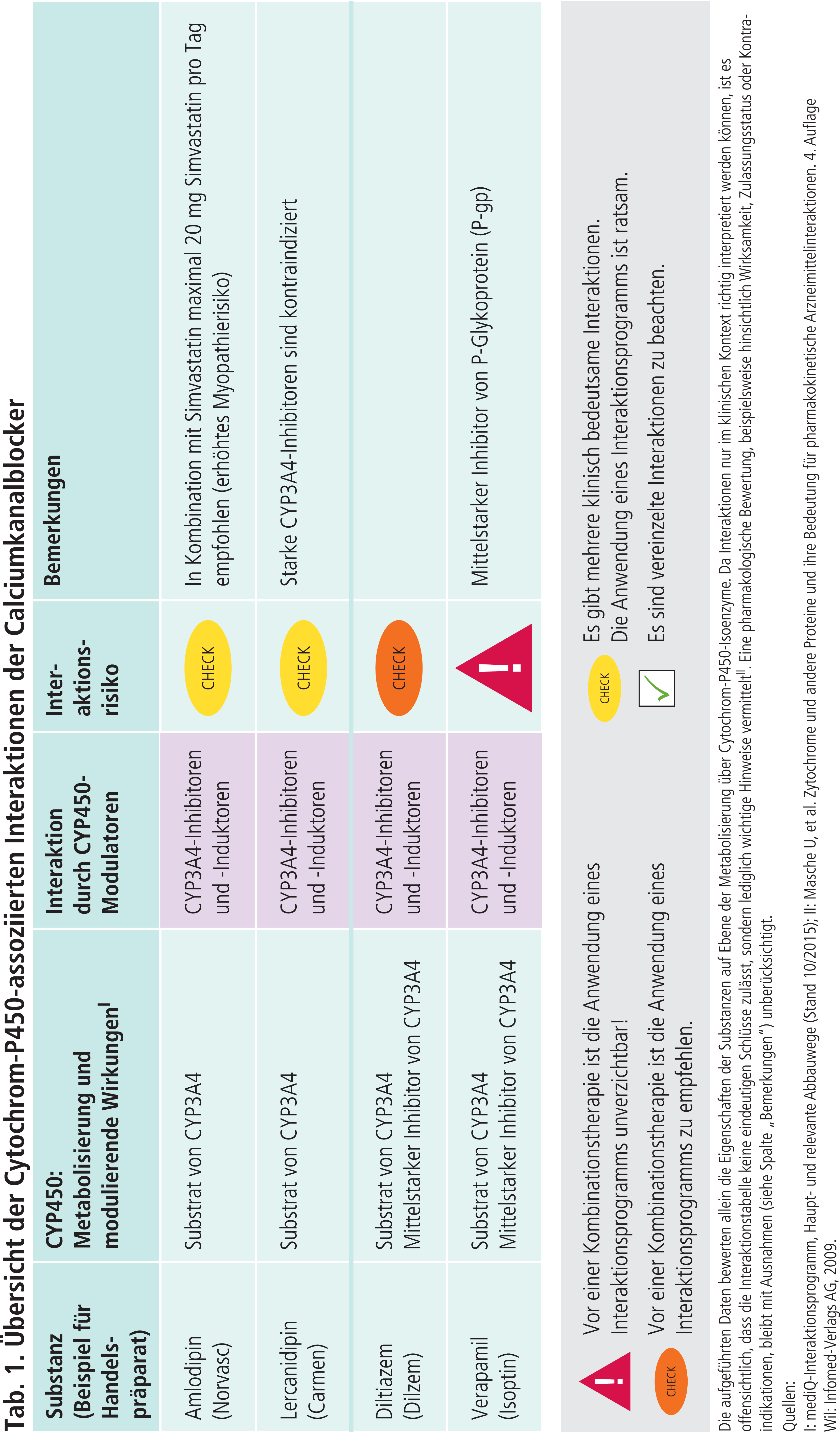
Verapamil ist zudem ein potenter Inhibitor des Efflux-Transporters P-Glykoprotein (P-gp) [11]. Dieser ist für die biliäre oder renale Ausscheidung wichtig. Die für den Efflux zuständigen Transporter kommen nicht nur in Leber und Niere vor, sondern auch in anderen Organen (Gastrointestinaltrakt, Blut-Hirn-Schranke, Plazenta, Hoden). Sie sollen den Körper vor Fremdstoffen (Xenobiotika) schützen [11]. Beispielsweise ist das Blutungsrisiko gesteigert, wenn Patienten unter Verapamil-Therapie das orale Antikoagulans Dabigatran erhalten. Dessen Prodrugester Dabigatranetexilat ist ein P-gp-Substrat und die Blockade des Efflux-Transporters durch Verapamil erhöht die Dabigatranexposition [6].
Pharmakokinetische Interaktionen sind auch möglich bei der gleichzeitigen Gabe von Verapamil mit dem hochpotenten Opioid-Analgetikum Buprenorphin. Tierexperimentell wurde nachgewiesen, dass der über CYP3A4 gebildete Metabolit Norbuprenorphin, im Gegensatz zur Muttersubstanz, Substrat von P-gp ist [2]. Norbuprenorphin besitzt eine schwache analgetische Wirkung. Wie beim P-gp-Substrat Loperamid werden keine suffizienten intrazerebralen Konzentrationen von Norbuprenorphin erreicht, da der Effluxtransporter das Opioid aus dem ZNS schleust. Bei P-gp-Knockout-Mäusen zeigt der Buprenorphin-Metabolit durch die Überwindung der Blut-Hirn-Schranke hingegen signifikante antinozizeptive Effekte. Mehr erwünschte und unerwünschte Opioid-Wirkungen von Buprenorphin beim Menschen könnten bei Komedikation mit dem P-gp-Inhibitor Verapamil auftreten [2, 10].
Calciumkanalblocker und Simvastatin
Simvastatin, der in Deutschland am häufigsten verschriebene HMG-CoA-Reductasehemmer, wird hauptsächlich über CYP3A4 abgebaut [1, 7, 12]. In deutschen Fachinformationen wird empfohlen, Simvastatin bei gleichzeitiger Anwendung mit Amlodipin, Diltiazem und Verapamil nicht höher als 20 mg pro Tag zu dosieren [7]. Dies, obwohl Amlodipin im Gegensatz zu Diltiazem und Verapamil nur ein schwacher Hemmer von CYP3A4 ist. Der unterschiedlichen CYP3A4-inhibierenden Potenz trägt die US-amerikanische Gesundheitsbehörde FDA Rechnung, indem bei Komedikation mit Diltiazem und Verapamil die Tagesdosis von Simvastatin auf 10 mg begrenzt wird [8].
Literatur
1. Böhm R, Reinecke K, Haen E, et al. Interaktionen mit CYP3A4. Dtsch Apo Ztg 2012;40:58–67.
2. Brown SM, et al. P-glycoprotein is a major determinant of norbuprenorphine brain exposure and antinociception. J Pharmacol Exp Ther 2012;343:53–61.
3. Fachinformation Carmen®. Stand: März 2008.
4. Fachinformation Durogesic®. Stand: Juli 2014.
5. Fachinformation Norvasc®. Stand: März 2015.
6. Fachinformation Pradaxa®. Stand: Dez 2014.
7. Fachinformation Zocor®. Stand: Juni 2015.
8. FDA Drug Safety communication 08.06.2011.
9. Garg AX, et al. Calcium-channel blocker – clarithromycin drug interactions and acute kidney injury. JAMA 2013;310:2544–53.
10. Megarbane B, Alhaddad H. P-glycoprotein should be considered as an additional factor contributing to opioid induced respiratory depression in paediatrics: the buprenorphine example. Br J Anaesth 2013; 110:842.
11. Reinecke K, Böhm R, Herdegen T, Cascorbi I. Arzneimittel und Transportproteine. Dtsch Apo Ztg 2015;16:44–51.
12. Schwabe U, Paffrath D. Arzneiverordnungs-Report 2015. Heidelberg: Springer, 2015.
*Nachdruck aus Krankenhauspharmazie 2015;36:557–9.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2015; 22(06)