Holger Petri, Bad Wildungen*
Als bisher einzige Leitsubstanz der Kassenärztlichen Bundesvereinigung (KBV) aus der Gruppe der selektiven Serotonin-Wiederaufnahmehemmer (SSRI) wurde Citalopram 2012 mit 340,9 Mio. definierten Tagesdosen am häufigsten verordnet [12]. Citalopram verlängert dosisabhängig das QT-Intervall und es drohen lebensbedrohliche polymorphe ventrikuläre Tachyarrhythmien, sogenannte Torsade de pointes. Citalopram ist daher bei Patienten mit bekannter QT-Intervallverlängerung oder angeborenem Long-QT-Syndrom kontraindiziert, ebenso bei gleichzeitiger Anwendung von Arzneimitteln, die bekannterweise das QT-Intervall verlängern [11]. Dieselben Gegenanzeigen gelten auch für sein Stereoisomer Escitalopram. Die anderen SSRI besitzen ein geringeres Risiko für Torsade-de-pointes-Arrhythmien [8].
Citalopram wird enantioselektiv über CYP2C19 abgebaut mit Bevorzugung des Abbaus von S-Citalopram. Durch CYP2C19-Inhibitoren wie Omeprazol geht diese Stereoselektivität verloren. Die Wirkspiegel von S-Citalopram steigen an, die von R-Citalopram nicht [10]. Dadurch besteht ein erhöhtes Risiko für kardiale Nebenwirkungen. Citalopram selbst weist nur schwache modulierende Eigenschaften auf.
SSRI als Modulatoren von CYP
Fluoxetin und Paroxetin sind starke Inhibitoren des Isoenzyms CYP2D6 (Tab. 1, Abb. 1). Bei Substanzen, die über diesen Weg weitgehend abgebaut werden, können die Plasmaspiegel klinisch signifikant steigen. Bei gleichzeitiger Einnahme des Betablockers Metoprolol ist mit einer Erhöhung der AUC (Fläche unter der Konzentrations-Zeit-Kurve) um 400 bis 600% zu rechnen [9]. Bei Substanzen, die über CYP2D6 bioaktiviert werden, vermindert sich die Bildung wirksamer Metaboliten mit der Gefahr des Therapieversagens. Beispielsweise blockieren starke CYP2D6-Hemmer die Biotransformation von Tamoxifen (Prodrug) zu seinem aktiven Metaboliten Endoxifen in einem klinisch relevanten Maß, sodass Fluoxetin und Paroxetin nicht zusammen mit dem Antiestrogen eingenommen werden sollen [1].
Citalopram, Escitalopram und Sertralin in der Standarddosis (50–100 mg) hemmen CYP2D6 geringfügig ohne klinische Bedeutung [15] und sind die besser geeigneten SSRI, wenn gleichzeitig Substanzen verordnet werden, deren Abbau wesentlich von CYP2D6 abhängt.
Fluvoxamin ist ein starker Inhibitor von CYP1A2 und CYP2C19. Es ist kontraindiziert bei Patienten unter Agomelatin-Therapie [6]. Im Einzelfall wird der hemmende Effekt auf die beiden Isoenzyme genutzt bei Patienten, die hohe Dosen des Antipsychotikums Clozapin benötigen. Unter Dosisanpassung und Kontrolle der Clozapin-Spiegel kann eine Kombination mit Fluvoxamin zu einer verbesserten Wirkung und Verträglichkeit der Clozapin-Therapie führen [13].
Neben der CYP2D6-Blockade (>150 mg/d) hemmt Sertralin CYP2B6 [14]. Über CYP2B6 werden unter anderem aus Cyclophosphamid (Prodrug) aktive Metaboliten gebildet [16]. Eine Abschwächung der zytostatischen Therapie durch Sertralin kann die Folge sein. Für nur wenige weitere Wirkstoffe ist eine klinisch relevante Hemmung dieses Isoenzyms denkbar (z.B. Clomethiazol, Methadon und Propofol) [14].
Auf Ebene der Cytochrom-P450-Isoenzyme erscheint das Interaktionsrisiko von Sertralin verglichen mit Fluoxetin, Fluvoxamin und Paroxetin geringer zu sein. Vor diesem Hintergrund ist es begrüßenswert, dass Sertralin für 2014 als weitere Leitsubstanz der KBV aufgenommen wurde.
Alle Serotonin-Wiederaufnahmehemmer können in Monotherapie ein Serotonin-Syndrom auslösen. Das Risiko steigt zum einen durch die Kombination mit Monoaminoxidase-(MAO-)Hemmern (z.B. Moclobemid), SNRI-Antidepressiva (z.B. Venlafaxin) und anderen serotonergen Substanzen (z.B. Tramadol), aber auch aufgrund eines Plasmaspiegelanstiegs der SSRI durch abbauhemmende Modulatoren. Vor dem Hintergrund dieser potentiell lebensbedrohlichen Notfallsituation sollte bei gleichzeitiger Verordnung eines SSRI mit einem anderen Medikament das Risiko für Wechselwirkungen geprüft werden, vorzugsweise mit einem Interaktionsprogramm.
Pharmakogenetik
Unabhängig von weiteren Medikamenten kann das klinische Ansprechen der Patienten unter Standard- dosen der selektiven Serotonin-Wiederaufnahmehemmer individuell stark abweichen. Dies liegt darin begründet, dass die Bildung funktionsfähiger CYP2C19- und CYP2D6-Enyzme genetisch bedingt variiert. Je nach Status unterscheidet sich die Eliminationsgeschwindigkeit bis um den Faktor 100 (bei CYP2D6-Substraten). Es werden abhängig von der Metabolisierungsaktivität vier Typen unterschieden.
- Langsame Metabolisierer (Poor metabolizer; PM) → stark reduzierter Stoffwechsel
- Intermediäre Metabolisierer (Intermediate metabolizer; IM) → reduzierter Stoffwechsel
- Extensive Metabolisierer (Extensive metabolizer; EM) → normaler Stoffwechsel
- Ultraschnelle Metabolisierer (Ultrarapid metabolizer; UM) → beschleunigter Stoffwechsel
In der Fachinformation von Citalopram findet sich der Hinweis, dass für Patienten, von denen eine verringerte Verstoffwechslung über CYP2C19 bekannt ist, die Maximaldosis auf 20 mg begrenzt ist [3].
Für Sertralin gibt die Fachinformation eine Plasmaspiegelerhöhung bei langsamen CYP2C19-Metabolisierern (PM) im Vergleich mit schnellen Metabolisierern (EM) an, ohne jedoch Vorgaben zur Dosisreduktion zu machen [7].
In der Fachinformation von Fluoxetin wird die Metabolisierung über das polymorphe CYP2D6 vermerkt [4]; welche klinischen Konsequenzen dies bei Patienten mit PM- oder UM-Status hat, wird nicht weiter ausgeführt. Auch in der Fachinformation von Paroxetin werden diesbezüglich keine Empfehlungen gegeben [5]. Für beide Substanzen werden bei Patienten mit PM-Status erhöhte Plasmaspiegel erwartet, wodurch das Risiko für unerwünschte Arzneimittelwirkungen (cave: Serotonin-Syndrom) steigt. Bei Patienten mit UM-Status können die Plasmaspiegel fallen mit der Gefahr des Therapieversagens [2].
Mit dem Aktionsplan 2008/2009 des Bundesministeriums für Gesundheit zur Verbesserung der Arzneimitteltherapiesicherheit (AMTS) in Deutschland sind Maßnahmen zur schwerpunktorientierten Verbesserung der Fachinformationen eingeleitet worden. Diese betreffen in erster Linie Anpassungen hinsichtlich Wechselwirkungen und Dosisanpassung bei Niereninsuffizienz. Wünschenswert wäre, wenn die Fachinformationen bei Wirkstoffen, deren Metabolisierung relevant abhängig ist vom genetischen Polymorphismus, stets Angaben zur Dosisreduktion und einer intensivierten Beachtung von unerwünschten Arzneimittelwirkungen enthielte.
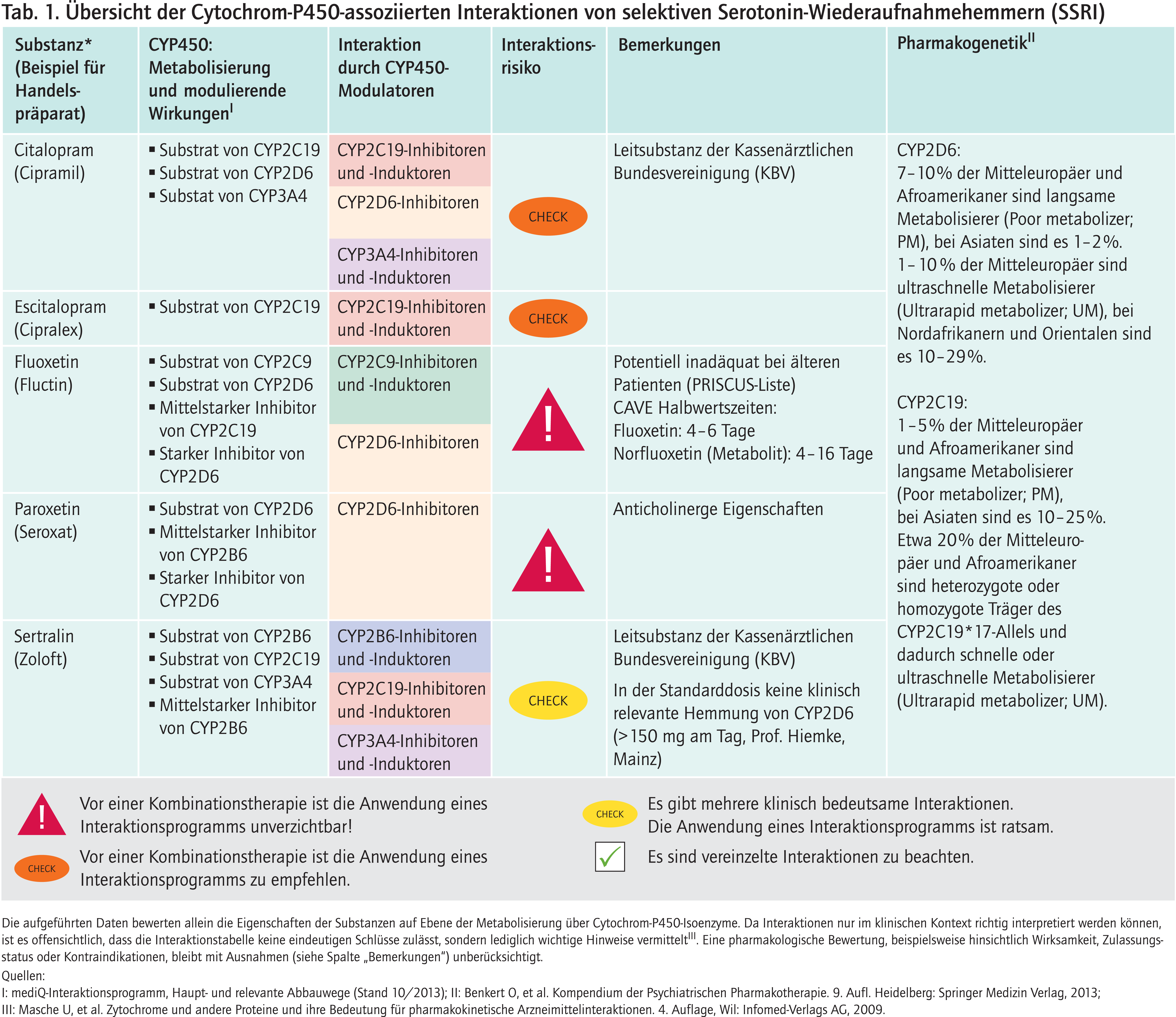
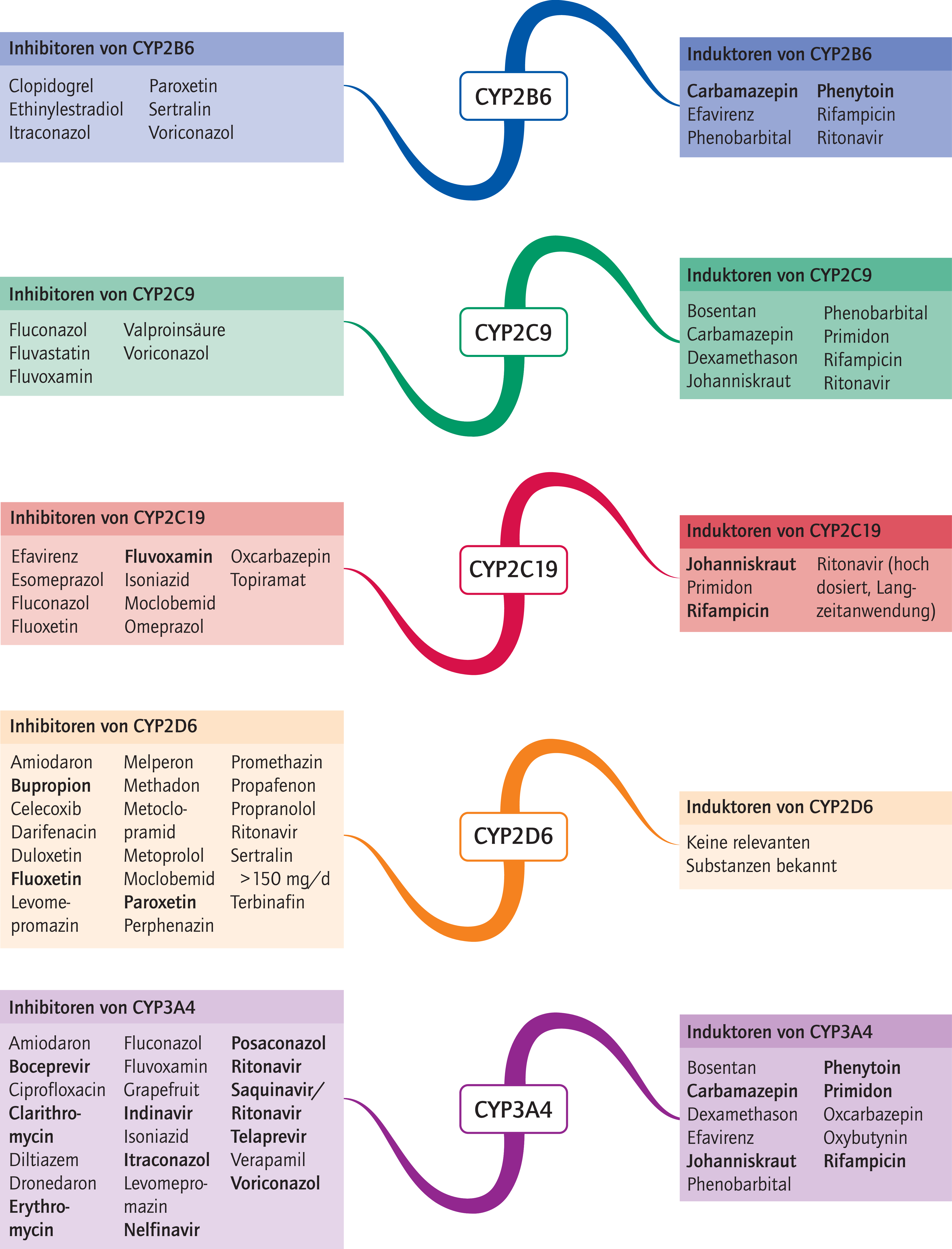
Abb. 1. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf einzelne CYP450-Isoenzyme (Stand: 10/2013) [Quelle: mediQ-Interaktionsprogramm]
Literatur
1. Bescheid BfArM vom 14.11.2012.
2. Charlier C et al. Polymorphisms in the CYP 2D6 gene: association with plasma concentrations of fluoxetine and paroxetine. Ther Drug Monit. 2003;25:738–42.
3. Fachinformation Cipramil®, Stand Januar 2013.
4. Fachinformation Fluctin®, Stand April 2012.
5. Fachinformation Seroxat®, Stand Juli 2012.
6. Fachinformation Valdoxan®, Stand April 2013.
7. Fachinfomation Zoloft®, Stand Juli 2013.
8. www.azcert.org/medical-pros/drug-lists/list-01.cfm (Zugriff am 14.09.2013).
9. Molden E et al. Interactions between metoprolol and antidepressants. Tidsskr Nor Laegeforen. 2011;131:1777–9.
10. Rocha A, Coelho EB, Sampaio SA, Lanchote VL. Omeprazole preferentially inhibits the metabolism of (+)-(S)-citalopram in healthy volunteers. Br J Clin Pharmacol 2010;70:43–51.
11. Rote-Hand-Brief Cipramil®, 31. Oktober 2011.
12. Schwalbe U, Paffrath D. Arzneimittelverordnungs-Report 2013. Heidelberg: Springer Verlag, 2013.
13. Schwarz M, et al. Therapeutisches Drug-Monitoring für die individualisierte Risikominimierung der Psychopharmakotherapie. Psychopharmakotherapie 2013;20:163–7.
14. Talakad JC, et al. Decreased susceptibility of the cytochrome P450 2B6 variant K262R to inhibition by several clinically important drugs. Drug Metab Dispos 2009;37: 644–50.
15. Waldschmitt C, et al. Das pharmakokinetische Interaktionspotenzial von Duloxetin. Psychopharmakotherapie 2009;16:14–8.
16. Zanger UM, et al. Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics 2007;8: 743–59.
*Nachdruck aus Krankenhauspharmazie 2013;34:519–22.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2013; 20(06)