Walter E. Müller, Frankfurt am Main
Die Neurobiologie der Alkoholabhängigkeit als pharmakologischer Angriffspunkt
Die Pathophysiologie (siehe der Beitrag von Spanagel und Kiefer) geht davon aus, dass im Rahmen der Abhängigkeitsentwicklung auf Alkohol (und andere Suchtstoffe) nicht nur das dopaminerge Belohnungssystem, sondern auch Opioidmechanismen involviert sind, die über My-Rezeptoren primär die euphorisierenden Effekte von Alkohol mit auslösen [4], aber auch über Kappa-Rezeptoren dysphorische Effekte gegenspielend initiieren, die eher eine Aversion gegen Alkohol bedeuten [74]. GABAerge und glutamaterge Mechanismen sind für die stimulierenden, aber auch sedierenden bis narkotischen Effekte von Ethanol sowie für die Entzugssymptome verantwortlich. Der Abhängigkeitsentwicklung wie Verstärkung der Einnahme, Belohnungsauslösung, Gier (craving), chronische Wiederaufnahme und Rückfall nach Abstinenz liegen dagegen dopaminerge und opioiderge Steuerungen zugrunde [4].
In einem vereinfachten Modell (Abb. 1) geht man davon aus, dass das für Abhängigkeitsentwicklungen besonders relevante dopaminerge Belohnungssystem, welches vom ventralen Tegment zum Nucleus accumbens führt, durch Opioidmechanismen verstärkt werden kann, die entweder direkt im Nucleus accumbens oder indirekt durch Hemmung von GABAergen Neuronen die Dopaminfreisetzung steigern. Die Opioidmechanismen gehen auf Beta-Endorphin-haltige Neuronen zurück, die hauptsächlich über My-Rezeptoren ihre Effekte auf die Targetzellen weiterleiten. Die Beta-Endorphin-haltigen Neuronen haben ihren Ursprung im Nucleus arcuatus und verbinden damit die Stressachse mit dem Belohnungssystem. Darüber hinaus werden GABAerge Strukturen im ventralen Tegment auch über Enkephalin-haltige Interneuronen wieder über My- und gegebenenfalls Delta-Rezeptoren gehemmt. Durch Blockade dieses Effekts durch Naltrexon und Nalmefen wird die reduzierende Wirkung von Opioidantagonisten auf Abhängigkeitsentwicklung und Abhängigkeitsstabilisierung erklärt. Der gegenspielende Effekt von Dynorphin, bedingt durch eine hohe Konzentration von Kappa-Rezeptoren in Nucleus accumbens, ventralem tegmentalem Areal (VTA) und präfrontalen Kortex (PFC), führt eher zu einer Senkung der Dopaminkonzentration. Die Dynorphin-haltigen Bahnen kommen unter anderem vom Striatum [74]. Während damit Acamprosat und die Opioidantagonisten regulierend in die Transmittersysteme eingreifen, welche die neurobiologische Grundlage der Alkoholabhängigkeit bilden, liegt der Wirkungsmechanismus von Disulfiram in einer Beeinflussung des Metabolismus von Alkohol und der unangenehmen Wirkung des unter Disulfiram kumulierenden Metaboliten Acetaldehyd.
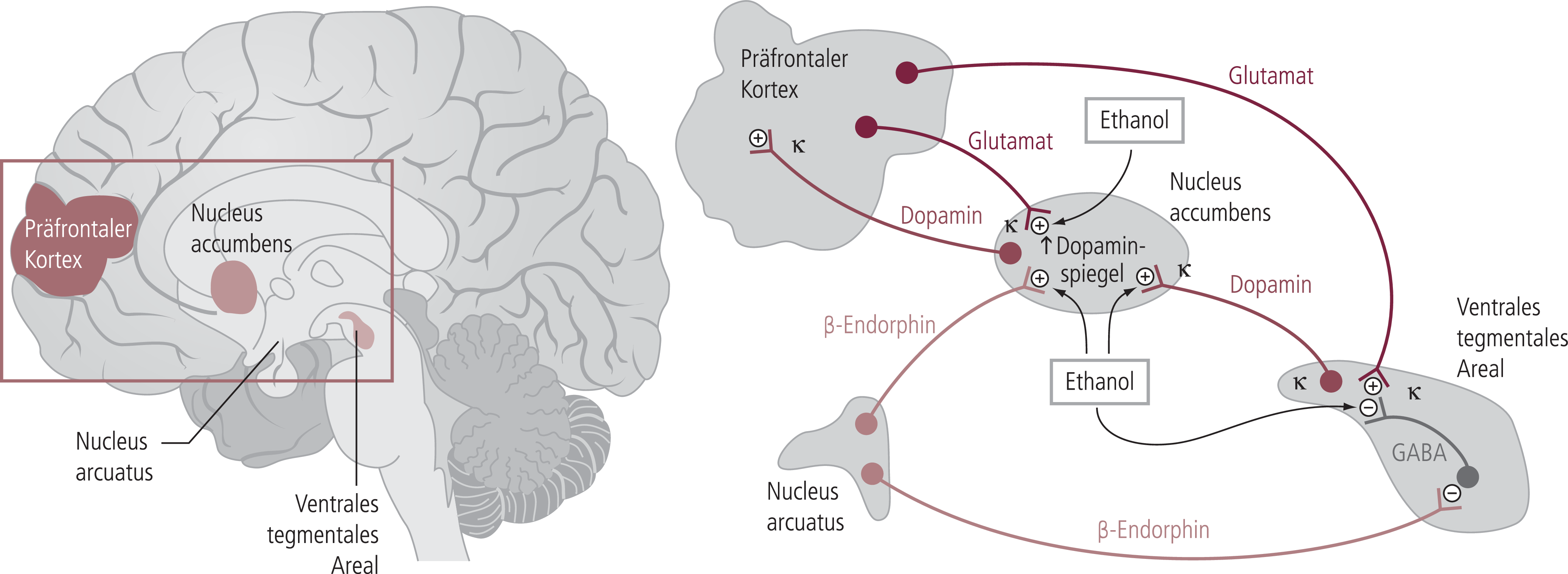
Abb. 1. Alkohol beeinflusst hemmend GABAerge und aktivierend glutamaterge und dopaminerge Mechanismen des Belohnungssystems. Beta-Endorphin (hauptsächlich über My-Opioidrezeptoren) und Dynorphin (über Kappa-Opioidrezeptoren) modellieren diesen Effekt [mod. nach 4, 75]
Disulfiram
Disulfiram ist seit über einem halben Jahrhundert in Deutschland zur Therapie alkoholabhängiger Patienten im Gebrauch. Die Substanz hemmt die zweite Stufe des Alkoholabbaus [63], der in der Oxidation des zunächst entstandenen Acetaldehyds über die Acetaldehyddehydrogenase besteht. Bei gleichzeitiger Einnahme von Disulfiram und Alkohol kommt es damit zu einer Kumulation von Acetaldehyd (Abb. 2) und gegebenenfalls zu einer sogenannten Alkohol-Disulfiram-Reaktion (ADR) (Tab. 1). Diese führt im günstigen Fall zu deutlich unangenehmen vegetativen Effekten, die den abstinenten Alkoholpatienten vor einem Rückfall zurückhalten sollen. Bei zu starker ADR kann es aber auch zu schweren Komplikationen hin bis zu lebensbedrohenden Situationen kommen. Zur aktuellen Bewertung von Disulfiram in der Therapie abstinenter alkoholabhängiger Patienten sei auf die Übersichten von Mutschler [58, 59] verwiesen. Neben der Auslösung einer milden und dann aversiven ADR wird für Disulfiram eine Hemmung der Dopamin-Beta-Hydroxylase diskutiert, die zu einem Anstieg von Dopamin und einer Verminderung von Noradrenalin in peripheren und zentralen Geweben führt. Man nimmt an, dass dieser Mechanismus auch eine Bedeutung für die Wirksamkeit der Substanz hat. Diese Hypothese geht parallel mit Befunden, dass die Aktivität der Dopamin-Beta-Hydroxylase selbst bei Alkoholabhängigen reduziert erscheint [43].
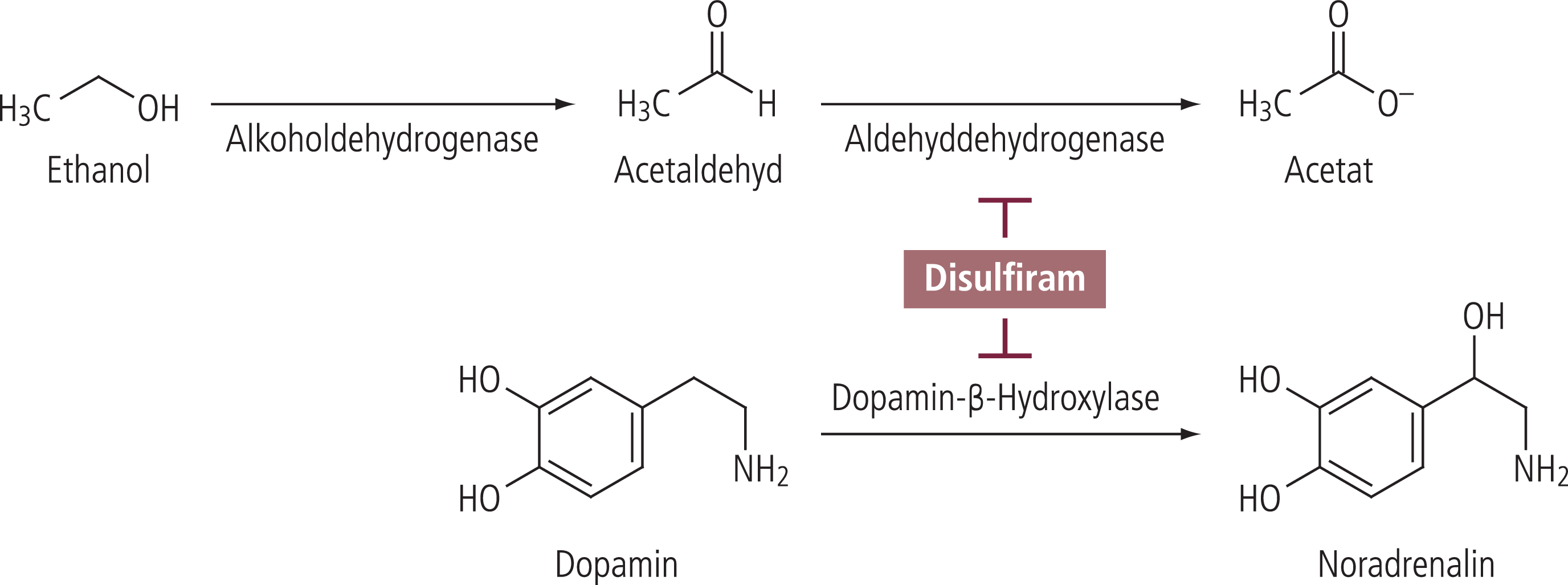
Abb. 2. Disulfiram hemmt die Aldehyddehydrogenease (Anstieg Acetaldehyd nach Alkoholeinnahme) und die Dopamin-Beta-Hydroxylase (Anstieg Dopamin)
Tab. 1. Die möglichen Symptome der Alkohol-Disulfiram-Reaktion, die bei Alkoholeinnahme unter Disulfiram-Therapie entstehen kann [59]
|
Alkohol-Disulfiram-Reaktion (ADR) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Für die Wirkung, besonders die Hemmung der Aldehyddehydrogenase scheint nicht Disulfiram selbst, sondern der aktive Metabolit Diethyldithiocarbamat und dessen Methylderivat verantwortlich zu sein. Letztere Struktur wird aus der Muttersubstanz über Cytochrom-P450-Enzyme in der Leber gebildet, besonders durch CYP2E1, CYP3A4 und CYP2A6 [49], während der erste Schritt nichtenzymatisch zum Teil schon im sauren Milieu des Magens erfolgt. Variationen der Aktivität dieser Enzyme beispielsweise durch Polymorphismen, aber auch durch induzierende bzw. inhibierende Komedikationen können daher die Wirkung von Disulfiram beeinflussen. Die Datenlage dazu ist allerdings sehr unübersichtlich.
Disulfiram wird nach oraler Gabe relativ langsam resorbiert, maximale Plasmaspiegel werden offensichtlich erst nach vielen Stunden erreicht. Die Eliminationshalbwertszeit liegt bei etwa sieben Stunden, sodass aktive Plasmaspiegel über lange Zeit nach oraler Einnahme bestehen [34]. Die ADR allerdings scheint in der Regel nach einigen Stunden in ihrer Ausprägung abzuklingen mit großen interindividueller Varianz. Dies ist für Maßnahmen bei Interventionen nach schwerer Ausprägung der ADR von Bedeutung. Während die Tagesdosierung heute meist bei unter einem halben Gramm liegt, hat man in früheren Jahren sehr viel höhere Dosen zwischen 1 und 3 Gramm eingesetzt, die häufiger schwerwiegende, im Einzelfall auch letale, unerwünschte Arzneimittelwirkungen auslösten. Die Pharmakokinetik zeigt relativ hohe Schwankungen, sodass die Wirkung zwar im Mittelwert nach wenigen Tagen verschwunden ist, im Einzelfall aber auch länger fortbestehen kann. Die geringe Beziehung zwischen der Pharmakokinetik und der pharmakodynamischen Wirkung wird zudem dadurch verständlich, dass neben der Ausgangsverbindung auch die beiden Hauptmetaboliten im Sinne einer Suizidhemmung (kovalente Bindung) die Aldehyddehydrogenase irreversibel inhibieren.
Aufgrund der nicht unerheblichen Toxizität von Disulfiram ist der Einsatz immer wieder kritisiert worden, dennoch scheint diese Therapieoption auch in Deutschland immer noch ihren Platz zu haben [58, 59].
Acamprosat
Acamprosat ist als erstes Arzneimittel in die Therapie der Alkoholerkrankung eingeführt worden, dessen Wirksamkeit auf eine Reduktion des Cravings, des Verlangens nach Alkohol, erklärt wird. Unter der Therapie mit Acamprosat kann daher Alkoholabstinenz leichter und länger aufrechterhalten werden, wenn auch die Effektstärke klein und die klinische Datenlage nicht sehr einheitlich ist [50].
Wirkungsmechanismus
Der Wirkungsmechanismus von Acamprosat ist immer noch nicht vollständig geklärt. Wegen der Strukturähnlichkeit zu den Neuromodulatoren Taurin bzw. Homotaurin hat man den Effekt von Acamprosat auf diese Substanzen untersucht, allerdings ohne hier eine klare Aussage treffen zu können [16]. Man geht heute eher davon aus, dass die Substanz eine kompensatorische Modulation glutamaterger Mechanismen während des Alkoholentzugs dämpft, wobei antagonistische Effekte an Glutamatrezeptoren vom N-Methyl-D-Aspartat-Typ und möglicherweise mGlu5-Typ eine Rolle spielen [47, 50, 73, 81] (Abb. 3).
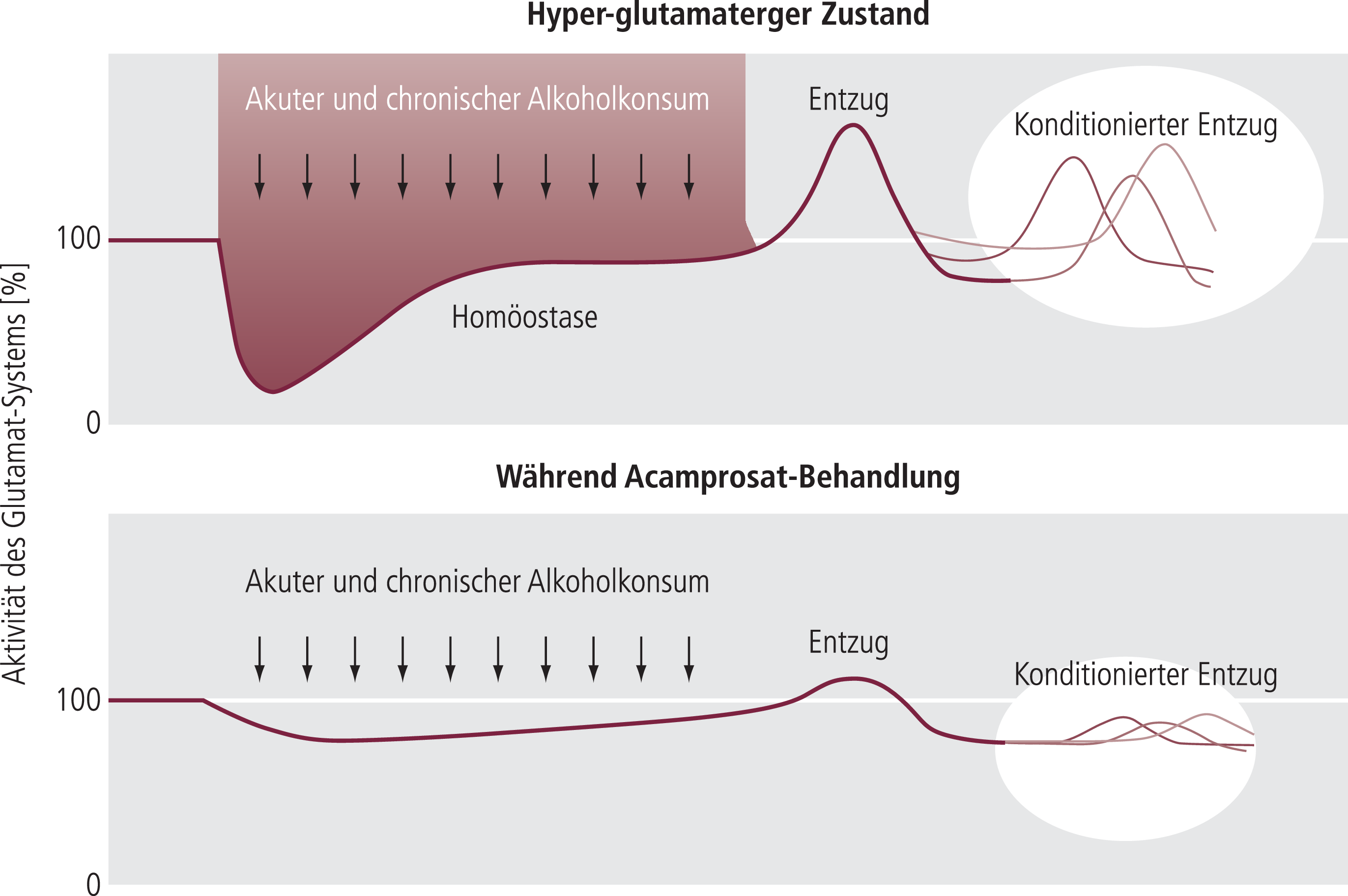
Abb. 3. Die heute gültige Modellvorstellung, wie Acamprosat über eine Modulation (aktivierend und hemmend) glutamaterger Mechanismen in die Neurobiologie der Alkoholabhängigkeit eingreifen kann [mod. nach 81]
Viele Daten weisen darauf hin, dass Acamprosat modulierend die Funktion des NMDA-Rezeptors beeinflusst, mit einer gewissen Aktivierung bei niedrigeren und einer gewissen Blockade bei hohen Konzentrationen. Der Angriffspunkt an dem komplexen NMDA-Rezeptorsystem ist allerdings nicht bekannt, die Substanz bindet weder direkt an die Transmitter- noch an die Kanalbindungsstelle, an der Substanzen wie Memantin, aber auch Phencyclin und MK801 angreifen. In früheren Untersuchungen wurde für Acamprosat eine Zielstruktur in Hirnmembranen gefunden, an die Acamprosat mit einer Dissoziationskonstanten von 120 µmol/l bindet. Da diese Bindungsstelle von Acamprosat besonders empfindlich war für Polyamine wie Spermidin [60] und da Polyamine wie Spermin und Spermidin eine negative Modulation am NMDA-Rezeptorkomplex bewirken können [68], hat man auch die Bindung von Acamprosat am NMDA-Rezeptor selbst über die Bindung von MK801 untersucht [60]. Tatsächlich war die Bindung biphasisch mit einer Verstärkung der MK801-Bindung bei niedrigeren und einer Hemmung bei höheren Konzentrationen. Diese Befunde wurden in der Arbeit von Al Qatari et al. [2] bestätigt, die darüber hinaus zeigen konnten, dass die Bindung unterschiedlich ist in Membranen von alkoholnaiven und chronisch alkoholbehandelten Tieren. Sie leiten daraus einen biphasischen Effekt von Acamprosat auf die NMDA-Rezeptorfunktion ab. Dieser Effekt wurde von Pierrefiche et al. [65] mit elektrophysiologischen Methoden bestätigt. Ausgehend von Befunden, die Änderungen der NMDA-Rezeptorbindungseigenschaften in Hirnmembranen von Ratten bei Alkoholentzug erkennen lassen, wurde der Effekt von Acamprosat an solchen Tieren weiter untersucht. Man geht davon aus, dass den Veränderungen des NMDA-Rezeptorkomplexes, die sich bei Alkoholentzug in Ratten ausbilden, durch Acamprosat entgegengesteuert werden kann [52]. Rammes et al. [69] lieferten ähnliche Befunde und ergänzten, dass unter Acamprosat auch die Expression von NMDA-Untereinheiten verändert ist. Über die Interaktion mit dem NMDA-Rezeptorkomplex hinaus scheint Acamprosat auch mit metabotropen mGlu5-Rezeptoren zu interagieren und über diesen Mechanismus gewisse neuroprotektive Effekte zu entwickeln, die wiederum mit dem Wirkungsmechanismus bei Alkoholabhängigkeit in Verbindung gebracht werden [29].
Obwohl diese Daten nicht in allen Punkten schlüssig sind, geben sie ein akzeptables Bild von der Wirksamkeit von Acamprosat bei Alkoholabhängigkeit [5]. Die bereits beschriebenen Angriffspunkte (NMDA-Rezeptor und mGlu5-Rezeptor) spielen hier eine wichtige Rolle und lassen die Hypothese zu, dass die biphasische Funktionsveränderung des glutamatergen Systems während des Alkoholkonsums (primäre Unterfunktion) und die mit dem Entzug verbundene Überfunktion (Abb. 3) durch Acamprosat gedämpft werden [48, 81].
Pharmakokinetik
Die Humanpharmakologie ist nur begrenzt untersucht [72, 91]. Die Substanz hat eine orale Bioverfügbarkeit von 11%, wobei die sehr langsame, wahrscheinlich parazellulär erfolgende Resorption erheblichen individuellen Schwankungen unterliegt. Dies erklärt möglicherweise, dass der Steady State erst nach sieben Tagen erreicht wurde und die terminale Halbwertszeit mit etwa 13 Stunden im Mittelwert angegeben wird. Die Substanz wird weitgehend unverändert über Urin und Fäzes ausgeschieden.
Effekt auf Alkoholeinnahme
Wesentliche Voraussetzung für die Verwendung von Acamprosat bei der Behandlung Alkohol-abhängiger Patienten waren Befunde, die in einer Reihe von tierexperimentellen Anwendungen auf eine reduzierte Alkoholaufnahme hinwiesen [47, 83]. Dieser Effekt ist primär bei Alkohol-abhängigen zumindest aber Alkohol-erfahrenen Tieren und eher nicht bei Alkohol-unerfahrenen Tieren zu beobachten [13, 79]. Im Sinne des Modells (Abb. 3) ist der Effekt mit einer Reduktion der glutamatergen Überstimulation zu Beginn der Trinkepisode verbunden, aber auch mit einer Reduktion der Alkohol-bedingten Erhöhung von Dopamin im Nucleus accumbens [61]. Darüber hinaus scheint die Substanz einen milden Effekt gegen motorische Entzündungssymptome zu haben [14].
Opioidantagonisten
Der schon lange in die Therapie von Opioidintoxikationen eingeführte Antagonist Naloxon ist wegen seiner kurzen Halbwertszeit und fehlenden oralen Bioverfügbarkeit zur Therapie der Alkohol-abhängigkeit nicht geeignet, hat aber in Tierversuchen unter chronischer Daueranwendung über Minipumpen eine Reduktion der Alkohol-Selbsteinnahme erreicht [56]. Wesentlich besser geeignet zur Therapie der Alkoholabhängigkeit erwiesen sich die beiden strukturell von Naloxon abgeleiteten Opioidantagonisten [27] Naltrexon und Nalmefen (Abb. 4).
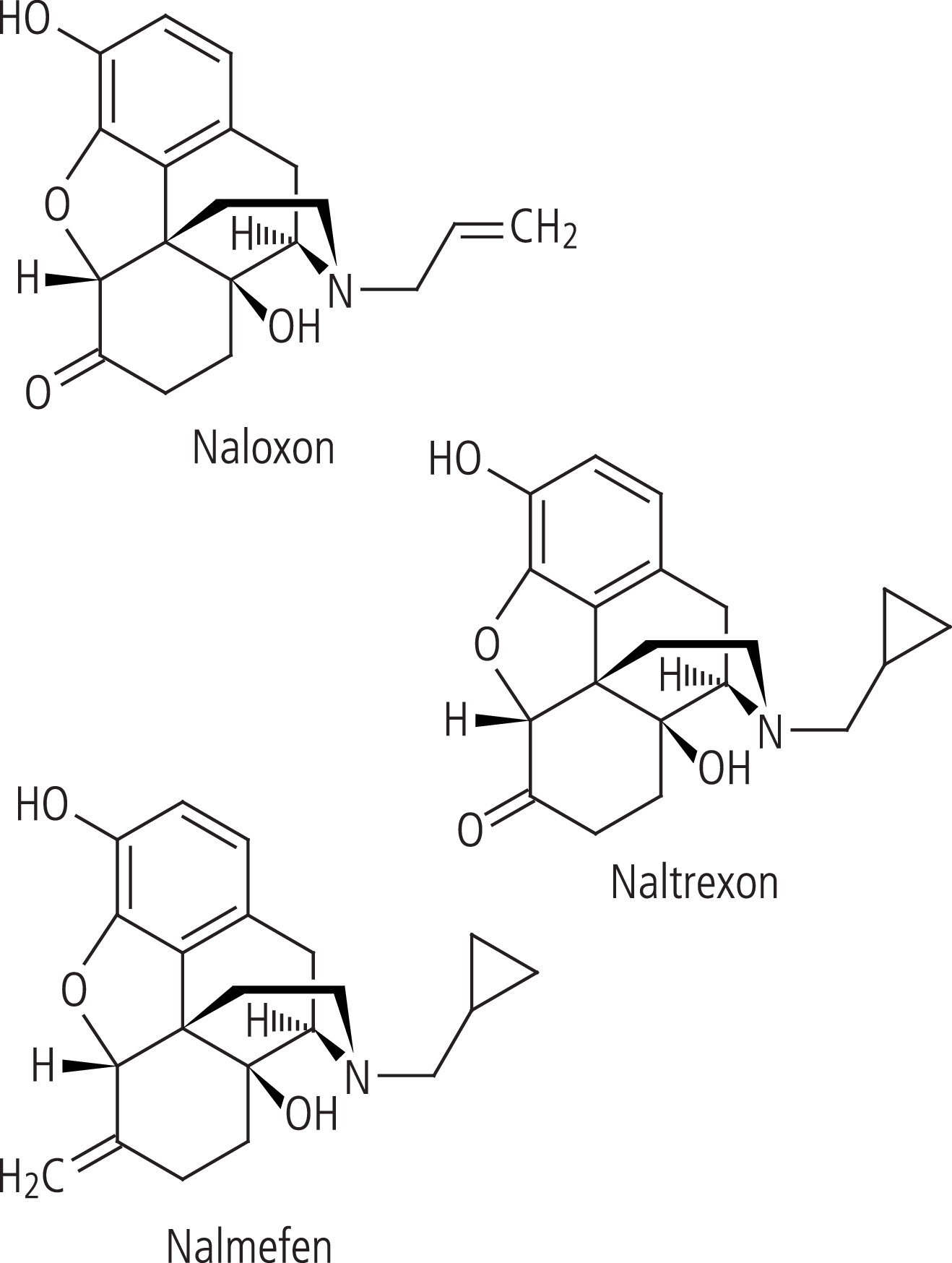
Abb. 4. Strukturformeln von Naloxon und der beiden zur Therapie der Alkoholabhängigkeit eingesetzten Opioidantagonisten Naltrexon und Nalmefen
Im Vergleich zu Naloxon mit einer Halbwertszeit von etwa einer Stunde ist bei Naltrexon die Verweildauer im Organismus deutlich länger mit einer mittleren Halbwertszeit von 4 bis 5 Stunden, aber weiterhin suboptimal [70]. Dies war zunächst ausschlaggebend für die Entwicklung von Nalmefen, einen Opioidantagonisten mit breitem Rezeptorspektrum, ähnlich wie Naloxon und Naltrexon, aber mit einer deutlich längeren Eliminationshalbwertszeit von über 8 Stunden [17, 18].
Naltrexon
Pharmakokinetik
Wie bereits erwähnt, war Naloxon aufgrund der ungünstigen Pharmakokinetik (kurze Halbwertszeit, fehlende orale Bioverfügbarkeit) zur Therapie der Alkoholabhängigkeit nicht geeignet. Naltrexon zeigte hier deutliche Vorteile (Tab. 2). Mit einer relativ schnellen Resorption (tmax ca. 1 h) und einer mittleren Halbwertszeit von 4 Stunden war die Substanz für die orale Anwendung bereits deutlich besser geeignet. Nachteilig gestaltete sich auch hier die noch sehr unbefriedigende Bioverfügbarkeit von 5%, die allerdings im individuellen Einzelfall hohen Schwankungen unterworfen zu sein scheint und angeblich bis zu 40% steigen kann [70]. Eine deutlich längere Eliminationshalbwertszeit hat der pharmakodynamisch ähnlich aktive Metabolit Beta-Naltrexol mit einer Halbwertszeit von 12 bis 13 Stunden. Beta-Naltrexol scheint darüber hinaus zu einem großen Teil für die Wirkung von Naltrexon verantwortlich zu sein, da es sehr viel höhere Plasmakonzentrationen als die Ausgangssubstanz erreicht [41]. Plasmaspiegel von Beta-Naltrexol korrelierten besser mit Nebenwirkungen wie Kopfschmerz, Erbrechen oder Ängstlichkeit als Plasmaspiegel von Naltrexon [41]. Der größte Teil von Naltrexon wird in Form von Beta-Naltrexol über den Urin und weniger in Form der Muttersubstanz ausgeschieden. Die renale Clearance von Muttersubstanz und Metabolit sind ähnlich [53]. Die bisher beschriebenen Eliminationshalbwertszeiten für Naltrexon und Beta-Naltrexol von 1 bzw. 12 Stunden können allerdings nicht die relativ lange Antagonisierung der Heroinwirkung (25 mg) durch orales Naltrexon erklären, die über 24 Stunden anhält [85]. In Analogie zu diesem sehr langen antagonistischen Effekt stehen Daten aus Positronen-Emissions-tomographischen Untersuchungen (PET), die auf eine ebenso lange Okkupation von Opioidrezeptoren (My-Typ) hinwiesen [44], und Befunde, dass sich im Eliminationsspektrum von Naltrexon noch eine dritte Eliminationskonstante von 36 Stunden bei sehr niedrigen Plasmakonzentrationen beschreiben ließ. Eine mögliche Erklärung wird von den Autoren darin gesehen, dass unmetabolisiertes Naltrexon in der Niere zurückresorbiert werden kann, nicht aber der hydrophilere Metabolit (Beta-Naltrexol). Wegen der immer noch relativ kurzen Pharmakokinetik verliert Naltrexon für die therapeutische Anwendung am Menschen an Bedeutung.
Tab. 2. Pharmakokinetische Eckdaten für Naltrexon am Menschen [1, 40, 53]
|
Naltrexon |
Beta- |
|
|
t1/2 |
1 |
2 |
|
tmax |
4 |
12 |
|
Elimination [l/h] |
94 |
– |
|
Bioverfügbarkeit [%] |
5 |
– |
|
Elrenal [ml/min] |
127 |
283 |
a Daten ermittelt nach oraler Gabe von Naltrexon
Untersuchungen zur Pharmakokinetik bei speziellen Patientenpopulationen liegen praktisch nicht vor [1]. Es gibt allerdings Hinweise darauf, dass bei Patienten mit schwerer Leberinsuffizienz die Plasmaspiegel von Naltrexon auf das 5- bis 10-Fache erhöht sein können.
Rezeptorpharmakologie
Analog zum Vorbild Naloxon verhält sich Naltrexon als unspezifischer Opioidrezeptorantagonist an den drei wichtigen Unterklassen (My, Delta, Kappa) (Tab. 3). Während die Bindungskonstanten an die entsprechenden Rezeptoren in Rattenhirnmembranen etwas abweichen, besonders im Hinblick auf die relativ geringe Affinität an Kappa-Rezeptoren, zeigt sich für die Bindungsdaten an rekombinante Humanrezeptoren eine deutliche Konsistenz. Naltrexon ist ein potenter Antagonist an humanen My- und Kappa-Rezeptoren mit Inhibitionskonstanten zwischen 1 und 2 nmol/l und verhält sich als deutlich schwächerer Antagonist an Delta-Rezeptoren. Das Bindungsspektrum des für die Wirkung ganz besonders relevanten Metaboliten Beta-Naltrexol ist dem der Muttersubstanz Naltrexon sehr ähnlich (Tab. 3). Inhibitionskonstanten gegen die Bindung eines spezifischen Rezeptorliganden bzw. Inhibitionskonstanten gegen die zelluläre Wirkung von Agonisten für die jeweiligen Opioidrezeptorenklassen ergeben das gleiche Bild für Naltrexon und Beta-Naltrexol [88]. Unter bestimmten Bedingungen zeigt Naltrexon, nicht aber Beta-Naltrexol invers-agonistische Eigenschaften am My-Rezeptor [67].
Tab. 3. Zusammenfassung publizierter Bindungskonstanten von Naloxon, Naltrexon und Beta-Naltrexol an Opioidrezeptorunterklassen
|
Substanz |
Ki [nmol/l] |
Gewebe [Lit.] |
||
|
My |
Delta |
Kappa |
||
|
Naloxon |
3,5 |
60,0 |
26,0 |
Ratte [54] |
|
Naltrexon |
0,9 |
10,0 |
10,0 |
|
|
Naloxon |
6,3 |
32,0 |
66,0 |
Ratte [55] |
|
Naltrexon |
0,6 |
6,0 |
6,0 |
|
|
Beta-Naltrexol |
2,1 |
213,0 |
7,4 |
Meerschwein [64] |
|
Naloxon |
1,4 |
68,0 |
2,5 |
Mensch [84] |
|
Naltrexon |
0,2 |
11,0 |
0,4 |
|
|
Naloxon |
7 |
8 |
4 |
Mensch [88] |
|
Naltrexon |
0,5 |
7 |
1 |
|
|
Beta-Naltrexol |
1,4 |
29 |
2 |
|
In einer PET-Rezeptor-Bindungsstudie an alkoholabhängigen Patienten konnte ein ähnlicher Unterschied zwischen My- und Delta-Rezeptor-Bindung für Naltrexon bestätigt werden. Unter Bedingungen einer fast 100%igen Okkupation unter Naltrexon am My-Rezeptor war die Delta-Rezeptor-Bindung mit durchschnittlich 25% deutlich geringer. Eine mögliche Erklärung könnte die Variation des Plasmaspiegels von Beta-Naltrexol sein, das deutlich schwächer an Delta-Rezeptoren bindet als die Muttersubstanz (Tab. 3) [90]. Daten über die Okkupation von Kappa-Rezeptoren sind nicht vorhanden, allerdings gibt es Hinweise, dass die Kappa-Okkupation unter therapeutischen Bedingungen geringer ist als die fast vollständige Blockade von My-Rezeptoren. Allerdings sind diese Unterschiede offensichtlich nicht gravierend, sodass Ray et al. [70] zusammenfassen, dass an den Wirkungen von Naltrexon bei Alkoholabhängigen nicht nur My-Rezeptoren, sondern auch Delta- und wahrscheinlich auch Kappa-Rezeptoren beteiligt sind.
Experimentelle Befunde zum Effekt auf die Alkoholaufnahme
Für Naltrexon und Beta-Naltrexol liegen viele experimentelle Daten über die Reduktion der Alkoholaufnahme im Tierversuch [56, 70], aber auch im Humanversuch [3] vor. In Übereinstimmung mit der großen Bedeutung von Opioid-Mechanismen für die Neurobiologie der Alkoholabhängigkeit (Abb. 1) konnten für Naltrexon Effekte auf die Alkoholaufnahme, das Verlangen (craving) und den mit einer Dopamin-Erhöhung im Nucleus accumbens verbundenen Alkohol-Effekt auf das Belohnungssystem gezeigt werden [7, 28].
Nalmefen – Entwicklungsgeschichte und Zulassungsstadium
Nalmefen wurde Anfang der 70er-Jahre als Derivat von Naloxon bzw. Naltrexon entwickelt, von denen es sich strukturchemisch nur gering unterscheidet (Abb. 4). Ziel war auch hier, einen Opioidantagonisten mit noch längerer Wirkung zur Verfügung zu haben.
Tab. 4. Pharmakokinetische Eckdaten von Nalmefen an gesunden Probanden
|
Parameter |
(1) |
(2) |
(3) |
(4A) |
(4B) |
(5) |
(6) |
|
t½ [h] |
8 |
10,5 |
13–15 |
10,8 |
9,4 |
12 |
|
|
VD [l] |
500 |
– |
600 |
600 |
|||
|
Cl [l/h] |
65 |
64 |
59 |
||||
|
tmax [h] |
– |
1,3 (50 mg) |
|||||
|
Cmax [ng/ml] |
– |
24 (50 mg) |
|||||
|
Bioverfügbarkeit [%] |
35–45 |
1) [17, 18] Gesunde männliche Probanden (19–46 Jahre), i.v. Applikation (2–24 mg)
2) [17] Gesunde männliche Probanden (19–44 Jahre), orale Applikation (50–300 mg)
3) [66] Gesunde männliche Probanden, i.v. Applikation, Asiaten (Chinesen)
4A) [23] Gesunde männliche Probanden (im Mittel 24 Jahre) i.v. Applikation
4B) [22] Gesunde männliche Probanden (im Mittel 70 Jahre), i.v. Applikation
5) [25] 50 mg orale Applikation
6) [45] Gesunde Probanden, i.v. Applikation, Asiaten (Chinesen)
Wie die beiden anderen Substanzen wurde Nalmefen als reiner Opioidantagonist eingeführt, mit höherer Potenz besonders für den My-Rezeptor, etwas breiterem Spektrum (auch Kappa-Rezeptoren werden sehr potent antagonisiert), aber mit längerer Halbwertszeit und damit verbunden eine länger währenden Wirkung im Vergleich zu Naloxon [26]. Die Substanz war einige Jahre in diversen Ländern unter verschiedenen Namen als Opioidantagonist am Markt, beispielsweise in den USA als Revex®. Nach Übernahme des Patents durch die Firma Lundbeck und Abschluss eines klinischen Entwicklungsprogramms wurde 2012 für Nalmefen die Zulassung zur Behandlung der Alkoholabhängigkeit, fokussiert auf eine Reduktion der Trinkmenge, durch die Europäische Kommission ausgesprochen.
Pharmakokinetik
Die Humanpharmakokinetik von Nalmefen wurde an gesunden Probanden, auch älteren, nach intravenöser und oraler Applikation und an Patienten mit Leber- oder Nierenfunktionsstörungen untersucht (Tab. 4 und 5).
Tab. 5. Pharmakokinetische Eckdaten von Nalmefen bei Patienten mit Leber- oder Nierenerkrankung
|
Parameter |
(1) |
(2) |
||
|
K |
P |
K |
P |
|
|
t½ [h] |
8 |
10,5 |
7,9 |
27,3 |
|
VD [l/kg] |
8,0 |
7,3 |
8,2 |
17,1 |
|
Cl [l/h/kg] |
0,9 |
0,61 |
0,96 |
0,68 |
1) [23] Gesunde Probanden (K) gegen Patienten mit Lebererkrankungen (P), i.v. Applikation
2) [51] Gesunde Probanden (K) gegen Patienten mit Niereninsuffizienz (P), i.v. Applikation
Ungeachtet der Applikation wird die Substanz mit einer mittleren terminalen Eliminationshalbwertszeit von 8 bis 10 Stunden ausgeschieden. Das Verteilungsvolumen ist mit etwa 600 Liter relativ hoch, das Gleiche gilt für die Clearance mit 65 Liter/Stunde. Nach oraler Applikation ist der maximale Plasmaspiegel mit Plasmakonzentrationen um 20 ng/ml bei einer oralen Dosis von 50 mg ungefähr nach 1½ Stunden erreicht. In allen Untersuchungen (i.v. bzw. oral) waren die pharmakokinetischen Daten linear, das heißt, sie haben sich nicht wesentlich mit der Dosis geändert. Bei älteren gesunden Probanden waren die pharmakokinetischen Eckdaten nicht verändert (Tab. 4). Das Gleiche gilt für Patienten mit Leberfunktionsstörungen, während bei Patienten mit Niereninsuffizienz die Clearance erniedrigt war, was zusammen mit einem erhöhten Verteilungsvolumen zu einer deutlichen Zunahme der Eliminationshalbwertszeit führte (Tab. 5).
Die Bioverfügbarkeit wurde in den vorangegangenen Untersuchungen nicht bestimmt. Sie wurde allerdings in der Arbeit von Gal et al. [25] mit 35 bis 45% nach oraler Gabe ermittelt (Tab. 4).
Metabolismus
Zum Metabolismus von Nalmefen liegt eine Untersuchung an Ratten und Hunden vor [57]. Im Hund wird Nalmefen fast ausschließlich zum Nalmefen-Glucuronid in einer Phase-II-Reaktion metabolisiert, wovon ein Teil noch zum Di-Glucuronid umgewandelt wird. Beide Metaboliten werden renal eliminiert. In der Ratte findet eine N-Dealkylierung zu Nornalmefen statt, das dann glucuroniert über die Niere ausgeschieden wird. Auch am Menschen scheint der größte Teil von Nalmefen als Konjugat über die Nieren eliminiert zu werden [17].
Sehr lange Rezeptorblockade
Im Gegensatz zur terminalen Eliminationshalbwertszeit von Nalmefen im Bereich von 8 bis 10 Stunden scheint die biologische Halbwertszeit vor allen Dingen im ZNS deutlich länger zu sein. Erste Hinweise lieferte eine Studie an gesunden Probanden [25], in der die Wirkung einer Testdosis Fentanyl auf die Atmung nach Gabe von 50 mg Nalmefen oral über die Zeit untersucht wurde. Die Plasmahalbwertszeit wurde auch hier zu ungefähr 11 Stunden ermittelt. Die Antagonisierung der durch Fentanyl ausgelösten Atemdepression war allerdings bis drei Tage nach Gabe von Nalmefen fast unverändert erhalten. Eine ähnlich lang anhaltende Wirkung des Opioidantagonisten im Falle einer oralen Dosis von Nalmefen (50 bis 100 mg) wurde auch von Jones et al. [37] beschrieben. Verschiedene pharmakologische Wirkungen von Morphin wie Auslösung von Miosis und die objektive Morphin-Wahrnehmung durch die Probanden (alles ehemalige Opioid-abhängige) wurden nach intravenöser Morphin-Gabe über die Zeit gemessen.
Hinweise auf eine sehr lange Persistenz von Nalmefen im ZNS aufgrund einer langsamen Dissoziation von zentralen Opioidrezeptoren konnten von Kim et al. [40] und später von Ingman et al. [33] wieder mithilfe von PET-Untersuchungen des Liganden [11C]Carfentanil bestätigt werden. Auch in dieser Untersuchung wurde eine Plasmaeliminationshalbwertszeit von Nalmefen von 13 Stunden ermittelt. Die mit dem Liganden ermittelte Okkupation der zentralen My-Rezeptoren wurde nach einmaliger (20 mg) bis 7-tägiger (20 mg/Tag) oraler Dosis von Nalmefen ermittelt und betrug in verschiedenen Hirnarealen kurz nach akuter Substanzeinnahme fast 100%. Nach einem Tag war die Rezeptorokkupation um ungefähr 20% abgefallen, nach zwei Tagen lag sie bei ungefähr 50%, nach drei Tagen bei ungefähr 30%, obwohl dazu die Plasmakonzentration mit der Eliminationshalbwertszeit von etwas über 10 Stunden abfiel (Abb. 5).
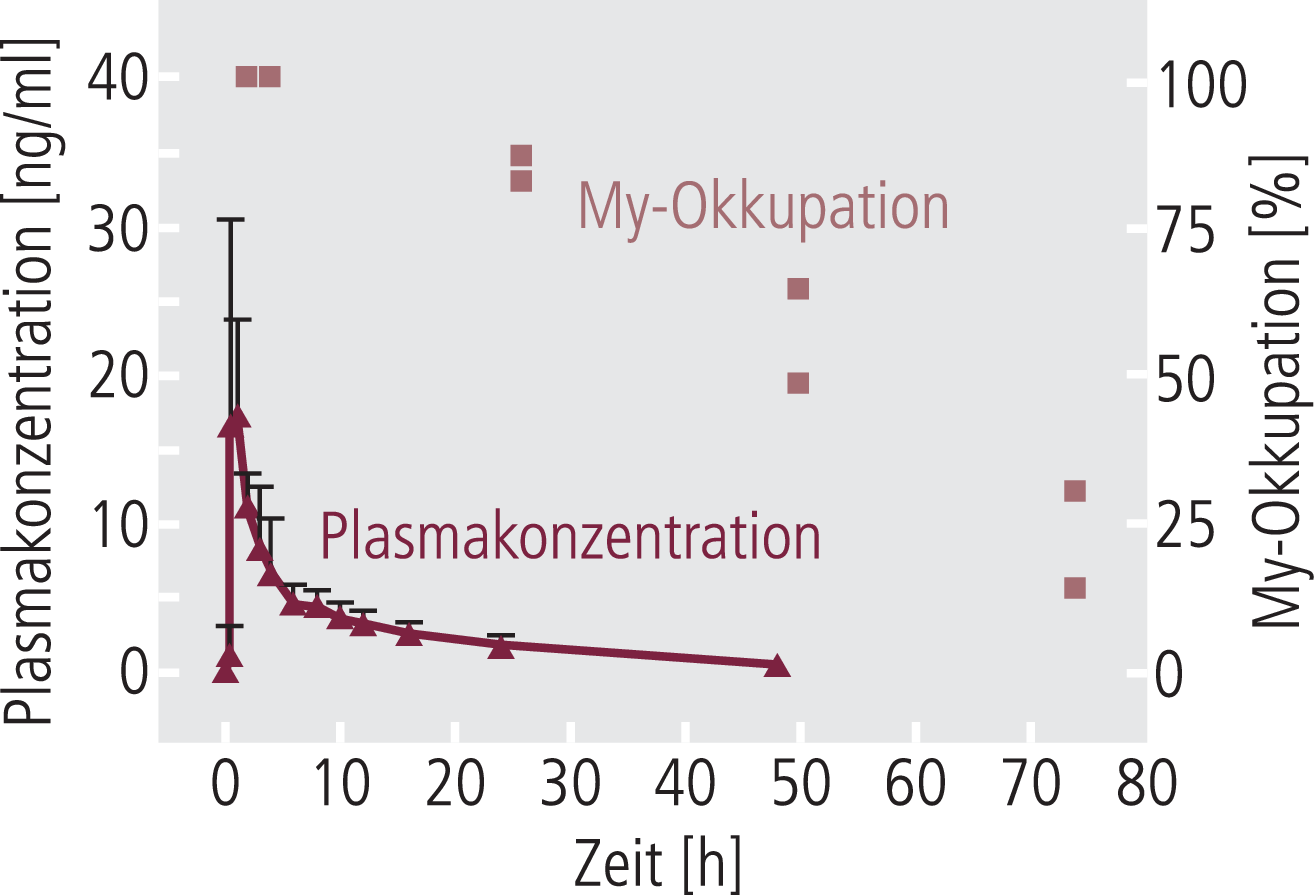
Abb. 5. Opioidrezeptorokkupation ([11C]Carfentanil-Bindung) nach einmaliger Gabe von Nalmefen im Vergleich zur Plasmakonzentration [mod. nach 33]
In-vitro-Rezeptorbindung
Die Bindung von Nalmefen an die drei relevanten Opioidrezeptorklassen wurde in vitro unter Verwendung von Ratten- bzw. Meerschweinchenhirnmembranen untersucht, aber auch an rekombinanten humanen Opioidrezeptoren (Tab. 6).
Tab. 6. Zusammenfassung publizierter Bindungskonstanten von Naltrexon und Nalmefen an Opioidrezeptorunterklassen
|
Substanz |
Ki [nmol/l] |
Gewebe [Lit.] |
||
|
My |
Delta |
Kappa |
||
|
Naltrexon |
0,9 |
10,0 |
10,0 |
Ratte [54] |
|
Nalmefen |
1,0 |
5,1 |
6,1 |
|
|
Naltrexon |
0,6 |
6,0 |
6,0 |
Ratte [55] |
|
Nalmefen |
0,3 |
3,4 |
2,0 |
|
|
Naltrexon |
0,6 |
1,4a |
0,5 |
Ratte [15] |
|
Nalmefen |
0,6 |
0,9a |
0,2 |
|
|
Naltrexon |
0,2 |
11,0 |
0,4 |
Mensch [84] |
|
Nalmefen |
0,3 |
7,3 |
0,3 |
|
|
Naltrexon |
– |
– |
– |
Mensch [6] |
|
Nalmefen |
0,2 |
16,0 |
0,08 |
|
a Die Daten wurden an neuronalen Membranen vom Meerschweinchen ermittelt
Trotz einer deutlichen Varianz ist erkennbar, dass sich die Rezeptoraffinitäten zwischen Rattenhirngewebe und menschlichem Hirngewebe relevant unterscheiden, vor allen Dingen in Bezug auf die Affinität zum Kappa-Rezeptor. Innerhalb der Affinitätsdaten zu den humanen Opioidrezeptoren sind die Daten konsistent dahingehend, dass beide Antagonisten etwas schwächer am Delta-Rezeptor wirken. Die Rezeptorprofile von Naltrexon und Nalmefen sind sonst ähnlich mit schwächerer Affinität zum Delta-Rezeptor und deutlich höherer Affinität zum My- und Kappa-Rezeptor. Mit einer leichten Tendenz bindet Nalmefen affiner an den Kappa-Rezeptor im Vergleich zum My-Rezeptor, allerdings sind die Unterschiede minimal. In der Untersuchung von Michel et al. [54] zeigten Nalmefen, Naltrexon und das hier ebenfalls geprüfte Naloxon den gleichen Natriumshift am My-Rezeptor, als möglichen Indikator für volle antagonistische Eigenschaften.
In-vivo-Aktivität als Opioidantagonist
Die Wirksamkeit von Nalmefen als Opioidrezeptorantagonist, das heißt die Aufhebung von verschiedenen Morphineffekten am Versuchtier, wurde in der Untersuchung von Osborn et al. [62] an Mäusen bestätigt. Auch die ältere Untersuchung von Culpepper-Morgan et al. [12] konnte die antagonistischen Eigenschaften von Nalmefen im Vergleich zu Naloxon und Naltrexon bestätigen. Von den drei Antagonisten war Nalmefen die potenteste Substanz in der Aufhebung einer durch einen Kappa-Agonisten ausgelösten Hemmung der Darmmotilität. Gegen eine durch Morphin ausgelöste Hemmung der Darmmotilität waren Naltrexon und Nalmefen gleich wirksam. Ausgeprägte Opioid-antagonistische Effekte am Menschen wurden von Fudala et al. [24] beschrieben.
Intrinsische Aktivität am Kappa-Opioidrezeptor
Naltrexon und Nalmefen binden ungefähr gleich stark an den My-Rezeptor und Kappa-Rezeptor von Menschen und Affen, mit einem leichten Affinitätsvorteil von Nalmefen für den Kappa-Opioidrezeptor, der auch in einigen funktionellen Tests gezeigt werden konnte. Während die pharmakologischen Daten keinen Zweifel daran lassen, dass die Substanz am My-Rezeptor sich wie ein reiner Antagonist verhält, gibt es im Rahmen der früheren Untersuchungen zu Nalmefen Hinweise darauf, dass die hochaffine Bindung an den Kappa-Rezeptor möglicherweise agonistische Komponenten enthält, sodass die Substanz als partieller Agonist oder partieller Antagonist am Kappa-Rezeptor einzuordnen wäre [84]. Naltrexon verhielt sich als reiner Antagonist am Kappa-Rezeptor.
Die Annahme, dass Nalmefen partial-agonistische Eigenschaften am Kappa-Rezeptor besitzen könnte, wurde unter anderem dadurch gefördert, dass in einem Diskriminierungsparadigma am Affen ein Kappa-Agonismus von Nalmefen gezeigt werden kann; im Gegensatz zu anderen Opioidantagonisten war Nalmefen in der Lage, nicht nur für Morphin, sondern auch für einen Kappa-Agonisten zu substituieren [92, 93]. Da die Bedeutung des Kappa-Rezeptors für die Gesamtwirkung von Nalmefen zu dieser Zeit nicht im Vordergrund stand, wurden die Daten zunächst nicht weiter verfolgt, bis Befunde über die freiwillig reduzierte Alkoholaufnahme durch den Einsatz von Kappa-Agonisten vorlagen [46]. Mit Blick auf eine sich anbahnende Therapie von Alkoholabhängigkeit mit Opioidantagonisten wurde das Interesse an einer möglichen partial-agonistischen Wirkung von Nalmefen am Kappa-Opioidrezeptor geweckt.
Ältere Befunde von Toll et al. [84] und Remmers et al. [71] weisen unter Zuhilfenahme einer In-vitro-Technik auf einen auf partial-agonistische Effekte am Kappa-Rezeptor hin. Der Assay beruht darauf, dass nach agonistischer Stimulation zur Koppelung der G-Proteine Guanosintriphosphat (GTP) an den Rezeptorkomplex gebunden wird. Reine Kappa-Agonisten wie Cyclazocin führen hier zu einer praktisch quantitativen Inkorporierung von GTP, während Nalmefen knapp 20% dieses Effekts erreichte, ein Hinweis auf eine partial-antagonistische Kappa-Komponente im Wirkungsspektrum von Nalmefen. Diese Befunde wurden später von Bart et al. [6] bestätigt. Unter Verwendung der gleichen Methode der Stimulation der GTP-Bindung konnten sie an mit humanen rekombinanten My- oder Kappa-Rezeptoren transfizierten CHO(Chinese hamster ovary)-Zellen zeigen, dass Nalmefen an den My-Rezeptor spezifisch exprimierenden CHO-Zellen keine Stimulation der GTP-Bindung auslöste, allerdings die durch einen My-Agonisten ausgelöste Stimulation konzentrationsabhängig vollständig inhibieren konnte (Abb. 6). Im Gegensatz dazu führte Nalmefen an stabil den Kappa-Rezeptor exprimierenden Zellen zu einer signifikanten, wenn auch nur mäßigen Stimulation der GTP-Bindung. Wie von einem Partialagonisten zu erwarten, konnten auch hier mit Nalmefen die Stimulation durch einen reinen Kappa-Agonisten nicht vollständig antagonisiert werden, sondern nur bis zu einer Reststimulation von etwa 40%, was sich relativ gut mit der Stimulation von Nalmefen allein deckt. Diese jüngeren Befunde zeigen zusammen mit den Daten von Remmers et al. [71] überzeugend eine partial-agonistische Komponente von Nalmefen am Kappa-Rezeptor mit einer intrinsischen Aktivität im Bereich zwischen 30% und 40%. Bart et al. [6] konnten darüber hinaus an gesunden Freiwilligen zeigen, dass Nalmefen zu einer Serum-Prolactin-Zunahme führte, ein Effekt, der über partial-agonistische Eigenschaften am Kappa-Rezeptor erklärt wurde. Dies wird von Walker and Koop [86] als Bestätigung der durch Alkohol ausgelösten Aktivitätssteigerung des Kappa-Opioid-Systems diskutiert. Einschränkend muss darauf hingewiesen werden, dass die Datenlage über Effekte von Kappa-Liganden auf Plasmaprolactin nicht einheitlich ist, wie bei Bart et al. [6] ausführlich diskutiert.
Befunde, dass Nalmefen bei übergewichtigen Mäusen zu einer vermehrten Aufnahme von hochkalorischer Nahrung führte, bei Reduktion der gesamten Nahrungsaufnahme, wurden auch über eine gewisse Kappa-Aktivität erklärt [6, 11]. Dieser Befund deckt sich mit älteren Untersuchungen am Menschen, wo Nalmefen bei freier Wahl primär die Aufnahme bevorzugter Nahrung erhöhte, nicht aber von Nahrung, die eher weniger Genusskomponenten hat. Naltrexon zeigte diesen selektiven Effekt eher nicht [94–96].
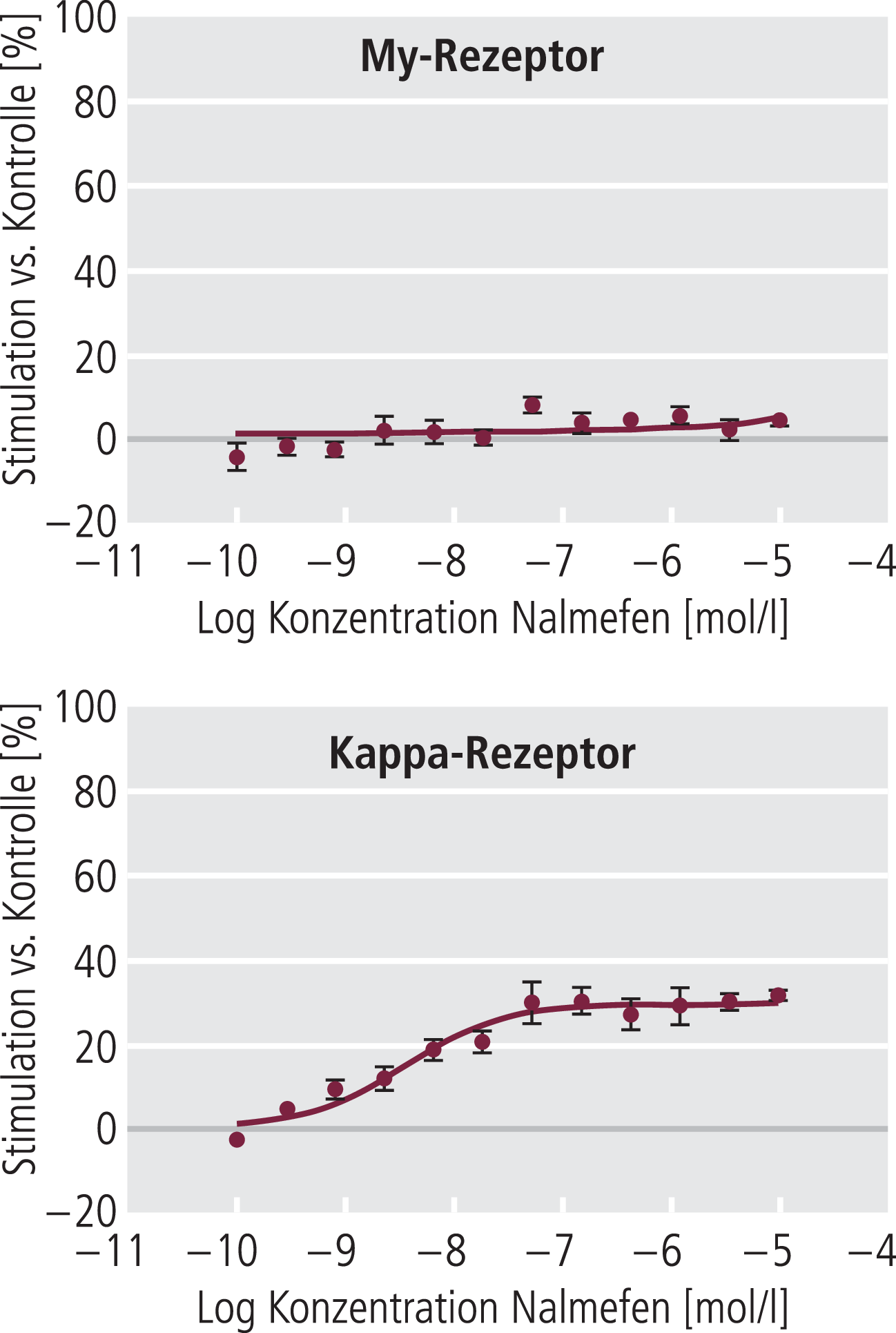
Abb. 6. An CHO-Zellen, die den My-Rezeptor exprimieren, zeigt Nalmefen keine Zunahme der GTP-Bindung, während an Kappa-Rezeptoren exprimierenden CHO-Zellen die GTP-Bindung um etwa 20% zunimmt. 100% Stimulation der GTP-Bindung entspricht dem jeweiligen Effekt eines vollen Agonisten. [mod. nach 6]
Zusammengefasst weisen die Daten darauf hin, dass sich Nalmefen im Gegensatz zu einem reinen My-Opioidrezeptor-Antagonismus am Kappa-Rezeptor als Partialagonist verhält. Dieser Befund hat in letzter Zeit an Bedeutung gewonnen, da man realisiert hat, dass physiologische Kappa-Rezeptoragonisten wie Dynorphin auf komplexe Weise in Abhängigkeitsentwicklung im Allgemeinen und Alkoholabhängigkeit im Speziellen eingreifen können, sodass die Applikation eines Partialagonisten im Gegensatz zu einem reinen Agonisten wünschenswert sein könnte.
Bedeutung des Kappa-Opioidrezeptors für die Alkoholabhängigkeit
Das relativ einfache Modell einer Interaktion opioiderger mit GABAergen und dopaminergen Mechanismen im Belohnungssystem (Abb. 1) wurde in den letzen Jahren dahingehend ergänzt, dass offensichtlich auch Kappa-Rezeptoren und der physiologische Agonist Dynorphin an der Regulation des Belohnungssystems beteiligt sind und ein mögliches Target bei der Behandlung von Substanzabhängigkeiten darstellen [30].
Unter nicht abhängigen Bedingungen stimuliert Ethanol präferenziell die Freisetzung von Beta-Endorphin (Abb. 7), das über My-Rezeptoren stimmungsverbessernde Effekte auslöst und damit die Wiederaufnahme von Alkohol verstärkt.
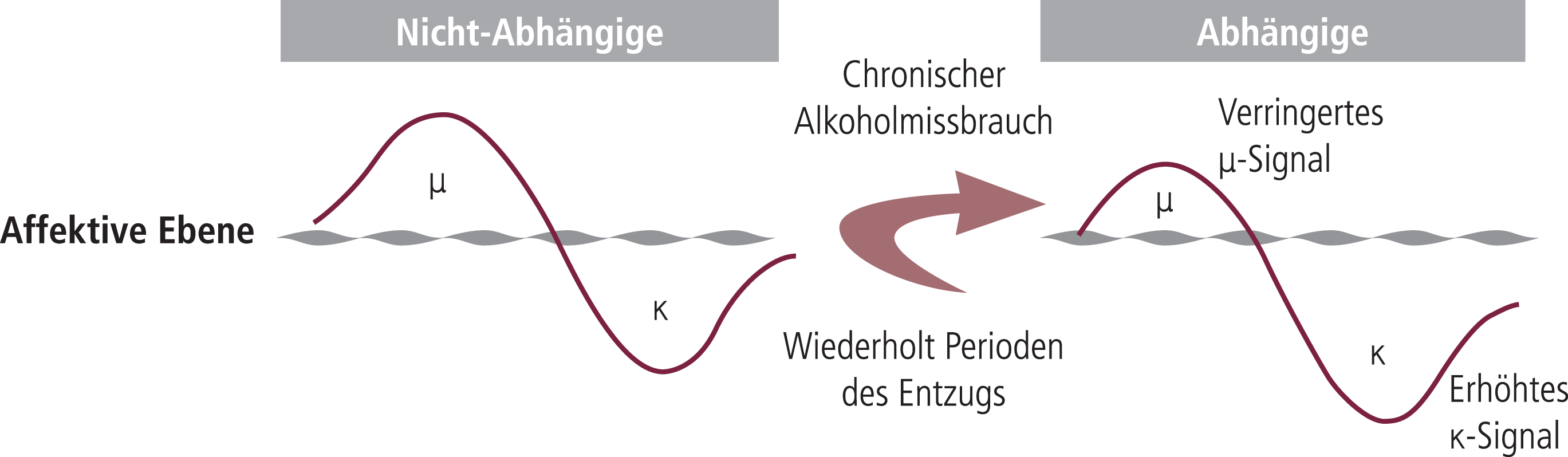
Abb. 7. Bei Nicht-Abhängigen führt Alkohol zu einer Verstärkung von My-Rezeptor-abhängigen Mechanismen (Euphorie) und einer Reduktion von Kappa-abhängigen Mechanismen und der damit verbundenen Dysphorie. Bei Abhängigen ist der My-Effekt abgeschwächt und der Kappa-Effekt verstärkt mit der Gefahr eines kompensatorischen Alkoholkonsums. [mod. nach 87]
Darüber hinaus wird aber auch Dynorphin freigesetzt, das über Kappa-Rezeptoren eher einen dysphorischen, anhedonen und Alkoholgenuss reduzierenden Effekt auslöst (Abb. 7). In abhängigen Personen ist das My-Rezeptorsystem eher herunterreguliert bzw. desensitiviert, während das Kappa-Opioidsystem aktiviert ist. Dynorphin und Kappa-Rezeptor haben damit eine duale Funktion; zum einen kann über diesen Mechanismus eine Dysphorie-auslösende, eher Alkoholaufnahme unterdrückende Wirkung ausgelöst werden, zum anderen scheinen auch Kappa-Mechanismen in die verstärkende Belohnungs-intensivierende Wirkung eingebunden zu sein. Therapeutisches Ziel sollte daher sein, dysphorische, aversive Effekte der Kappa-Opioidaktivierung nach Alkoholeinnahme nicht zu blockieren, sondern nur die auch vorhandene Verstärkung des Belohnungssystems. Darüber hinaus kann eine zu starke aversive Komponente der Kappa-Opioidaktivierung nach Ethanol zu einer kompensatorischen vermehrten Alkoholaufnahme führen (Abb. 8).

Abb. 8. Die duale Rolle von Dynorphin als endogenen Liganden von Kappa-Rezeptoren als negativen Wiederaufnahmeverstärker (Reinforcer), dessen reduzierender Effekt auf die Alkoholaufnahme allerdings auch zur kompensatorischen Mehraufnahme führen kann [mod. nach 9]
Das heißt, die dysphorische Komponente nach Alkoholeinnahme sollte teilweise, die Belohnung-verstärkende Wirkung des Kappa-Opioidsystems ganz durch einen optimalen Opioid-Antagonisten zur Behandlung blockiert werden. Damit erscheint für Substanzen, die auch am Kappa-Rezeptor angreifen, eine korrekte Balance zwischen agonistischen und antagonistischen Effekten sinnvoll [89].
Kappa-Liganden scheinen zudem ausgeprägte Effekte auf affektive Symptome zu haben, mit antidepressiven und anxiolytischen Eigenschaften für Kappa-Antagonisten, antimanischen Eigenschaften für Agonisten und Stimmungs-stabilisierenden Eigenschaften für partielle Antagonisten [10, 42]. Ob hier Nalmefen einen relevanten Effekt auf affektive Symptome im Rahmen der Behandlung der Alkoholabhängigkeit hat, ist nicht bekannt.
Experimentelle Befunde über den Effekt von Nalmefen: Unterschiede zu Naltrexon
Aufgrund seiner langen Halbwertszeit hat man Nalmefen von Anfang an als geeignet zur Therapie der Alkoholabhängigkeit angesehen. Erste Untersuchungen über den Effekt von Nalmefen auf die Alkoholaufnahme von an Alkohol gewöhnten Ratten wurde von Hubbell et al. [37] publiziert. Ratten wurde eine mit Saccharin gesüßte Alkohol-Wasser-Verdünnung angeboten, die sie nach einer gewissen Zeit mit einer konstanten Trinkmenge pro Tag aufnahmen. Wurden diese Ratten mit Nalmefen behandelt, sah man eine Dosis-abhängige Abnahme der Alkoholaufnahme. Nalmefen wurde in diesen Experimenten subkutan verabreicht, was die sehr niedrigen wirksamen Dosen erklärt. Maximale Effekte wurden bei 1 mg/kg Nalmefen gesehen. Die Wirkung von Nalmefen zeigte sich schon am ersten Tag und blieb im Vergleich zu Plazebo, das keinen Effekt hatte, über den gesamten Versuchsverlauf von einer Woche konstant mit einer mittleren Reduktion der Alkoholaufnahme von etwa 50%. Mit Blick auf zwei andere pharmakologische Interventionsstrategien (Fluoxetin und Pimozid) war in dieser Versuchsanordnung Nalmefen die wirksamste Substanz im Hinblick auf die Reduktion der Alkoholaufnahme [32].
In der Untersuchung von June et al. [38] wurden Ratten daran gewöhnt, entweder eine Alkohol/Wasser-Verdünnung oder eine Saccharin/Wasser-Lösung (hedonische Kontrolle) zu präferieren.
Nalmefen in subkutaner Applikation reduzierte die Alkoholaufnahme schon in einem Konzentrationsbereich von 0,01 bis 0,1 mg/kg, noch bevor die Präferenz für Saccharin beeinflusst wurde (Abb. 9). Erst in Dosen von 0,25 mg/kg und höher, als der Effekt auf die Alkoholpräferenz schon stabil war, wurde auch eine signifikante Reduktion der Saccharin-Präferenz gesehen. Die spezifische Reduktion der Alkohol-Präferenz bei niedrigeren Dosierungen konnte für Naltrexon, das erst bei einem Bereich von 0,1 mg/kg parallel die Alkohol- und Saccharin-Aufnahme reduzierte, dagegen nicht beobachtet werden. Wegen der unterschiedlichen Bioverfügbarkeit (40% Nalmefen vs. 5% Naltrexon) sind die Dosen für beide Substanzen bei subkutaner Applikation an der Ratte eher vergleichbar als bei oraler Gabe. Wichtig ist an diesen Befunden, dass für die Reduktion der Alkohol-Präferenz ein Niedrigdosiseffekt für Nalmefen beobachtet wurde, ohne Wirksamkeit auf die Saccharin-Präferenz, während dies bei Naltrexon nicht der Fall war.
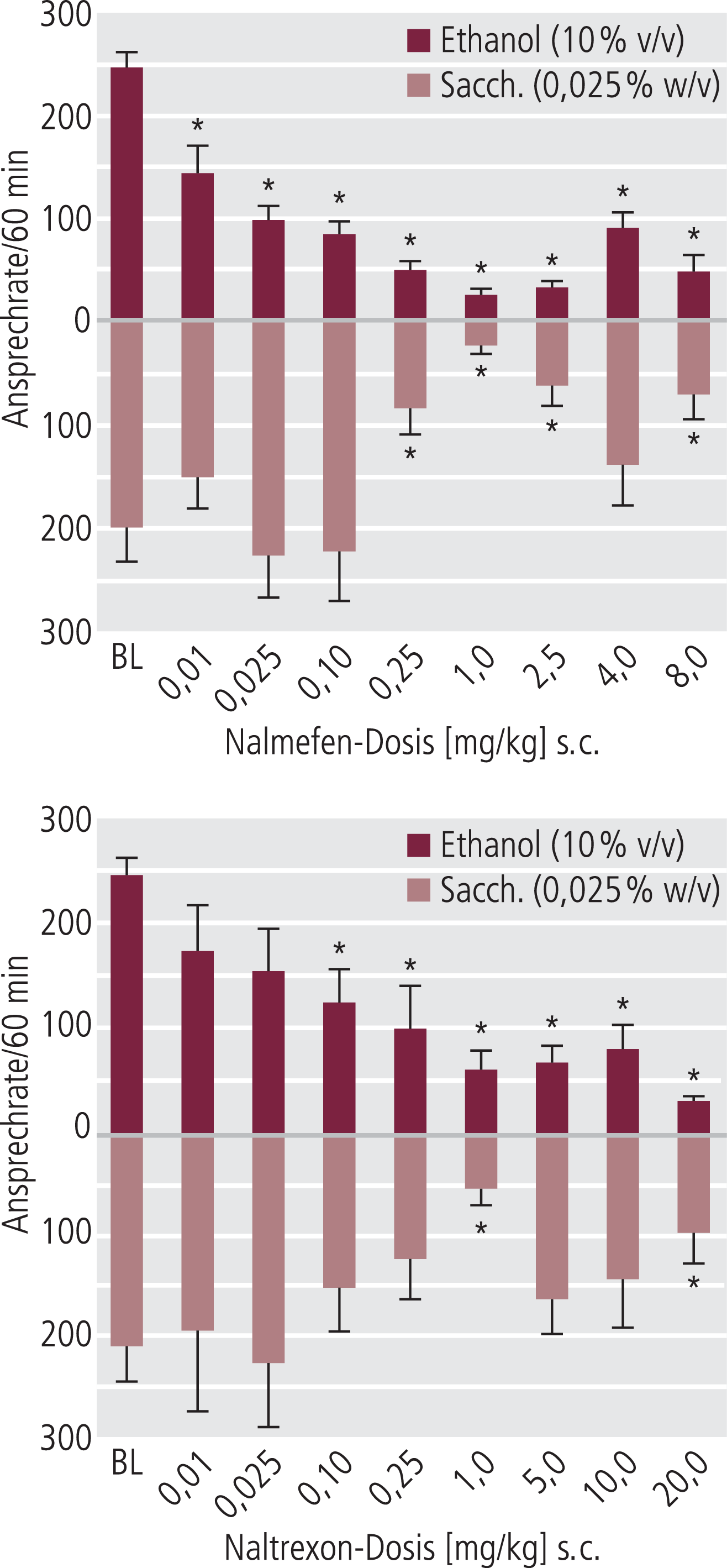
Abb. 9. Ratten wurden für Alkohol- bzw. Saccharin-Präferenz (als hedonische Kontrolle) trainiert. Nalmefen reduziert die Alkoholpräferenz schon bei niedrigen Dosen, während ein Effekt auf die Saccharin-Präferenz erst bei höheren Dosen gesehen wird. Naltrexon reduziert die freiwillige Aufnahme für beide Präferenzen eher gleich stark, aber erst bei höheren Dosen. [mod. nach 38] * signifikanter Unterschied zwischen Alkohol- und Saccharin-Präferenz (p<0,05)
Einen etwas anderen Ansatz verfolgten Walker and Koob [86]. Ratten wurden hier durch konstante Applikation von Alkohol in der Atemluft abhängig gemacht und wurden dann in einem Akutversuch im Hinblick auf Alkohol-Selbstadministration nach Nalmefen-Gabe untersucht, und zwar die alkoholabhängigen versus die nicht alkoholabhängigen Tiere (Abb. 10).
Nalmefen hatte bereits in niedrigen Dosen einen sehr deutlichen Effekt auf die Alkohol-Selbstapplikation der alkoholabhängigen Tiere (Abb. 10). Eine leichte Reduktion der Alkoholaufnahme in den nicht abhängigen Tieren wurde erst bei höheren Dosen gesehen. Naltrexon reduzierte in sehr hohen Dosen die Alkoholaufnahme der nicht abhängigen Tiere, um dann auch zu einer verminderten Selbstaufnahme bei den alkoholabhängigen Tieren zu führen (Abb. 10). Vergleicht man die Wirksamkeit beider Substanzen bei 0,1 mg/kg s. c., sieht man für Nalmefen einen deutlich stärkeren Effekt bei den Alkohol-gewöhnten Tieren, während beide Substanzen sich bei den Kontrolltieren nicht unterschieden (Abb. 11).
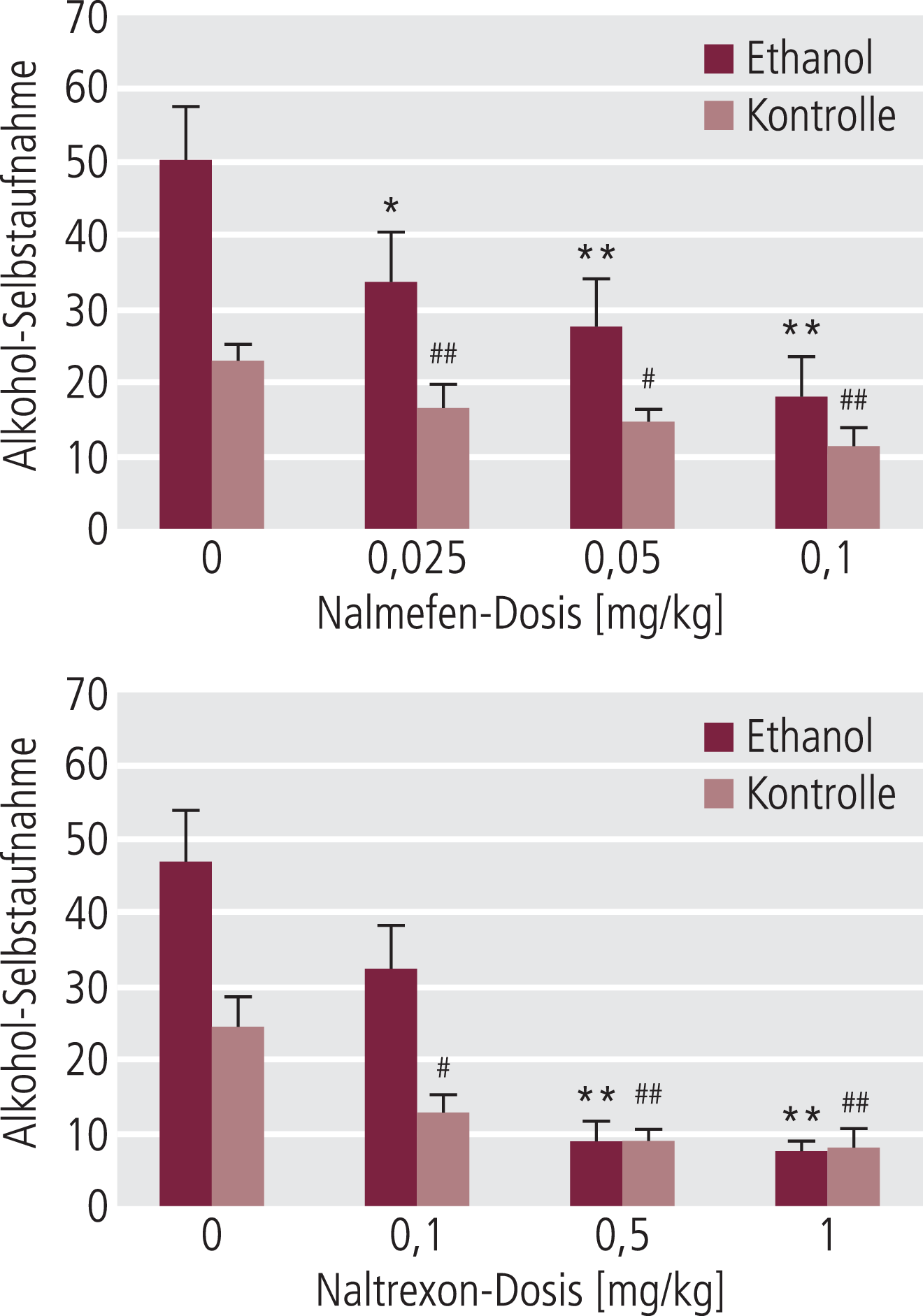
Abb. 10. Nach subkutaner Applikation (hier sind die Dosen vergleichbar) hat Nalmefen einen deutlich stärkeren Effekt auf die Alkohol-Selbstaufnahme bei abhängigen Ratten (subchronische Applikation über Atemluft) als bei Kontrolltieren bei akutem Entzug. Naltrexon ist generell etwas weniger potent und zeigt diese Selektivität eher nicht. [mod. nach 86] *signifikant im Vergleich zu den abhängigen Tieren ohne Nalmefen bzw. Naltrexon (*p<0,05; **p<0,01) #signifikant im Vergleich zu den Kontrolltieren (nicht abhängig) ohne Nalmefen bzw. Naltrexon (#p<0,05; ##p<0,01)
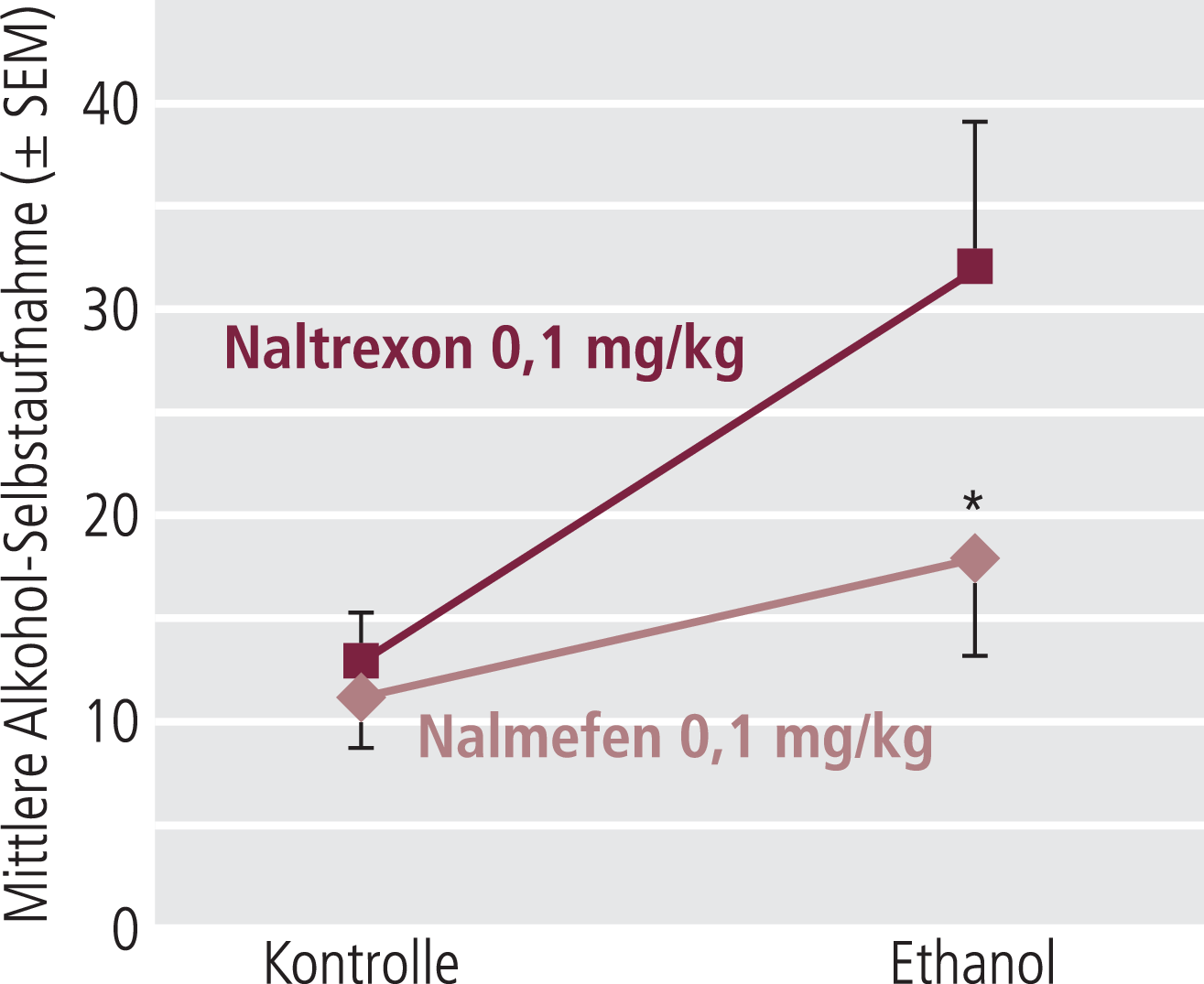
Abb. 11. Bei 0,1 mg/kg s.c. ist der Effekt von Nalmefen und Naltrexon auf die Alkohol-Selbstaufnahme der Kontrollratten (nicht abhängig) gleich, während bei den Ethanol-exponierten Tieren (abhängig) Nalmefen die Alkohol-Selbstaufnahme deutlich stärker reduziert. [mod. nach 86] * signifikanter Unterschied zwischen Nalmefen und Naltrexon (p<0,05)
Die Autoren vermuten, dass die gewisse Selektivität von Nalmefen für die Reduktion der Alkoholaufnahme bei den alkoholabhängigen Tieren über die relativ starke Kappa-antagonistische Eigenschaft vermittelt sein könnte. Zur Klärung wurden alkoholabhängige Tiere mit dem Kappa-Antagonisten Norbinaltorphimin (nor-BNI) untersucht.
Tatsächlich zeigte der Kappa-Antagonist ein ähnliches Wirkungsbild wie Nalmefen, nämlich eine deutliche Reduktion der Alkohol-Selbstapplikation bei den abhängigen Tieren (Abb. 12). Der Kappa-Antagonist hatte hier keinen Effekt bei den nicht abhängigen Tieren im Hinblick auf die Alkohol-Selbstapplikation, sodass die Reduktion dieses Parameters durch Nalmefen und Naltrexon bei höheren Dosierungen möglicherweise nicht über Kappa-Rezeptoren, sondern vermutlich über einen anderen Mechanismus, beispielsweise den My-Rezeptor, vermittelt wird [86].
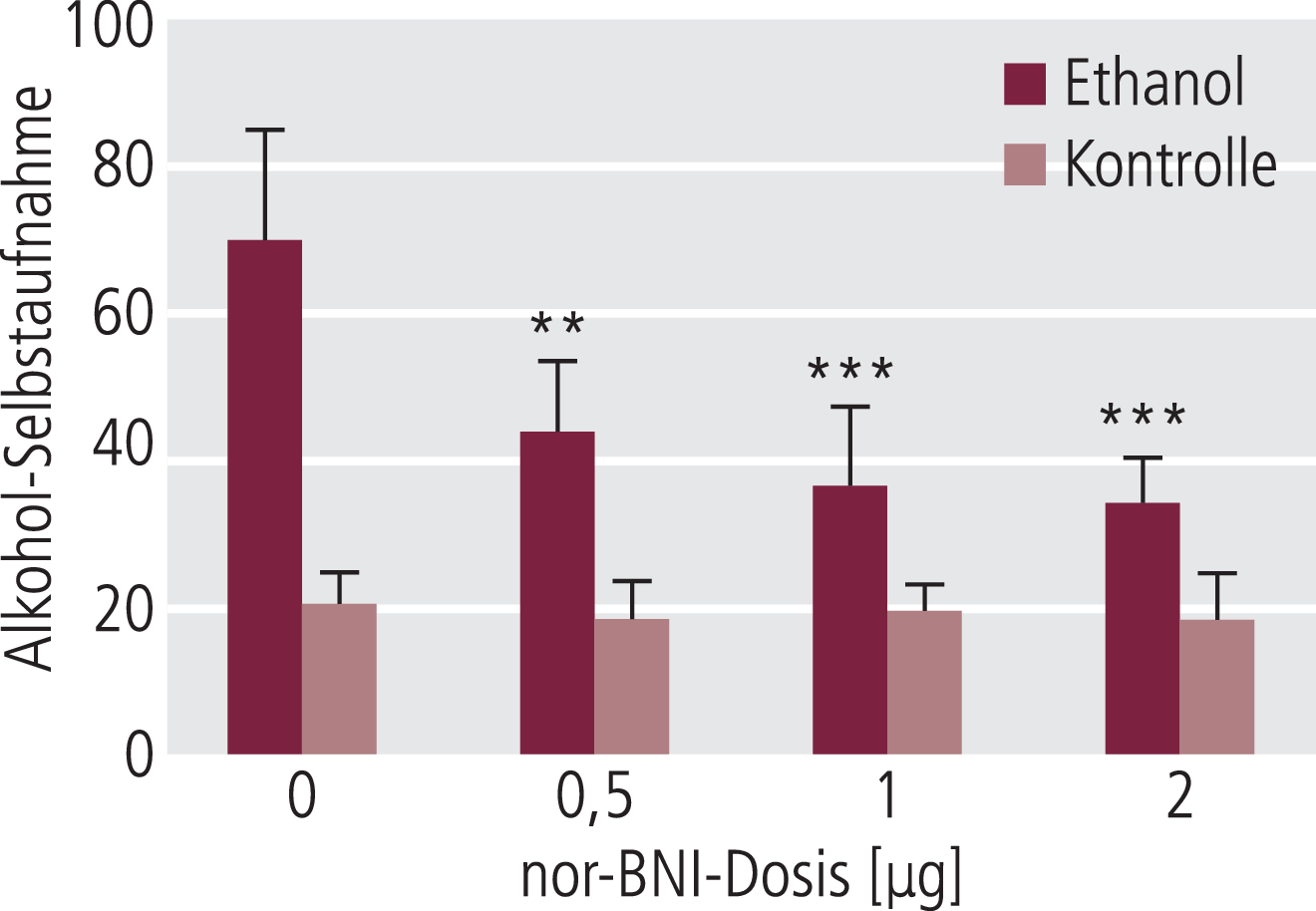
Abb. 12. Der Kappa-Antagonist nor-BNI (nor-Binaltorphimin) reduziert nur bei abhängigen, nicht aber bei nicht-abhängigen Ratten die Alkohol-Selbstaufnahme nach Entzug. Methode wie Abb. 10. [mod. nach 86] * signifikanter Effekt im Vergleich zu den abhängigen Kontrolltieren (*p<0,05; **p<0,01; ***p<0,001)
Klinisch-pharmakologische Untersuchungen am Menschen
In klinischen Laboruntersuchungen wurden von Drobes et al. [19, 20] an jeweils 100 alkoholabhängigen und 100 nicht alkoholabhängigen Alkoholtrinkern (Gelegenheitstrinkern) der Effekt von Naltrexon (50 mg/Tag) bzw. Nalmefen (40 mg/Tag) auf das Trinkverhalten beobachtet. Die Probanden wurden sieben Tage lang mit den Substanzen behandelt. Für die ersten fünf Tage gab es keine Vorgaben hinsichtlich Trinkverhalten, aber sie wurden angewiesen, am Tag 6 und 7 nicht zu trinken. In der ersten der beiden Publikationen [19] konnten die Autoren zeigen, dass beide Substanzen bei den gelegentlichen Alkoholtrinkern in der dem eigentlichen Versuch vorangehenden mehrtägigen Behandlungsphase einen geringen Effekt auf die Alkoholaufnahme zeigten, während bei den Alkoholikern die Anzahl der in dieser Zeit getrunkenen Alkoholmenge signifikant reduziert war. Naltrexon und Nalmefen erzielten dabei vergleichbare Ergebnisse. Auf der anderen Seite wurde untersucht, wie viele der Probanden in der angewiesenen alkoholfreien Zeit (Tage 6 und 7) doch Alkohol zu sich nahmen. Hier hatten beide Substanzen auch einen Effekt bei den gelegentlichen Trinkern, reduzierten aber die Regelverstöße signifikant bei den Alkoholikern bei einem leichten numerischen Vorteil von Nalmefen.
Beide Substanzen reduzierten sowohl bei den Alkoholikern als auch bei den gelegentlichen Alkoholtrinkern das Gefühl von Craving nach Alkoholeinnahme, wobei beide Gruppen deutliche Unterschiede zeigten (Abb. 13). Hier war Nalmefen nicht nur numerisch, sondern auch statistisch leicht besser wirksam als Plazebo (Abb. 13).
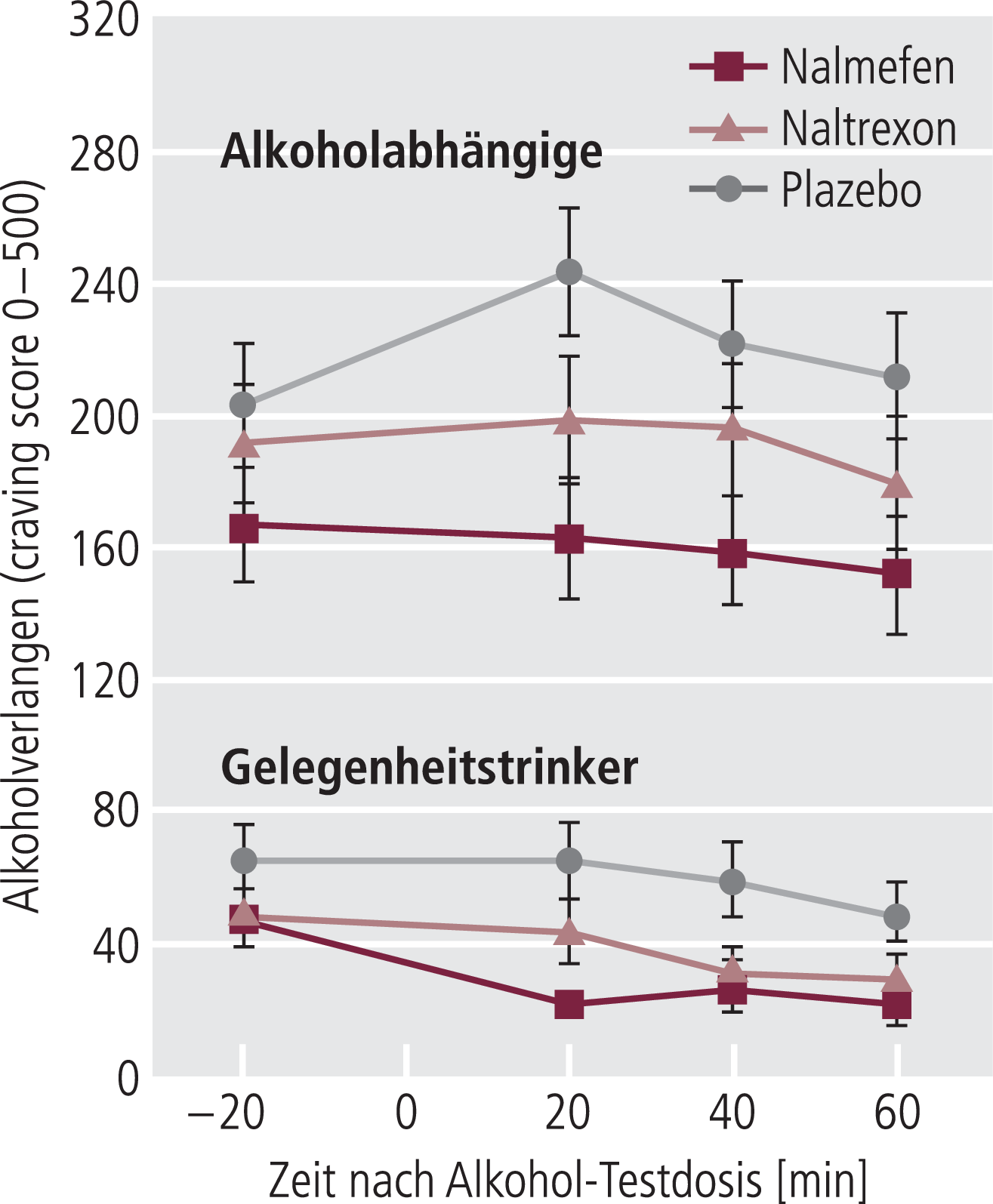
Abb. 13. Das Gefühl von „craving“ nach einer Alkohol-Testdosis am Tag 8 der Untersuchung war höher in der Plazebo-Gruppe als in den Gruppen mit Nalmefen oder Naltrexon-Therapie (7 Tage 50 mg Naltrexon bzw. 40 mg Nalmefen täglich). Die Unterschiede von Naltrexon und Nalmefen gegen Plazebo waren unterschiedlich ausgeprägt, wobei Naltrexon nur 20 min nach Einnahme von Alkohol, Nalmefen sich dagegen 20, 40 und 60 min nach Alkoholeinnahme signifikant von Plazebo (p<0,05) unterschieden. Bei Gelegenheitstrinkern hatten beide keinen Effekt. [mod. nach 19, 20]
Diese Daten kann man dahingehend interpretieren, dass durch den My-Antagonismus bei Nalmefen und Naltrexon die Aufrechterhaltung einer Alkoholabhängigkeit reduziert wird, ein Effekt, zu dem auch der Kappa-Antagonismus beiträgt. Hat die Substanz aber noch eine gewisse agonistische Aktivität am Kappa-Rezeptor, könnte auf dem Boden einer gewissen Dysphorie, ausgelöst durch die partial-agonistischen Effekte, die Entwicklung eines Belohnungsverhaltens reduziert werden. Daher wird heute diskutiert, dass ein partieller Kappa-Rezeptor-Antagonismus im Zusammenhang mit My- und gegebenenfalls Delta-Antagonismus positive Zusatzeffekte im Hinblick auf die Behandlung der Alkoholabhängigkeit haben könnte.
Zusammenfassung
Von den drei zur Therapie der Alkoholabhängigkeit zur Verfügung stehenden Substanzen bzw. Substanzklassen hat Disulfiram eine Sonderstellung, da es nicht in die Neurobiologie der Alkoholerkrankung, sondern in den Metabolismus des Alkohols auf der Stufe des Acetaldehyd eingreift. Die Kumulation von Acetaldehyd führt zur unangenehmen Alkohol-Disulfiram-Reaktion, die den Patienten vom Alkoholgenuss abhalten soll. Die Substanz hat heute nur noch einen begrenzten Stellenwert. Pharmakologisch interessanter sind Acamprosat und die beiden Opioidantagonisten Naltrexon und Nalmefen, die unseren heutigen Vorstellungen nach relativ spezifisch neuronale Mechanismen im Rahmen des Belohnungssystems und damit innerhalb der Neurobiologie der Alkoholerkrankung beeinflussen. Während Acamprosat eine modulierende Wirkung auf glutamaterge Mechanismen zeigt, wirken die beiden Opioidantagonisten zunächst einmal über eine Blockade von My-Opioidrezeptoren hemmend innerhalb des Belohnungssystems. Darüber hinaus beeinflussen sie auch Kappa-Opioidrezeptoren, wobei Nalmefen insofern eine Sonderstellung einnimmt, da es an den Kappa-Rezeptoren als partieller Agonist wirkt, was den neurobiologischen Vorstellungen nach Vorteile in der Reduktion der Alkoholabhängigkeit haben kann. Wichtig für die praktische Anwendung ist die Tatsache, dass Acamprosat auf der einen und die beiden Opioidantagonisten auf der anderen Seite an unterschiedlichen Stellen der neurobiologischen Mechanismen, die zur Alkoholabhängigkeit führen, eingreifen. Es hat daher pharmakologisch Sinn, sie zu kombinieren, was auch den aktuellen klinischen Daten nach mit einem zusätzlichen therapeutischen Gewinn verbunden zu sein scheint [39].
Interessenkonflikterklärung
WEM hat Beraterhonorare und Honorare für Vorträge, Stellungnahmen oder Artikel von Lundbeck, Schwabe und UCB sowie Forschungsbeihilfe von Schwabe und UCB erhalten.
Literatur
Das Literaturverzeichnis finden Sie im Internet (www.ppt-online.de) unter „Archiv“ → „Literatur“ als PDF-Datei sowie bei der HTML-Version dieses Beitrags.
Prof. Dr. Walter E. Müller, Pharmakologisches Institut für Naturwissenschaftler, Biozentrum der Goethe-Universität Frankfurt, Campus Riedberg, Max-von-Laue-Straße 9, 60438 Frankfurt/M, E-Mail: pharmacolnat@em.uni-frankfurt.de
Pharmacology and pharmacokinetics of the drugs available in Germany to treat alcohol dependence
For the treatment of alcohol dependence three strategies are available in Germany. Beside the aversive drug disulfiram, acamprosate and the opioid antagonists naltrexone and nalmefene can be used. Disulfiram has limited use due to the bad compliance and because it is not liked by patients. Nevertheless, it still seems to have a place mainly for highly motivated patients. In contrast to the aversive therapy by disulfiram, acamprosate reduces the craving for alcohol. Abstinence is better obtained under acamprosate therapy than by therapy without the drug. The third option to treat alcohol dependence are the opioid antagonists naltrexone and nalmefene whereof only nalmefene is licensed to reduce drinking.
Key words: Alcohol dependence, drug treatment,
disulfiram, acamprosate, naltrexone, nalmefene
Psychopharmakotherapie 2013; 20(05)