Rainer Hellweg, Berlin, und Christoph Goemann, Hamburg
Depression ist die häufigste psychiatrische Erkrankung; gleichzeitig ist sie die Erkrankung, die für die Betroffenen die größte Behinderung im Alltagsleben darstellt, berechnet in der Einheit DALY (Disability adjusted life years). Laut einer Hochrechnung der World Health Organization (WHO) wird Depression bis zum Jahr 2030 weltweit die größte volkswirtschaftliche Belastung unter den Krankheiten darstellen [57].
Depression ist charakterisiert durch eine negative Wahrnehmung der Welt, der eigenen Person und der Zukunft. Die kognitiven Prozesse sind gekennzeichnet durch die Betonung negativer Informationen und die Unfähigkeit, sich von negativen Gedankengängen zu lösen, daher sind depressive Personen besonders anfällig für negatives Feedback aus ihrer Umgebung. Zusätzlich wird bei der Depression eine verminderte Reaktion auf erwartete oder tatsächliche Belohnung beobachtet, was eine kognitive Erklärung für eines der Kernsymptome der Depression, die Anhedonie, darstellt [56]. Kognitive Einschränkungen wie insbesondere Konzentrationsstörungen, aber auch Angst und Schlafstörungen können bei Depressiven zum Teil auch unabhängig von der depressiven Kernsymptomatik (wie der Anhedonie) auftreten [1, 38, 39, 43, 53].
Ätiologie der Depression
Epidemiologische Studien zeigen, dass etwa 40 bis 50% des Risikos, an Depression zu erkranken, genetisch bedingt sind. Dies macht Depression zu einer erblichen Krankheit, vergleichbar mit einigen anderen komplexen Erkrankungen, wie Diabetes mellitus Typ 2, Bluthochdruck, Asthma bronchiale oder einige Formen von Krebs, die häufig als genetisch bedingt angesehen werden. Allerdings konnte bis heute kein spezifisches Gen sicher mit der Entstehung von Depression in Verbindung gebracht werden. Dies liegt wahrscheinlich daran, dass Depression ein komplexes Phänomen ist, an dem viele Gene beteiligt sind und bei dem jedes einzelne Gen nur geringen Einfluss hat. Dazu kommt die Möglichkeit, dass in verschiedenen Familien unterschiedliche Gene für die Krankheit verantwortlich sind [36].
Neben den genetischen gibt es aber noch andere Faktoren, die als Auslöser einer Prädisposition für Depression diskutiert werden. Hierzu gehören so unterschiedliche Einflüsse wie Stress, emotionales Trauma (insbesondere in der Kindheit), virale Infektionen (z.B. Borna-Virus) oder zufällige Ereignisse während der Embryonalentwicklung des Gehirns. Depressive Syndrome, die alle Kriterien einer Major Depression erfüllen, werden auch im Zusammenhang mit vielen somatischen Erkrankungen beobachtet, beispielsweise Schilddrüsenfehlfunktionen, Morbus Parkinson, Asthma bronchiale oder Schlaganfall [36, 56].
Der Faktor Stress bedarf eines besonderen Kommentars: Er spielt bei depressiven Erkrankungen eine wichtige Rolle; häufig wird Depression sogar als eine stressbedingte Erkrankung angesehen. Aber Stress alleine verursacht keine Depression; die meisten Menschen werden auch nach äußerst stressvollen Erlebnissen nicht depressiv. Umgekehrt erkranken andere Menschen an Depression, nachdem sie relativ milden Stress erlebt haben. Dies unterstreicht die Annahme, dass Depression bei den meisten Patienten durch ein Zusammenspiel zwischen Prädisposition und Umweltfaktoren verursacht wird. Auf welche Weise Stress vor dem Hintergrund einer individuellen Vulnerabilität oder Resilienz doch in die Entstehung von Depression involviert sein könnte, wird weiter unten dargestellt [56].
Neurobiologische Mechanismen
Die Frage nach dem neurobiologischen Mechanismus der Entstehung von Depression wurde lange Zeit von einer Hypothese dominiert: der Monoamin-Hypothese. Diese Theorie geht davon aus, dass Depression verursacht wird durch ein Ungleichgewicht oder einen Mangel an Neurotransmittern, in erster Linie den Monoaminen Serotonin, Dopamin und Noradrenalin. Die Theorie entstand aus der Untersuchung des Wirkungsmechanismus der ersten Antidepressiva, der trizyklischen Antidepressiva (TZA) und Monoaminoxidase(MAO)-Hemmer, die in den Synapsen den verfügbaren Spiegel an Monoaminen erhöhen. Ein Nachteil dieser Hypothese ist die Annahme eines einzigen Mechanismus, der für die Entstehung von Depression verantwortlich gemacht wird und der den Medikamenten zu ihrer Behandlung zugrunde liegt. Demnach sollte nach der Behandlung mit Antidepressiva der Normalzustand des Gehirns wieder hergestellt sein. Dies ist aber nicht der Fall. Das Gehirn eines mit Antidepressiva behandelten Patienten ist nicht in einem vergleichbaren Zustand zu dem eines nichtdepressiven Menschen. Dafür spricht auch die hohe Rate an Rückfällen oder an Patienten, die gar nicht auf Antidepressiva ansprechen [56].
Die wichtige Rolle, die Stress offenbar bei der Entstehung von Depression spielt, bot einen Ansatzpunkt für die Suche nach alternativen neurobiologischen Mechanismen bei der Entstehung der Depression. Die Hypothalamus-Hypophysen-Nebennieren(HPA)-Achse ist der zentrale neuroendokrine Schaltkreis des Körpers für die Stressverarbeitung. Unter Stress wird im Hypothalamus das Corticotropin-Releasing-Hormon (CRH) freigesetzt, welches die Produktion des adrenocorticotropen Hormons (ACTH) in der Hypophyse verstärkt. ACTH wiederum stimuliert in der Nebennierenrinde die Ausschüttung des Glucocorticoids Cortisol. Cortisol unterbindet in dem Kreislauf daraufhin die eigene Ausschüttung durch eine negative Rückkopplung. Diese Rückkopplungsschleife umfasst verschiedene Gehirnregionen, unter anderem den Hippocampus und die Amygdala. Eine Hypothese zur Entstehung der Depression nimmt an, dass es hierbei zu einer Fehlfunktion von Glucocorticoid-Rezeptoren kommt, insbesondere im Hippocampus, wo diese Rezeptoren sehr zahlreich sind. Dies führt zum Ausfall der negativen Rückkopplung in der HPA-Achse und einem dauerhaft ungedämpften Cortisolspiegel, mit weitreichenden Folgen.
In Tierstudien konnte durch Antidepressiva die Exprimierung von Glucocorticoid-Rezeptoren erhöht und der negative Rückkopplungsmechanismus in der HPA-Achse wiederhergestellt werden [36, 45, 52].
Eine andere Hypothese stellt die Plastizität der Neurone in den Mittelpunkt. Die Strukturen von Neuronen sind anpassungsfähig und reagieren ständig auf interne und externe Reize. Zu dieser neuronalen Plastizität trägt unter anderem die Fähigkeit der subgranularen Zone des Hippocampus bei, auch im Erwachsenenalter neue Neurone zu bilden und auf verschiedene Weise auszudifferenzieren. Sowohl Daten von Patienten mit Depression als auch Studien an Tiermodellen der Depression zeigen eine Volumenverringerung des Hippocampus, die durch Behandlung mit Antidepressiva reversibel ist, was für eine stimulierende Wirkung der antidepressiven Behandlung auf die Neuroplastizität spricht. Dies gab den Anstoß zu der Theorie, dass Depression auch durch verminderte Neurogenese und verminderte neuronale Plastizität im Hippocampus mitverursacht wird. Als Auslöser werden chronischer Stress, Fehlfunktion der Glucocorticoid-Rezeptoren im Hippocampus und Hypercortisolämie angenommen. In der Tat wurde in Tierversuchen unter solchen Bedingungen neuronale Apoptose, Atrophie von Dendriten und Unterdrückung der Neurogenese im Hippocampus beobachtet [4]. Jüngere Daten zeigen, dass in präklinischen Modellen der Depression und deren antidepressiver Behandlung auch Veränderungen der neuronalen Spine-Regulation und der Neuritogenese maßgeblich beteiligt sind [11].
Eine wichtige Rolle für Neurogenese und neuronale Plastizität spielen die sogenannten „Neurotrophine“ [52], insbesondere der „brain-derived neurotrophic factor“ (BDNF) im Hippocampus, der Amygdala und im zerebralen Kortex. Neurotrophine fördern Wachstum und Differenzierung von Neuronen, sind aber auch wichtig für Plastizität und Überleben ausgewachsener Neurone und Gliazellen.
Eine weitere Hypothese zur Neurobiologie der Depression geht davon aus, dass es unter Stress zu einem Mangel an neurotrophen Faktoren kommt und daraufhin zur neuronalen Atrophie im Hippocampus. Ein verringerter BDNF-Spiegel wurde sowohl bei Patienten mit Depression [32] als auch in Tiermodellen der Depression [45] beobachtet und könnte eine verminderte Resilienz bzw. erhöhte Vulnerabilität widerspiegeln [51, 52]. Dieser Zustand kann durch Gabe von Antidepressiva behoben werden, wobei dieser Effekt wahrscheinlich durch Serotonin vermittelt wird. Eine Reihe von Studien haben gezeigt, dass serotonerge Neurotransmission die Expression von BDNF beeinflusst [27].
Ein weiterer möglicher Pathomechanismus für die Entstehung von Depression ist die Störung des zirkadianen Rhythmus. Eine Verbindung zwischen Stimmungslage und zirkadianem Rhythmus lässt sich leicht herstellen durch die häufig beobachteten Fälle von Depression bei Lichtmangel oder Schichtarbeit. Eine mögliche Verbindung zu den oben dargestellten molekularen Mechanismen könnte darin liegen, dass sowohl HPA-Achse und Cortisolspiegel, die Synthese und/oder Sekretion von Neurotransmittern, als auch Neurogenese und Neurotrophine einem zirkadianen Rhythmus unterliegen [2].
Die hier nur kurz skizzierten Hypothesen zu möglichen Pathomechanismen der Depression beruhen vielfach auf Tierversuchen und nicht alle Forschungsergebnisse sind frei von Widersprüchen. Trotzdem lässt sich feststellen, dass sich in den letzten etwa zehn Jahren unser Bild von den pathologischen Vorgängen bei Depression enorm erweitert hat. Die Monoamin-Hypothese ist nicht obsolet, aber sie ist Teil eines größeren und komplizierteren Bildes geworden.
Behandlung der Depression
So wie Depression nicht nur eine einzige Ätiologie besitzt, ist auch deren Diagnose nicht scharf definiert. Als Hauptsymptome zur Diagnose einer Major Depression nennt die Leitlinie der Deutschen Gesellschaft für Psychiatrie und Psychotherapie, Psychosomatik und Nervenheilkunde (DGPPN)„gedrückte, depressive Stimmung“, „Interessenverlust, Freudlosigkeit“ und „Antriebsmangel, erhöhte Ermüdbarkeit“. Daneben gibt es noch eine Gruppe von Zusatzsymptomen, die unter anderem kognitive Störungen, Ess- und Schlafstörungen und Suizidgedanken beinhaltet [15]. Eine Major Depression liegt vor, wenn zumindest je zwei Symptome aus beiden Gruppen für mindestens zwei Wochen auftreten; bei einer schweren Depression sind alle Hauptsymptome und mindestens vier der Zusatzsymptome vorhanden.
Die medikamentöse Behandlung der Depression gilt in erster Linie den affektiven Symptomen. Ansatzpunkt der Medikamente ist die Erhöhung der verfügbaren Menge der Neurotransmitter Serotonin, Dopamin oder Noradrenalin über unterschiedliche Wirkungsmechanismen. Die ältesten unter diesen Medikamenten sind die trizyklischen Antidepressiva (TZA) und die Monoaminoxidase(MAO)-Hemmer. Während die TZA die Serotonin- und Noradrenalin-Wiederaufnahme aus dem synaptischen Spalt hemmen, erhöhen die MAO-Hemmer die Neurotransmitterkonzentration, indem sie durch Hemmung des Enzyms Monoaminoxidase den Abbau von Serotonin, Dopamin und Noradrenalin verhindern. Beide Substanzklassen sind allerdings für ihre zahlreichen Nebenwirkungen bekannt.
Die Substanzen beider Gruppen besitzen mehrere Wirkungsmechanismen, nicht alle davon therapeutisch erwünscht: TZA weisen eine zusätzliche Affinität zu Muscarin-, Histamin-H1- und Adrenozeptoren auf und das Enzym Monoaminoxidase baut nicht ausschließlich Neurotransmitter ab.
Da die Nebenwirkungen dieser Substanzen zum großen Teil mit den nicht erwünschten Wirkungsmechanismen in Verbindung stehen, galt die Forschung im Bereich Antidepressiva lange Zeit der Entdeckung von hochselektiven Medikamenten. Das Ergebnis waren die heute am häufigsten eingesetzten Serotonin- (SSRI) bzw. Serotonin/Noradrenalin-Wiederaufnahmehemmer (SNRI). Diese haben jeweils nur einen pharmakologischen Wirkungsmechanismus, die Hemmung des Serotonin-Transporters (SERT) bzw. der Serotonin- und Noradrenalin-Transporter (NET) [37]. Die neuen Klassen von Antidepressiva haben jedoch keine grundsätzlich neuen Wirkungsmechanismen, sie stellen lediglich eine Verfeinerung der Mechanismen älterer Antidepressiva dar [56].
Somit ist es nicht verwunderlich, dass es nach wie vor in der medikamentösen Behandlung depressiver Störungen eine ganze Reihe von Problemen gibt. Ältere wie neuere Antidepressiva haben gemeinsam, dass sich erst nach längerer Einnahme eine Wirkung zeigt und auch die neueren SSRI und SNRI haben störende Nebenwirkungen, insbesondere Gewichtszunahme und gestörte Sexualfunktion, was zu häufigen Therapieabbrüchen führt.
Nach wie vor wirken Antidepressiva nur bei einem Teil der Patienten: Nur etwa 50% der Patienten mit Major Depression sprechen auf den ersten Behandlungsversuch mit einem Antidepressivum an [48, 54] und viele der ansprechenden Patienten erreichen keine Remission ihrer Erkrankung [54]. Etwa 50% der Patienten berichten Restsymptome [28], unter anderem Schlafstörungen, Angststörungen oder kognitive Störungen – Symptome, die nicht das therapeutische Ziel der Antidepressiva sind. Unter diesen finden insbesondere die kognitiven Symptome zunehmend Beachtung, da sich gezeigt hat, dass diese mehr sind als nur eine Konsequenz von depressiver Stimmung und Antriebsmangel [30].
Die häufigste Strategie bei Versagen der First-Line-Therapie ist der Wechsel zu einem anderen Antidepressivum. Auch die Kombination mit einem weiteren Medikament aus einer anderen Substanzklasse oder einem anderen Antidepressivum mit unterschiedlichem Wirkungsmechanismus kann sinnvoll sein [17] und wird zunehmend verwendet [31]. Allerdings bringt das zusätzliche Medikament eine neue Palette von Nebenwirkungen ins Spiel, abgesehen von dem Risiko der Interaktionen. Nur für wenige der Kombinationen gibt es kontrollierte klinische Studien, die Sicherheit und Wirksamkeit untermauern [31].
Dieses Dilemma gab der Entwicklung von Antidepressiva eine neue Richtung: Gesucht sind jetzt Substanzen, die mehrere therapeutische Wirkungsmechanismen besitzen, ohne das Nebenwirkungspotenzial der Substanzkombinationen oder der älteren Antidepressiva. Für diese Art von Medikamenten hat sich die Bezeichnung „multimodal“ durchgesetzt [37].
Neurobiologische Wirkorte von Vortioxetin
Eines dieser multimodalen Antidepressiva ist Vortioxetin. Die Strategie bei der Entwicklung von Vortioxetin war es, die Aktivität an SERT beizubehalten und um weitere Wirkungsmechanismen zu ergänzen, die sich in präklinischen Studien als vielversprechend dargestellt hatten.
Vortioxetin besitzt Affinität zu SERT, zusätzlich jedoch auch zu den Serotonin-Rezeptoren 5-HT1A, 5-HT1B, 5-HT1D, 5-HT3 und 5-HT7. Es blockiert SERT und wirkt am 5-HT1A-Rezeptor als Agonist, an 5-HT1B als partieller Agonist und an den Rezeptoren 5-HT3, 5-HT7 und 5-HT1D als Antagonist [8, 55].
Der Hintergrund für die Wahl dieser Rezeptoren sind Ergebnisse aus zahlreichen In-vitro- und Tierstudien, die Hinweise auf die Arbeitsweise des Serotonin-Systems geben.
Serotonerge Neurone befinden sich hauptsächlich in Zellgruppen im Bereich des Hirnstamms; ihre Axone reichen jedoch in fast alle Gehirnregionen und interagieren mit anderen Neurotransmittersystemen. Im wachen Zustand haben die serotonergen Neurone eine langsame, gleichmäßige Feuerrate. Ihre Aktivität wird einerseits kontrolliert durch die Interaktion mit anderen Neurotransmittersystemen, andererseits selbst-regulatorisch durch negative Rückkopplung [6, 26].
Eine besonders gut untersuchte Gruppe von Serotonin-Rezeptoren ist die Familie der 5-HT1-Rezeptoren. Die Rezeptoren 5-HT1A, 5-HT1B und 5-HT1D sind in den somatodendritischen und axonalen Regionen der serotonergen Neurone lokalisiert. Sie sind wichtige Effektoren in Regelkreisen, die über negative Rückkopplung sowohl Feuerrate der Neurone als auch Serotonin-Freisetzung im Gleichgewicht halten. Die Aktivierung des präsynaptischen Autorezeptors 5-HT1A bewirkt einerseits eine Verlangsamung der Feuerrate des Neurons, gleichzeitig sorgt 5-HT1A im Zusammenspiel mit SERT für das Ende der synaptischen Aktion des Serotonins: Während SERT durch Serotonin-Wiederaufnahme den Neurotransmitter aus dem synaptischen Spalt entfernt, bewirkt die serotonerge Aktivierung des Autorezeptors, dass die weitere Freisetzung von Serotonin gestoppt wird. Der Subtyp 5-HT1A ist an somatodendritischen Synapsen lokalisiert, während 5-HT1B eine ähnliche Funktion an serotonergen Axonen hat [6].
Es wird vermutet, dass der verzögerte Wirkungseintritt der SSRI zumindest teilweise durch diese negative Rückkopplung zu erklären ist. Erst nach einer Phase der Desensitivierung der Autorezeptoren können SSRI für einen erhöhten Serotonin-Spiegel im synaptischen Spalt sorgen. Entsprechend kann der Wirkungseintritt von SSRI durch zusätzliche Gabe von 5-HT1A-Antagonisten beschleunigt werden [6]. Dies ist aber auch bei Einsatz eines zusätzlichen 5-HT1A-Agonisten der Fall, was wahrscheinlich zu einer schnelleren Desensitivierung der Autorezeptoren führt [7].
Substanzen mit Wirkung auf den 5-HT1A-Rezeptor haben aber nicht nur eine unterstützende Funktion für SSRI; die Aktivierung postsynaptischer 5-HT1A-Rezeptoren spielt eine eigene wichtige Rolle für den antidepressiven Effekt [6]. Darüber hinaus wurde nach längerer Aktivierung von 5-HT1A-Rezeptoren im Gyrus dentatus, wie sie bei längerer Behandlung mit Antidepressiva stattfindet, vermehrte adulte Neurogenese im Hippocampus beobachtet. Diese Neurogenese im Hippocampus spielt wahrscheinlich nicht nur eine Rolle bei Depression, sondern auch für kognitive Funktionen [6, 44].
Die 5-HT1-Rezeptoren fungieren nicht nur als Autorezeptor, sondern beeinflussen als Heterorezeptoren andere Neurotransmittersysteme. So moduliert 5-HT1B als terminaler Heterorezeptor die Abgabe verschiedener Neurotransmitter wie Dopamin, Glutamat, GABA und Acetylcholin. 5-HT1A-Heterorezeptoren auf nichtserotonergen Neuronen sind im Gehirn weit verbreitet, finden sich aber besonders zahlreich im präfrontalen Kortex. Es wird daher vermutet, dass 5-HT1-Rezeptoren nicht nur eine wichtige Rolle bei der Kontrolle von Stimmung und Emotionen spielen, sondern auch für kognitive Prozesse [12, 44].
Die 5-HT3-Rezeptoren bewirken als Ligand-gesteuerte Ionenkanäle eine schnelle Depolarisation des Neurons. In präklinischen Studien konnte gezeigt werden, dass 5-HT3-Antagonisten im Vorderhirn die Freisetzung von Noradrenalin und Acetylcholin fördern, indem sie inhibitorische 5-HT3-Heterorezeptoren auf GABA-ergen Interneuronen blockieren [58]. Die frontokortikalen GABA-ergen Interneurone und die Pyramidenzellen sind wichtige Schnittstellen zwischen den verschiedenen Neurotransmittersystemen, die Emotionen und Kognition beeinflussen [30]. Daher wird vermutet, dass diese Wirkung von 5-HT3-Antagonisten für die in Tierversuchen beobachteten positiven Effekte auf Stimmung, Lern- und Gedächtnisleistung sowie exekutive Funktionen eine wichtige Rolle spielt [6, 46].
Die 5-HT7-Rezeptoren sind im Zentralnervensystem an der Steuerung des zirkadianen Rhythmus und des Schlafens, sowie an der Thermoregulierung beteiligt [22]. Dieser Rezeptortyp ist im Gehirn weit verbreitet, aber besonders zahlreich in Thalamus, Hypothalamus, Hippocampus und Kortex [47]. Eine Reihe von Antidepressiva und Antipsychotika haben Affinität zum 5-HT7-Rezeptor und auch präklinische Studien mit 5-HT7-Rezeptorantagonisten oder Knock-out-Mäusestämmen zeigen, dass 5-HT7-Rezeptoren in Verbindung mit Depression und Angststörungen stehen. Eine Studie lässt sogar vermuten, dass die antidepressive Wirkung von Amisulprid auf seiner Eigenschaft als 5-HT7-Rezeptorantagonist beruht [6, 22]. Vor allem haben aber zahlreiche Tierstudien gezeigt, dass der 5-HT7-Rezeptor in Lern- und Gedächtnisleistungen involviert ist [47].
Neue Ergebnisse in der Antidepressiva-Forschung, insbesondere die schnelle und andauernde antidepressive Wirkung des N-methyl-D-aspartat(NMDA)-Rezeptorantagonisten Ketamin, zeigen, dass die glutamaterge Neurotransmission eine wichtige Rolle für die Stimmung spielt. Die Bedeutung der glutamatergen Transmission für die Kognition ist schon länger untersucht; sie ist ein Hauptziel der Forschung nach kognitionsfördernden Substanzen [42]. Da Serotonin-Mangel die Wirkung von Ketamin aufhebt, wird vermutet, dass die glutamaterge Neurotransmission durch das Serotonin-System moduliert wird. Dabei scheinen die Rezeptoren 5-HT1A, 5-HT1B, 5-HT3 und 5-HT7 von besonderem Interesse zu sein. Viele der oben beschriebenen Einzelergebnisse zu diesen Rezeptortypen lassen sich in einem Bild integrieren, wenn man das Glutamat-System in den Mittelpunkt stellt (Abb. 1) [42].
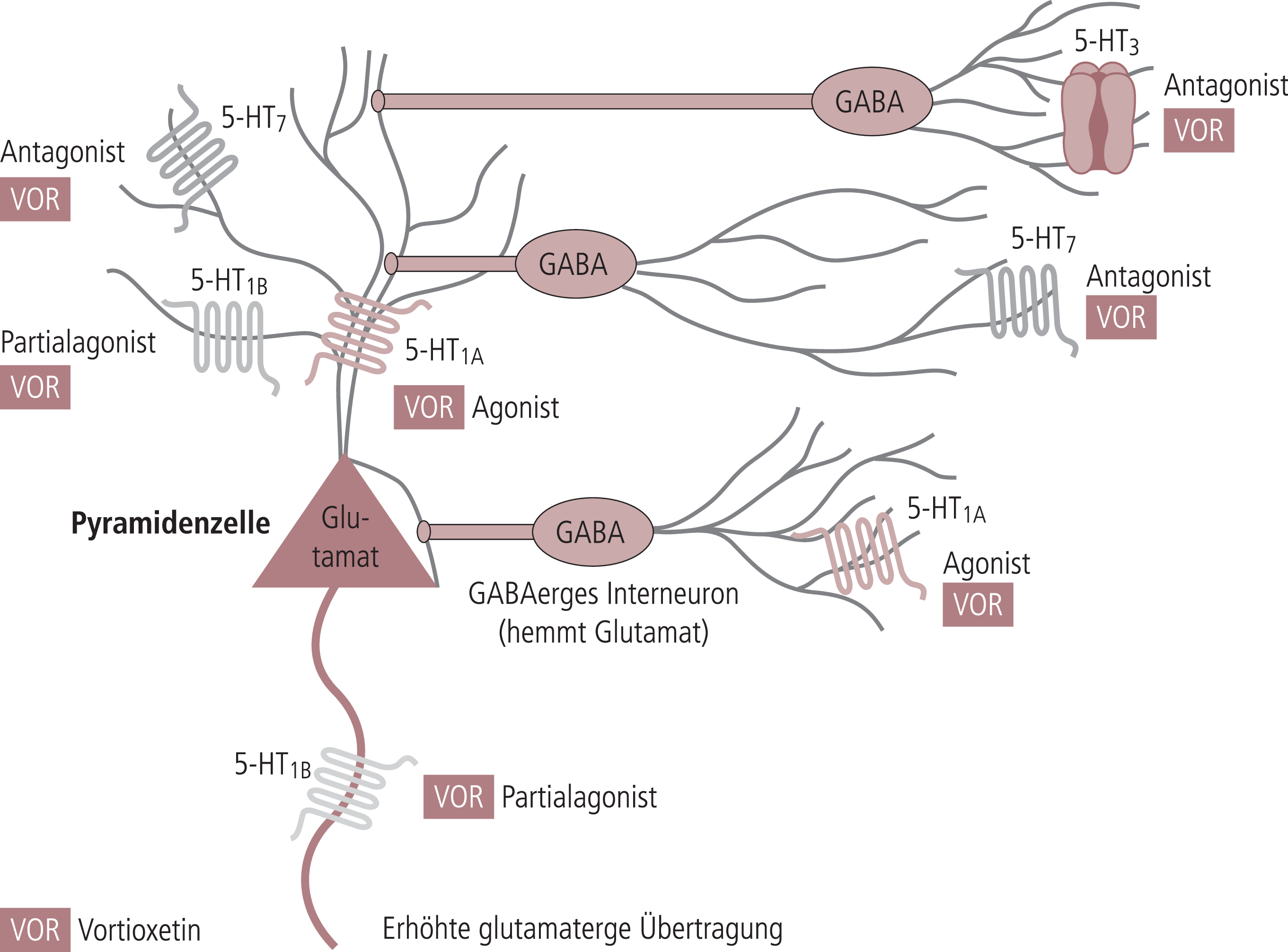
Abb. 1. Die frontokortikalen GABAergen Interneurone und die Pyramidenzellen sind wichtige Schnittstellen zwischen den verschiedenen Neurotransmittersystemen. Serotonin(5-HT)-Rezeptoren modulieren beide Zelltypen auf verschiedenen Wegen. GABA: Gammaaminobuttersäure
Neben der glutamatergen spielt auch die cholinerge Neurotransmission eine wichtige Rolle für kognitive Funktionen. Cholinerge Neurone im basalen Vorderhirn projizieren in den Hippocampus und präfrontalen Kortex und gelten als Hauptquelle cholinerger Transmission im ZNS. Defekte im cholinergen System sind mit kognitiven Störungen in Verbindung gebracht worden [50]. Die Aktivität der basalen Neurone des Vorderhirns wird durch das Neurotrophin BDNF beeinflusst [13], welches seinerseits vermutlich durch das Serotonin-System moduliert wird [27].
Vortioxetin vereint somit eine Reihe von vielversprechenden Wirkungsmechanismen: Einerseits die klassische Serotonin-Wiederaufnahmehemmung an SERT, zum anderen Aktivitäten an verschiedenen Serotonin-Rezeptoren, die mit Depression, Kognition, Angst, sowie der zirkadianen Rhythmik und Schlaf involviert sind. Von einigen dieser Serotonin-Rezeptor-Subtypen konnte gezeigt werden, dass sie modulierend auf andere Neurotransmittersysteme einwirken wie Dopamin, Glutamat, GABA und Acetylcholin [12, 42, 44, 58]. Auf diesem indirekten Weg könnte Vortioxetin, das hauptsächlich an Serotonin-Rezeptoren bindet, möglicherweise auch andere Neurotransmittersysteme beeinflussen, insbesondere die glutamaterge und cholinerge Neurotransmission.
Präklinische Erfahrungen mit Vortioxetin
Vortioxetin bindet mit hoher Affinität an die Serotonin-Rezeptoren 5-HT1A (Ki =15 nmol/l), 5-HT1B (Ki =33 nmol/l), 5-HT1D (Ki =54 nmol/l), 5-HT3A (Ki =3,7 nmol/l), 5-HT7 (Ki =19 nmol/l) sowie an SERT (Ki =1,6 nmol/l) [8].
Präklinische Studien zeigen, dass die Effekte von Vortioxetin an unterschiedlichen Elementen des Serotonin-Systems tatsächlich, wie erhofft, auf interessante Weise zusammenspielen. Bereits nach drei Tagen Behandlung mit Vortioxetin konnte bei Ratten ein deutlicher Anstieg des extrazellulären Serotonin-Spiegels im Hippocampus gemessen werden, wobei die SERT-Belegung nur 41% betrug [5, 34]. Auch in klinischen Studien wurde eine therapeutisch wirksame Plasmakonzentration von Vortioxetin bei einer SERT-Belegung von etwa 50% erreicht [3, 5]. Dagegen haben SSRI oder SNRI eine SERT-Belegungsrate von etwa 80% bei Dosierungen, die eine bessere antidepressive Wirkung zeigen als Plazebo [23, 29].
In Studien zur Feuerrate von serotonergen Neuronen an Ratten und Mäusen löste die akute Behandlung mit Vortioxetin und dem SSRI Fluoxetin, wie erwartet, eine Unterdrückung der Aktionspotenziale aus. Allerdings kehrten die Neurone der mit Vortioxetin behandelten Tiere bereits nach einem Tag zu Normalwerten zurück, während die mit Fluoxetin behandelten Tiere dafür mindestens 14 Tage brauchten [9, 49]. Mithilfe eines 5-HT1A-Agonisten konnte festgestellt werden, dass unter Vortioxetin tatsächlich eine frühe Desensitivierung des 5-HT1A-Rezeptors stattgefunden hatte, nach nur drei Tagen, was allerdings nicht die Wiederaufnahme der ursprünglichen Feuerrate nach nur einem Tag erklärt. Weitere Untersuchungen in dieser Studie zeigten, dass diese schnelle Erholung zumindest teilweise auf den 5-HT3-Antagonismus von Vortioxetin zurückzuführen ist [9].
Die Behandlung mit Vortioxetin bewirkte bei Ratten eine Dosis-abhängige Erhöhung der Konzentration von Serotonin, Noradrenalin, Dopamin, Histamin und Acetylcholin im Gehirn. Diese Konzentrationserhöhung von Neurotransmittern war unterschiedlich und spezifisch für einzelne Gehirnregionen, insbesondere für solche, die bei Depression und Kognition involviert sind [8, 33, 34, 41]. In Mikrodialysestudien an Ratten kam es nach Behandlung mit Vortioxetin zur Erhöhung des Serotonin-Spiegels im ventralen Hippocampus (VH), medialen präfrontalen Kortex (MPK) und Nucleus accumbens (NA); die Konzentration von Dopamin und Noradrenalin war nur im VH und MPK erhöht, nicht jedoch im NA. Im MPK kam es unter Vortioxetin ebenfalls zur Erhöhung der Spiegel von Acetylcholin und Histamin [33].
In einer Studie mit Mäusen kam es nach Behandlung mit Vortioxetin zu einem Anstieg des BDNF-Spiegels im Hippocampus, nicht jedoch nach Behandlung mit Fluoxetin [25]. Es wird vermutet, dass ein erhöhter BDNF-Spiegel zu einem Anstieg der neuronalen Plastizität im präfrontalen Kortex beiträgt. Auch die Aktivität anderer Gene, die mit Neuroplastizität im präfrontalen Kortex im Zusammenhang stehen, war bei Ratten nach Vortioxetin erhöht, nicht jedoch nach Gabe von Fluoxetin [16].
Auch die Neurogenese wurde in Tierstudien durch Vortioxetin positiv beeinflusst. Die Behandlung von Mäusen mit Vortioxetin förderte die Zellteilung im Hippocampus [18, 19]. Außerdem kam es zu längeren Dendriten und zahlreicheren dendritischen Kontaktpunkten, als ohne Behandlung, was vermuten lässt, dass Vortioxetin die Reifung von Neuronen unterstützt [18].
Verhaltensstudien an Tiermodellen für Depression oder kognitive Defizite bestätigen die vielversprechenden Ergebnisse der molekularbiologischen, histologischen und elektrophysiologischen Experimente. Ähnlich wie das SSRI Fluoxetin zeigte Vortioxetin antidepressive Wirkung im forced swim test mit Mäusen [18, 49]. In einem anderen Tiermodell für Depression, hervorgerufen durch Progesteron-Entzug, hatte jedoch nur Vortioxetin, nicht aber Escitalopram oder Duloxetin eine antidepressive Wirkung [49].
Da depressive Patienten häufig eine Störung der Rapid-Eye-Movement(REM)-Schlafphase aufweisen, wurde die Wirkung von Vortioxetin, Escitalopram und Duloxetin in einer Schlaf-EEG-Studie an Ratten untersucht. Unter allen Medikamenten kam es zu einer Verkürzung der REM-Phase im Vergleich zu unbehandelten Tieren, aber nur Vortioxetin und Duloxetin verkürzten die Non-REM-Phase und verlängerten die Vigilanzphase. Allein unter Vortioxetin waren Theta-, Alpha- und Gamma-Wellen während der Vigilanzphase dosisabhängig verstärkt; der Unterschied zu unbehandelten Tieren war am deutlichsten im präfrontalen Kortex zu sehen [24].
Der Novel-Object-Recognition(NOR)-Test wird in Tierstudien zur Prüfung der Gedächtnisleistung eingesetzt. In diesem Test zeigten mit Vortioxetin behandelte Ratten eine verbesserte Gedächtnisleistung im Vergleich zu unbehandelten Tieren [19, 33]. Die Verbesserung war vergleichbar mit der unter Donepezil, das bekannt ist für seinen positiven Einfluss auf kognitive Leistungen [19]. Testergebnisse nach zusätzlichem Einsatz von 5-HT3- oder 5-HT7-Antagonisten lassen vermuten, dass die positive Wirkung von Vortioxetin auf das Gedächtnis durch Effekte an den Rezeptoren 5-HT3 und 5-HT7 vermittelt werden [19].
In einer Studie mit unterschiedlich alten Mäusen verbesserte die Gabe von Vortioxetin die schlechtere Gedächtnisleistung alter Mäuse beim Object-Placement-Test. In derselben Studie kam es nach Gabe von Vortioxetin bei allen Mäusen zu einer Erhöhung des BDNF-Spiegels im präfrontalen Kortex. Fluoxetin dagegen hatte weder Einfluss auf die Gedächtnisleistung noch auf den BDNF-Spiegel. Die Autoren postulierten, dass der erhöhte BDNF-Spiegel die Neuroplastizität erhöht und damit die verbesserte Gedächtnisleistung bewirkt [25].
Diskussion
Medikamente mit mehreren Wirkungsmechanismen bergen den Nachteil, für zahlreiche Nebenwirkungen verantwortlich zu sein. Dies führte in der Vergangenheit zu großen Anstrengungen, möglichst spezifisch wirkende Medikamente mit nur einem Mechanismus zu entwickeln, wie die SSRI oder SNRI. Diese neuen Klassen von Antidepressiva besitzen jedoch keine grundsätzlich neuen Wirkungsmechanismen, sie stellen lediglich eine Verfeinerung der älteren Antidepressiva dar. Die Wirksamkeit der modernen Antidepressiva ist nicht höher als die der älteren Medikamente, auch wenn die Verträglichkeit deutlich besser ist. Antidepressiva wirken nach wie vor nur bei einem Teil der Patienten und viele der ansprechenden Patienten erreichen keine Remission. Etwa 50% der Patienten berichten Residualsymptome [28], unter anderem Schlafstörungen, Angststörungen oder kognitive Störungen; Symptome, die auch nicht das therapeutische Ziel der heutigen Antidepressiva sind.
Zunehmend Beachtung finden die kognitiven Defizite, die in Verbindung mit Depression beobachtet werden. Dabei kann es sich um ganz unterschiedliche Ausprägungen handeln, wie Störungen der Konzentrationsfähigkeit, des Gedächtnisses, der Lernfähigkeit, der Psychomotorik, bis hin zu Störungen der exekutiven Funktionen wie Planen und Entscheidungen treffen. Solche Symptome werden inzwischen weitgehend als charakteristisch für die akute Phase einer Depression angesehen. Obwohl dies alles Symptome sind, die das Alltagsleben des Patienten erheblich beeinträchtigen, sind sie nicht primär Ziel der antidepressiven Therapie. Lange Zeit wurde angenommen, dass kognitive Dysfunktion eine direkte Folge der Stimmungsstörungen sei und somit bei erfolgreicher Therapie „automatisch“ abklingen würde [14, 20].
Studien zeigen jedoch, dass kognitive Symptome auch zwischen akuten Phasen der Erkrankung oder in Remission persistieren. Patienten, die mit neueren Antidepressiva erfolgreich behandelt wurden, schnitten zwar bezüglich Kognition besser ab als unbehandelte Patienten, aber schlechter als gesunde Personen aus einer Kontrollgruppe [20, 21]. Es wurde sogar postuliert, dass sich die kognitive Leistungsfähigkeit bei Patienten mit rezidivierender Depression mit jeder akuten Episode verschlechtert [20]. Gleichzeitig trage kognitive Dysfunktion erheblich zum Grad der Behinderung im täglichen Leben bei, die Patienten mit Depression erfahren [35].
Die Rückfallrate bei Depression ist sehr hoch und das Vorhandensein von Restsymptomen wurde als wichtiger Faktor und Prädiktor für einen Rückfall identifiziert [28, 40]. Kognitive Störungen gehörten dabei zusammen mit Schlafstörungen und persistierenden Stimmungsstörungen zu den am häufigsten berichteten Symptomen [28].
In den letzten etwa zehn Jahren haben neue Forschungsergebnisse unsere Sicht der pathologischen Vorgänge bei Depression stark erweitert. Die aufgestellten Theorien bedürfen weiterer Forschungsarbeit, aber klar ist, dass die Monoamin-Hypothese nur ein Teil eines größeren und komplizierteren Bildes ist, in dem unter anderem die HPA-Achse und die Prozesse der neuronalen Plastizität in Gehirnregionen des limbischen Systems wichtige Rollen spielen. Entsprechend „einseitig“ stellen sich unsere heutigen Antidepressiva dar, die ja in enger Beziehung zur Monoamin-Hypothese stehen. Auch die Vorstellung, dass Depression in erster Linie eine Stimmungsstörung sei, ist zu eng geworden. Affektive Symptome gelten zurzeit als die „Hauptsymptome“ der Depression [15], denen auch in erster Linie die Behandlung gilt. Aber die kognitiven Symptome und die Störungen im Bereich von Grundbedürfnissen wie schlafen oder essen sind nicht nur „Zusatzsymptome“, sondern eigenständige Symptome von Störungen in verschiedenen Gehirnregionen, die durch neue Forschungsergebnisse zur Depression in Beziehung gesetzt wurden. Auch diese Symptome sind behandlungsbedürftig, bleiben aber bei der heutigen Therapie mit Antidepressiva häufig als Restsymptome zurück, selbst wenn der Patient auf die Behandlung der „Hauptsymptome“ anspricht [28].
Aus diesen neuen Einblicken sollten Antidepressiva resultieren, die tatsächlich neue Wirkungsmechanismen besitzen. Auch spricht einiges dafür, Medikamente mit mehreren Wirkungsmechanismen anzustreben. Depression hat nicht nur eine Ätiologie oder einen neurobiologischen Mechanismus. Eine große Anzahl von Faktoren, die von genetischen bis zu sozialen Einflüssen reichen, interagieren auf unterschiedliche Weise und führen zu den Symptomen, die wir unter der Diagnose „Major Depression“ zusammenfassen. Dabei sind eine Reihe von Gehirnregionen und Neurotransmittersystemen involviert. Medikamente, die an mehreren Stellen in diese komplexen Vorgänge eingreifen können, sollten eine bessere Chance auf Erfolg bei einer großen Anzahl von Patienten haben als spezialisierte Medikamente.
Das multimodale Antidepressivum Vortioxetin eröffnet die Aussicht, durch multiple Wirkungsmechanismen Depressionen unterschiedlicher Ätiologie behandeln zu können. Darüber hinaus hat Vortioxetin durch seine individuelle Kombination von Aktivitäten an SERT sowie den Rezeptoren 5-HT1A, 5-HT1B, 5-HT3, 5-HT7 und 5-HT1D und seine zusätzlichen Effekte auf Neuroplastizität und Neurogenese das Potenzial, möglicherweise auch kognitive Dysfunktionen positiv zu beeinflussen.
Literatur
1. Aarsland D, Taylor JP, Weintraub D. Psychiatric issues in cognitive impairment. Mov Disord 2014;29:651–62.
2. Albrecht U. The circadian clock, reward, and memory. Front Mol Neurosci 2011;4:41.
3. Alvarez E, Perez V, Dragheim M, Loft H, et al. A double-blind, randomized, placebo-controlled, active reference study of Lu AA21004 in patients with major depressive disorder. Int J Neuropsychopharmacol 2012;15:589–600.
4. Anacker C, Cattaneo A, Luoni A, et al. Glucocorticoid-related molecular signaling path ways regulating hippocampal neurogenesis. Neuropsychopharmacology 2013;38:872–83.
5. Areberg J, Luntang-Jensen M, Søgaard B, Nilausen DØ. Occupancy of the serotonin transporter after administration of Lu AA21004 and its relation to plasma concentration in healthy subjects. Basic Clin Pharmacol Toxicol 2012;110:401–4.
6. Artigas F. Serotonin receptors involved in antidepressant effects. Pharmacol Ther 2013;137:119–31.
7. Assié MB, Lomenech H, Ravailhe V, Faucillon V, et al. Rapid desensitization of somatodendritic 5-HT1A receptors by chronic administration of the high-efficacy 5-HT1A agonist, F13714: a microdialysis study in the rat. Br J Pharmacol 2006;149:170–8.
8. Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl] piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem 2011;54:3206–21.
9. Bétry C, Pehrson AL, Etiévant A, Ebert B, et al. The rapid recovery of 5-HT cell firing induced by the antidepressant vortioxetine involves 5-HT3 receptor antagonism. Int J Neuropsychopharmacol 2013;16:1115–27.
10. Blier P, Bergeron R, de Montigny C. Selec tive activation of postsynaptic 5-HT1A receptors induces rapid antidepressant response. Neuropsychopharmacology 1997;16:333–8.
11. Castrén E, Hen R. Neuronal plasticity and antidepressant actions. Trends Neurosci 2013;36:259–67.
12. Celada P, Puig MV, Artigas F. Serotonin modulation of cortical neurons and networks. Front Integr Neurosci 2013;7:25.
13. Chourbaji S, Hellweg R, Brandis D, et al. Mice with reduced brain-derived neurotrophic factor expression show decreased choline acetyltransferase activity, but regular brain monoamine levels and unaltered emotional behavior. Brain Res Mol Brain Res 2004;121:28–36.
14. Cowen P, Sherwood AC. The role of serotonin in cognitive function: evidence from recent studies and implications for understanding depression. J Psychopharmacol 2013;27:575–83.
15. DGPPN, BÄK, KBV, AWMF, AkdÄ, BPtK, BApK, DAGSHG, DEGAM, DGPM, DGPs, DGRW (Hrsg.) für die Leitliniengruppe Unipolare Depression. S3-Leitlinie/Nationale VersorgungsLeitlinie Unipolare Depression – Kurzfassung. 1. Auflage 2009. Berlin, Düsseldorf: DGPPN, ÄZQ, AWMF, 2009. Version 1.3, Januar 2012. www.dgppn.de, www.versorgungsleitlinien.de, www.awmf-leitlinien.de.
16. Du Jardin KG, Liebenberg N, Müller H, et al. Single dose vortioxetine or ketamine but not fluoxetine increases expression of neuroplasticity related genes in the rat prefrontal cortex. Poster presented at the 26th Congress of the European College of Neuropsychopharmacology (ECNP), 5–9 October, 2013, Barcelona, Spain.
17. Garcia-Toro M, Medina E, Galan JL, Gonzalez MA, et al. Treatment patterns in major depressive disorder after an inadequate res ponse to first-line antidepressant treatment. BMC Psychiatry 2012;12:143.
18. Guilloux JP, Mendez-David I, Pehrson A, Guiard BP, et al. Antidepressant and anxiolytic potential of the multimodal antidepressant vortioxetine (Lu AA21004) assessed by behavioural and neurogenesis outcomes in mice. Europharmacology 2013;73C:147–59.
19. Haddjeri N, Etiévant A, Pehrson A, Sanchez C, et al. Effects of the multimodal antidepressant Vortioxetine (Lu AA21004) on rat synaptic and cellular hippocampal plasticity and memory recognition. Poster presented at the 25th Congress of the European College of Neuropsychopharmacology (ECNP), 13–17 October, 2012, Vienna, Austria.
20. Hammar A, Ardal G. Cognitive functioning in major depression – a summary. Front Hum Neurosci 2009;3:26.
21. Hasselbalch BJ, Knorr U, Hasselbalch SG, Gade A, et al. Cognitive deficits in the remitted state of unipolar depressive disorder. Neuropsychology 2012;26:642–51.
22. Hedlund PB. The 5-HT7 receptor and disorders of the nervous system: an overview. Psychopharmacology (Berl) 2009;206:345–54.
23. Larsen AK, Brennum LT, Egebjerg J, Sánchez C, et al. Selectivity of (3)H-MADAM binding to 5-hydroxytryptamine transporters in vitro and in vivo in mice; correlation with behavioural effects. Br J Pharmacol 2004;141:1015–23.
24. Leiser SC, Robichaud PJ, Pehrson AL, Sanchez C. Vortioxetine (Lu AA21004) effects on attention and vigilance as measured by EEG activity in the rat. Poster presented at the 22nd Neuropharmacology Conference, 11–12 October 2012. New Orleans, LA, USA.
25. Li Y, Sanchez C, Gulinello M. Memory impairment in old mice is differentially sensitive to different classes of antidepressants. Poster presented at the 26th Congress of the European College of Neuropsychopharmacology (ECNP), 5–9 October, 2013, Barcelona, Spain.
26. Maejima T, Masseck OA, Mark MD, Herlitze S. Modulation of firing and synaptic transmission of serotonergic neurons by intrinsic G protein-coupled receptors and ion channels. Front Integr Neurosci 2013;7:40.
27. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology 2008;33:73–83.
28. McClintock SM, Husain MM, Wisniewski SR, Nierenberg AA, et al. Residual symptoms in depressed outpatients who respond by 50% but do not remit to antidepressant medication. J Clin Psychopharmacol 2011;31:180–6.
29. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J Psychiatry Neurosci 2007;32:86–102.
30. Millan MJ, Agid Y, Brüne M, Bullmore ET, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 2012;11:141–68.
31. Mojtabai R, Olfson M. National trends in psychotropic medication polypharmacy in office-based psychiatry. Arch Gen Psychiatry 2010;67:26–36.
32. Molendijk ML, Spinhoven P, Polak M, et al. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (n=9484). Mol Psychiatry 2014;19:791–800.
33. Mørk A, Montezinho LP, Miller S, Trippodi-Murphy C, et al. Vortioxetine (Lu AA21004), a novel multimodal antidepressant, enhances memory in rats. Pharmacol Biochem Behav 2013;105:41–50.
34. Mørk A, Pehrson A, Brennum LT, Nielsen SM, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther 2012;340:666–75.
35. Naismith SL, Longley WA, Scott EM, Hickie IB. Disability in major depression related to self-rated and objectively-measured cognitive deficits: a preliminary study. BMC Psychiatry 2007;7:32.
36. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron 2002;34:13–25.
37. Nutt DJ. Beyond psychoanaleptics – can we improve antidepressant drug nomenclature? J Psychopharmacol 2009;23:343–5.
38. Nyer M, Farabaugh A, Fehling K, et al. Relationship between sleep disturbance and depression, anxiety, and functioning in college students. Depress Anxiety 2013;30:873–80.
39. Paradiso S, Duff K, Vaidya JG, et al. Cognitive and daily functioning in older adults with vegetative symptoms of depression. Int J Geriatr Psychiatry 2010;25:569–77.
40. Paykel ES, Ramana R, Cooper Z, Hayhurst H, et al. Residual symptoms after partial remission: an important outcome in depression. Psychol Med 1995;25:1171–80.
41. Pehrson AL, Cremers T, Bétry C, van der Hart MG, et al. Lu AA21004, a novel multimodal antidepressant, produces regionally selective increases of multiple neurotransmitters – a rat microdialysis and electrophysiology study. Eur Neuropsychopharmacol 2013;23:133–45.
42. Pehrson AL, Sanchez C. Serotonergic modulation of glutamate neurotransmission as a strategy for treating depression and cognitive dysfunction. CNS Spectr 2013 Aug 1:1–13. [Epub ahead of print]
43. Peters KR, Rockwood K, Black SE, et al. Neuropsychiatric symptom clusters and functional disability in cognitively-impaired-not-demented individuals. Am J Geriatr Psychiatry 2008;16:136–44.
44. Polter AM, Li X. 5-HT1A receptor-regulated signal transduction pathways in brain. Cell Signal 2010;22:1406–12.
45. Ridder S, Chourbaji S, Hellweg R, et al. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci 2005;25:6243–50.
46. Robbins TW, Roberts AC. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cerebral Cortex 2007;17:151–60.
47. Roberts AJ, Hedlund PB. The 5-HT7 receptor in learning and memory. Hippocampus 2012;22:762–71.
48. Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006;163:1905–17.
49. Sanchez C, Pehrson AL, Betry C, David D, et al. Vortioxetine (Lu AA21004), an investigational multimodal antidepressant: Differentiation from currently used antidepressants in rodent models. Poster presented at the 166th Annual Meeting of the American Psychiatric Association, May 18–22, 2013, San Francisco, CA, USA.
50. Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci 2005;6:48–56.
51. Schulte-Herbrüggen O, Braun A, Rochlitzer S, et al. Neurotrophic factors – a tool for therapeutic strategies in neurological, neuropsychiatric and neuroimmunological diseases? Curr Med Chem 2007;14:2318–29.
52. Schulte-Herbrüggen O, Hellweg R, Chourbaji S, et al. Differential regulation of neurotrophins and serotonergic function in mice with genetically reduced glucocorticoid receptor expression. Exp Neurol 2007;204:307–16.
53. Spiegelhalder K, Regen W, Nanovska S, et al. Comorbid sleep disorders in neuropsychiatric disorders across the life cycle. Curr Psychiatry Rep 2013;15:364.
54. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163:28–40.
55. Westrich L, et al. In vitro and in vivo effects of the multimodal antidepressant vortioxetine (Lu AA21004) at human and rat targets. Int J Psychiatry Clin Pract 2012;16(Suppl 1):47 (P 71). 12th International Forum on Mood and Anxiety Disorders (IFMAD), Barcelona, Spain, 7–9 November, 2012.
56. Willner P, Scheel-Krüger J, Belzung C. The neurobiology of depression and antidepressant action. Neurosci Biobehav Rev 2013;37:2331–71.
57. World Health Organization. The global burden of disease: 2004 update. 2008.
58. Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Mol Neurobiol 2002;26:203–16.
Prof. Dr. med. Rainer Hellweg, Geschäftsführender Oberarzt, Klinik für Psychiatrie und Psychotherapie Charité – Universitätsmedizin Berlin, Campus Charité Mitte (CC15), Charitéplatz 1, 10117 Berlin, E-Mail: Rainer.Hellweg@charite.de
Dr. med. Christoph Goemann, Lundbeck GmbH, Ericusspitze 2, 20457 Hamburg
Effect of vortioxetine on depressive symptoms of major depression. Preclinical data and the potential relationship to the etiology of depression.
Today’s antidepressive therapy with highly selective drugs is confronted with a number of unmet needs: only about half of the patients respond to therapy, a considerable latency period, and symptoms like cognitive deficits are rarely treatment targets. However, cognitive deficits are part of the acute phase of depression, persist during remission, and contribute to the high relapse rate in major depression. The new generation of multimodal antidepressives like vortioxetine has several synergistic modes of action and thus open the prospect of treating depression of varying etiology and reaching a larger number of molecular targets. Vortioxetine with its combination of activities at the serotonin transporter (SERT) and the receptors 5-HT1A, 5-HT1B, 5-HT1D, 5-HT3, and 5-HT7 has the potential to act fast and safely. In addition, it may improve cognitive deficits.
Key words: Major depression, multimodal antidepressives, cognitive deficits, vortioxetine
Psychopharmakotherapie 2014; 21(04)