Rainer Spanagel und Falk Kiefer, Mannheim
Die Alkoholabhängigkeit geht mit einer komplexen Störung der Gehirnfunktion einher. Diese beginnt mit der exzessiven chronischen Einnahme von Alkohol (Ethanol), dessen pharmakologischer Effekt zu tiefgreifenden Veränderungen in Kognition und Verhalten führt. Es entsteht eine Präferenz für Stimuli und Verhaltensweisen, die mit Alkohol assoziiert sind. Im Verlauf werden Verhaltensmuster, die in Bezug zur Alkoholeinnahme stehen, zwanghaft-repetitiv eingeengt. Die Fähigkeit, auf Alkohol zu verzichten, geht trotz zumeist schwerwiegender negativer Konsequenzen verloren. Welcher pathophysiologische Mechanismus liegt dem zugrunde? Um an diese Thematik heranzuführen, werden im Folgenden zuerst die spezifischen molekularen Effekte von Alkohol im zentralen Nervensystem (ZNS) betrachtet (primäre Angriffsstellen des Alkohols), um dann auf die für die Suchtentstehung maßgeblichen neurochemischen Funktionssysteme und Signalwege (sekundäre Veränderungen) einzugehen. Hierbei werden insbesondere molekulare und physiologische Veränderungen im mesokortikolimbischen System diskutiert, da hier die Datenlage besonders fundiert ist, wohl wissend, dass auch andere Gehirnsysteme bei der Ausprägung von Sucht eine Rolle spielen [30].
Die primären Angriffsstellen des Alkohols
Welche molekularen Mechanismen sind primär dafür verantwortlich, dass Ethanol auf das ZNS einwirkt? Erst in letzter Zeit hat sich die sogenannte Proteintheorie, die besagt, dass Ethanol in erster Linie Membranproteine angreift, gegen die Lipidtheorie durchgesetzt [34].
Lipidtheorie
Bis in die 90er-Jahre postulierte die Lipidtheorie, dass die Wirkung von Ethanol dadurch zustande kommt, dass er eine Störung bei den Membranlipiden der Nervenzellen des ZNS verursacht. Insbesondere soll dabei Ethanol direkt die Membranfluidität stören und dadurch auch die Ordnung des Lipidzustands von Membranen. Durch diese primäre Wirkung von Ethanol auf Membranlipide könnte auch eine unspezifische Wirkung auf eingelagerte Membranproteine wie Ionenkanäle erklärt werden. Diese Hypothese hat jedoch wesentliche Einschränkungen: so ist es bisher nur möglich, die Entstehung von Membranstörungen durch Ethanol bei Konzentrationen nachzuweisen, die deutlich über 500 mg/dl (4,06‰) Blutalkohol und somit nahe an der lethalen Dosis von Ethanol beim Menschen liegen. Diese Tatsache und viele weitere Befunde führten zu der Schlussfolgerung, dass die beobachtete Wirkung von Alkohol auf Membranfluidität und -organisation als biophysikalisches Phänomen zwar nachweisbar ist, aber für die pharmakologische Wirkung von Alkohol auf das ZNS im psychopathologisch relevanten Dosisbereich unterhalb einer komaassoziierten Konzentration vermutlich nicht von Bedeutung ist.
Proteintheorie
Im Gegensatz dazu sagt die Proteintheorie voraus, dass Ethanol auf spezifische Membranproteine (Rezeptoren und Ionenkanäle) einwirkt – dies sind demnach die primären Angriffsstellen für Ethanolmoleküle [44]. Bereits bei sehr niedrigen Blutalkohol-Konzentrationen von<0,1‰ kann direkt die Funktion verschiedener Ionenkanäle und Rezeptoren gestört werden. In einer Schlüsselpublikation haben David Lovinger und Mitarbeiter [25] gezeigt, dass die Funktion von N-Methyl-D-aspartat-(NMDA-)Glutamatrezeptoren bereits von einer sehr geringen Ethanolkonzentration gehemmt wird. Überdies besteht zwischen der Fähigkeit verschiedener Alkohole zur Inhibierung des durch den NMDA-Rezeptor aktivierten Calciumstroms und der berauschenden Wirkung ein linearer Zusammenhang. Dieser Befund legt nahe, dass die durch Ethanol hervorgerufene Hemmung des NMDA-Rezeptors zu den neuronalen und kognitiven Veränderungen beitragen kann, die mit einer Alkoholintoxikation assoziiert sind. Aber wie ist es möglich, dass Ethanol die Funktion des NMDA-Rezeptors direkt beeinflusst?
Ethanol-Bindungsstellen am NMDA-Rezeptor
Der NMDA-Rezeptor ist ein Liganden-gesteuerter Ionenkanal, der sich aus NR1-, NR2- (A bis D) und NR3-Untereinheiten zusammensetzt und der weitverbreitetste exzitatorische Rezeptor im Säugerhirn ist. Die NR1-Untereinheit ist entscheidend für die Kanalfunktion, die NR2-Untereinheiten enthalten die Liganden-Bindungsstelle für Glutamat, und die NR3-Untereinheiten modulieren unter pathologischen Bedingungen die Kanalaktivität. Elektrophysiologische Untersuchungen haben gezeigt, dass Ethanol mit Transmembran-Domänen (dem in der Membran verankerten Bereich des Rezeptorproteins) interagiert, welche die Kanalfunktion beeinflussen. Bei der Suche nach einer möglichen Bindungsstelle für Ethanol am NMDA-Rezeptor wurden gezielte Mutagenese-Untersuchungen durchgeführt, die zu einem Aminosäurenaustausch in den Transmembran-Domänen der NR1-Untereinheit führten. Mit dieser Vorgehensweise konnten potenzielle Bindungsstellen für Ethanolmoleküle identifiziert werden. Zum Beispiel führte ein Aminosäurenaustausch von Phenylalanin durch Alanin in einer Transmembran-Domäne der NR1-Untereinheit zu einer stark verminderten Empfindlichkeit gegenüber Ethanol [38]; NMDA-Rezeptoren, die durch diese gezielte Mutagenese verändert wurden, konnten nur noch durch extrem hohe Alkoholkonzentrationen gehemmt werden.
Ethanol-Bindungsstellen am GABAA-Rezeptor
Neben dem NMDA-Rezeptor zeigen noch weitere im ZNS vorkommende Rezeptoren bzw. Ionenkanäle potenzielle Bindungsstellen für Alkohol. Insbesondere wird die Funktion von GABAA-Rezeptoren durch Ethanol verstärkt. Der GABAA-Rezeptor ist der bedeutendste inhibierende Neurotransmitter-Rezeptor im Säugerhirn, er ist ebenfalls ein Liganden-gesteuerter Ionenkanal, der sich wiederum auch aus verschiedenen Untereinheiten zusammensetzt. Mit derselben methodischen Vorgehensweise – Aminosäurenaustausch mithilfe gezielter Mutagenese – konnte eine Region in den Transmembran-Domänen der Alpha- und Beta-Untereinheit des GABAA-Rezeptors gefunden werden, die an der Wirkung von Ethanol beteiligt ist [28].
Weitere Rezeptoren für Ethanol
Neben NMDA- und GABAA-Rezeptoren konnten noch weitere Rezeptoren und Ionenkanäle mit potenziellen Bindungsstellen für Ethanol identifiziert werden [44]. Man spricht von potenziellen Bindungsstellen, da es zurzeit noch keine biophysikalischen Methoden gibt, mit denen man direkt die Bindung von Ethanolmolekülen an diesen Rezeptoren oder Ionenkanälen messen kann, weil das Ethanolmolekül zu klein ist und eine sehr geringe Bindungsenergie aufweist. Diese Eigenschaften schließen mit den zurzeit verfügbaren Methoden eine direkte Bestimmung einer Ethanol bindenden Proteinregion aus. Andererseits hat die Entdeckung des LUSH-Proteins in der Fruchtfliege Drosophila melanogaster ermöglicht, ein Modell zu generieren, das aufzeigt, wie bestimmte Transmembran-Domänen eine spezifisch Ethanol bindende „Proteintasche“ bilden können. Die Arbeitsgruppe von David Jones erhielt hochauflösende Kristallstrukturen von einem Komplex aus LUSH mit verschiedenen kurzkettigen Alkoholen [20] und konnte damit zeigen, dass die Struktur von LUSH tatsächlich eine spezifische Bindungsstelle für Alkohol enthält und es nach Bindung von Ethanol zu einer Konformationsänderung kommt; dies ist eine wichtige Voraussetzung für eine funktionale Bindungsstelle. Eine spezifische Gruppe von Aminosäuren bildet dabei ein Netzwerk von Wasserstoffbrücken zwischen Protein- und Ethanol-Molekül und erzeugt damit ein strukturelles Motiv, mit dem die Affinität für die Bindung von Ethanol in dieser Position verstärkt wird. Dieses strukturelle Motiv ist in Säugern bei NMDA- und GABAA-Rezeptoren und anderen primären Angriffsstellen für Ethanolmoleküle konserviert, weshalb der Schluss naheliegt, dass die Ethanolbindungsstelle im LUSH-Protein ein allgemeines Modell für potenzielle Ethanolbindungsstellen bei Proteinen wie NMDA- oder auch GABAA-Rezeptoren darstellt. Ein weiterer interessanter Aspekt zum Schluss: Alkohol ist im Sinnesspektrum von Fruchtfliegen ein wichtiges Geruchssignal. Die Fähigkeit von Fruchtfliegen, Ethanolgeruch zu detektieren, ist wichtig für die Chemotaxis in Richtung auf Nahrungsquellen, beispielsweise vergorene Früchte. LUSH stellt dabei eine Proteinmutante dar, die besonders gut Alkohol detektiert. Fruchtfliegen reagieren auf Alkohol in einem Konzentrationsbereich der dem des Menschen ähnlich ist. So kann es zur Alkoholintoxikation mit einer Sedierung bis hin zum Koma und Tod kommen. Diese verschiedenen Grade der Alkoholintoxikation lassen sich sehr exakt mit einem sogenannten Inebriometer messen und so stellt Drosophila ein brauchbares Tiermodell für das Verständnis von Intoxikations- und Toleranzphänomenen dar [29].
Vergleich mit anderen Drogen
Insgesamt betrachtet zeigen die Daten der letzten zwanzig Jahre, dass Ethanol direkt Membranrezeptoren und Ionenkanäle angreift. Dies untermauert die sogenannte Proteintheorie, und gegenwärtig wird angenommen, dass Ethanol einige primäre Angriffsstellen wie NMDA- und GABAA-Rezeptoren besitzt. Hier findet sich eine Gemeinsamkeit mit anderen Drogen. Alle psychoaktiven Substanzen haben primäre Angriffsstellen im ZNS: Opiate wirken an Opioid-Rezeptoren im ZNS und entfalten so ihre primäre psychotrope Wirkung, Cannabis-Produkte wirken an Cannabinoid-Rezeptoren, Nicotin an nicotinischen Acetylcholin-Rezeptoren, usw. Während diese spezifischen Drogen-Rezeptor-Interaktionen oft bereits im nanomolaren Bereich stattfinden, zeigt sich eine spezifische Ethanol-Rezeptor-Interaktion erst ab dem unteren millimolaren Bereich; zudem verfügt Ethanol über mehrere Rezeptor- und Ionenkanal-Interaktionspartner im ZNS.
Psychotrope Effekte bereits in geringen Konzentrationen
Hier sei noch einmal betont, dass molekulare Interaktionen bereits bei Blutalkoholkonzentrationen von 0,05‰ eintreten und somit schon geringste Mengen an konsumiertem Alkohol auf unser ZNS einwirken. Diese neuen wissenschaftlichen Erkenntnisse sind besonders wichtig für schwangere Frauen und Kinder, da hier schon geringste Mengen an konsumiertem Alkohol langanhaltende, wenn nicht gar persistierende Veränderungen im ZNS bewirken können. Obwohl unser Wissen über die potenziellen Bindungsstellen von Ethanol an Membranproteinen, wie am NMDA-Rezeptor, kontinuierlich wächst, sind die funktionellen Konsequenzen dieser primären Ethanol-Rezeptor-Interaktionen noch nicht völlig geklärt. Fest steht jedoch, dass ab Blutalkoholkonzentrationen von 0,15‰ erste psychotrope Effekte beobachtet werden und hierbei die primären Ethanol-Rezeptor-Interaktionen eine essenzielle Rolle spielen. Dies belegen Diskriminationsuntersuchungen mit Ethanol. Bei einem Diskriminationstest fragt der Experimentator: „Fühlen Sie sich, als wenn Sie Alkohol getrunken hätten?“ In der Tat entspricht der diskriminative Ethanolstimulus weitgehend der subjektiven Wirkung bei Gelegenheitstrinkern und tritt bereits bei Blutalkoholkonzentrationen von 0,15‰ auf [16]. Eine Vielzahl an Diskriminationsuntersuchungen zeigen eine eindeutige Beteiligung von NMDA- und GABAA-Rezeptoren bei der Ausprägung von psychotropen Wirkungen, somit liegt der Schluss nahe, dass primäre Angriffsstellen für Ethanolmoleküle im ZNS subjektive Alkoholwirkungen und Intoxikation bewirken.
Neurochemische Funktionssysteme, die bei der Suchtentstehung involviert sind
Bereits das erste Auftreffen von Ethanol auf die spezifischen Zielstrukturen im Hirn – sprich, die primären Angriffsstellen – führt zu den typischen akuten subjektiven Wirkungen, aus denen die Eigenschaften des diskriminativen Stimulus dieser Droge bestehen.
Zusätzlich zu diesen psychotropen Wirkungen zeigt Alkohol bzw. die Alkoholintoxikation Auswirkungen, die sich mit ansteigender Alkoholkonzentration von Enthemmung über Sedierung bis hin zur Hypnose erstrecken können.
Dem schließt sich eine zweite Welle indirekter Wirkungen auf eine Reihe von Neurotransmitter-/Neuropeptid-Systemen an [46]. Man nimmt an, dass diese zweite Welle, an der in erster Linie Monoamine (Dopamin) und das endogene Opioidsystem beteiligt sind, entscheidend für die verstärkenden und belohnenden Eigenschaften von Alkohol ist. Molekulare Interaktionen von primären Angriffsstellen und den genannten neurochemischen Funktionssystemen können zu lang anhaltenden physiologischen Veränderungen innerhalb des mesokortikolimbischen Systems führen und als Folge süchtiges Verhalten bedingen.
Das mesokortikolimbische dopaminerge Belohnungssystem
Die Befunde von James Olds, der 1954 zerebrale Selbststimulationsexperimente an Ratten durchführte, stellen einen Meilenstein in der Entschlüsselung neurobiologischer Mechanismen der Sucht dar [32]. James Olds ist einer der bedeutendsten Psychologen des 20. Jahrhunderts und hat maßgeblich zu unserem Wissen über neuronale Schaltkreise und deren funktionelle Bedeutung, insbesondere auf der Verhaltensebene, beigetragen. Die Entdeckung des Belohnungssystems stellt dabei einen herausragenden Beitrag dar. Olds implantierte feine Elektroden in verschiedene Hirnregionen von Ratten, die dann die Möglichkeit erhielten, über den Druck eines Hebels die jeweilige Region selbst zu stimulieren. Hierbei stieß er auf eine anatomisch eng umgrenzte Region im Mittelhirn, deren Stimulation mit einer so ausgeprägten Verhaltensverstärkung einherging, dass die Selbststimulierung bis zur körperlichen Erschöpfung unter Vernachlässigung essenzieller Bedürfnisse fortgeführt wurde. Schnell wurde klar, dass hier ein für Verhaltensgenerierung, Lernen und positive Verstärkung (Belohnung) wichtiges Zentrum entdeckt wurde, dessen Fehlsteuerung offensichtlich mit einem Verhalten einhergeht, dass man klar als süchtig, eingeengt, zwanghaft und repetitiv bezeichnen kann. Das Faszinierende für die Suchtforschung war nun, dass der gleiche Effekt, der für die elektrische Stimulation gezeigt werden konnte, nämlich die wiederholte Selbststimulierung, auch bei direkter zerebraler Applikation von Suchtstoffen nachweisbar war. Die Infusion von Cocain, Amphetamin, Heroin, Alkohol und weiteren Stoffen mit Abhängigkeitspotenzial in diese anatomische Region bewirkte eine Fortsetzung der Selbstapplikation bis zur körperlichen Erschöpfung [50].
Das System, das aufgrund dieser Befunde als Belohnungs- oder Verstärkungssystem bezeichnet wurde, wurde in den letzten Jahren mit modernen retrograden Neuromarkierungsmethoden anatomisch sehr gut charakterisiert [21]. Es handelt sich um dopaminerge Bahnen, die ihren Ausgangspunkt im ventralen Tegmentum (VTA, ventral tegmental area) im Mittelhirn nehmen und in den Nucleus accumbens (NAC) als Hauptbestandteil des ventralen Striatums projizieren und eng mit dem präfrontalen Kortex und anderen Zielregionen verknüpft sind. In der Gesamtheit wird es als mesokortikolimbisches System bezeichnet. Je nach Zielort können die dopaminergen Bahnen unterschiedliche Funktionen erfüllen [21], insbesondere geht jedoch eine stimulusabhängige Aktivierung von einem großen Anteil dieser Neuronen mit einer phasischen Dopamin-Ausschüttung im NAC einher, dem offensichtlich zentralen Verschaltungspunkt für die beobachtbaren Verhaltenseffekte. Eine dopaminerge Aktivierung im NAC kann zur positiven Verstärkung des Verhaltens führen, das zeitlich mit der Aktivierung einhergeht. Ein Merkmal von dopaminergen Neuronen ist ihre tonische Aktivität, das heißt die regelmäßige, spontane extrasynaptische Entladung. Mit einer phasischen Entladung hingegen wird die synaptische Freisetzung großer Mengen von Dopamin infolge kurzer Salven (Dopaminburst) von Aktionspotenzialen bezeichnet. Der Dopaminburst ist somit eine Antwort auf eine Reizung dopaminerger Neuronen im VTA, beispielsweise in Reaktion auf Belohnungssignale [12]. Aber welche Informationen werden durch die phasische dopaminerge Transmission kodiert?
Rolle von Dopamin
Galt Dopamin früher noch als hedones Signal bzw. „Glücksbotenstoff“, nimmt man heute an, dass es einerseits ein Lernsignal für unkonditionierte appetitive Reize darstellt und anderseits in Folge von klassischer Konditionierung als ein belohnungsankündigendes und aufmerksamkeitslenkendes Signal auf konditionierte appetitive Reize fungiert [4, 41]. Nach der unerwarteten Verabreichung einer Belohnung (z.B. eine Zuckerlösung bei Ratten = unkonditionierter Stimulus) kommt es somit zu einem phasischen Dopaminsignal. Dabei tritt der Dopaminburst mit einer zeitlichen Verzögerung von wenigen hundert Millisekunden nach der Verabreichung der Belohnung ein. Wird anschließend die Belohnung mit spezifischen Reizen gepaart (z.B. einem Ton), so tritt der Dopaminburst mit zunehmender Konditionierung in zeitlicher Folge mit dem konditionierten Reiz auf, jedoch nicht mehr nach erfolgter Belohnung. Das phasische Dopaminsignal kündigt somit dem Gehirn an, dass es zu einer Belohnung kommen kann. In manchen Individuen können die konditionierten Reize und das damit verbundene phasische Dopaminsignal zu einer zunehmenden Anreizmotivation (incentive salience) führen, sprich ein konditionierter Reiz kann ein nicht kontrollierbares Appetenzverhalten auslösen; solch ein mehr oder weniger ausgeprägter Impulskontrollverlust ist aller Wahrscheinlichkeit nach genetisch moduliert [48].
Dopaminerge Neuronen, die zum NAC projizieren, haben im Zusammenspiel mit den primären Angriffsstellen für Ethanolmoleküle eine essenzielle Bedeutung für die Heranbildung der von Alkohol hervorgerufenen Verstärkungsprozesse. Bei der Vermittlung der hedonistischen Aspekte des Belohnungsgefühls durch Ethanol spielt Dopamin hingegen wahrscheinlich keine Rolle. Wahrscheinlicher ist, dass ein phasisches Dopaminsignal im NAC als Lernsignal für den nicht konditionierten appetitiven Reiz des Alkoholkonsums dient [43]. Es ist dagegen bekannt, dass das endogene Opioidsystem ein neurochemisches Substrat darstellt, das an der Vermittlung positiver Gemütszustände beteiligt ist [22] und somit auch die hedonistischen Aspekte des Belohnungsgefühls durch Ethanol vermitteln könnte.
Die Rolle des endogenen Opioidsystems
Opioid-Rezeptoren, spezifische Bindungsstellen für Morphin und andere Opioide, konnten erstmals 1973 anhand von Bindungsstudien in Extrakten von Säugerhirnen nachgewiesen werden. Erst 20 Jahre später wurde die tatsächliche Existenz von multiplen Opioid-Rezeptoren molekularbiologisch nachgewiesen. Drei Opioid-Rezeptoren sowie einige Subtypen sind heute kloniert und sequenziert: My-, Kappa- und Delta-Opioid-Rezeptoren, wobei Morphin insbesondere an My-Opioid-Rezeptoren bindet. Die Entdeckung der Opioid-Rezeptoren warf natürlich die Frage nach ihrer physiologischen Bedeutung auf, da nicht anzunehmen war, dass der Organismus diese Bindungsstellen ausschließlich für exogene Stoffe bereithält. Tatsächlich gelang dem deutsch-britischen Pharmakologen Hans Walter Kosterlitz 1975 der Durchbruch: Gemeinsam mit John Hughes isolierte er aus über zweitausend Schweinegehirnen eine Substanz, die er „Encephalin“ benannte.
Endogene Opioidpeptide
Der Nachweis der endogenen Opioid-Rezeptor-Liganden mit der Isolierung der Pentapeptide Met- und Leu-Enkephalin [38] führte zu einem enormen Aufschwung der gesamten Opioidforschung. Mit über 20 weiteren endogenen Opioidpeptiden, die in der Zwischenzeit noch entdeckt wurden und denen allen die N-terminale Sequenz Tyr-Gly-Gly-Phe-Met/Leu gemeinsam ist, zählt das Opioidsystem zu einem der am besten charakterisierten neurochemischen Systeme. Der Grund für diese Vielzahl an Opioidpeptiden liegt in einem gewebespezifischen, posttranslationalen Prozessieren dreier Opioidpeptidvorläufer-Moleküle. Diese verschiedenen Vorläuferpeptide, die als Proopiomelanocortin (POMC), Proenkephalin und Prodynorphin bezeichnet werden, sind im Aufbau ähnlich, entwicklungsgeschichtlich miteinander verwandt und wahrscheinlich durch Genduplikation entstanden. POMC ist das gemeinsame Vorläufermolekül des adrenocorticotropen Hormons (ACTH), des Alpha- und Beta-Melanotropins, des Met-Enkephalins, des Beta- und Gamma-Lipotropins aber auch des endogenen Opioids Beta-Endorphin, das bevorzugt an My-Opioid-Rezeptoren bindet. Proenkephalin enthält mehrere Met-und Leu-Enkephalin-Sequenzen sowie eine Anzahl längerkettiger Opioide, die vornehmlich an den Delta-Opioid-Rezeptor binden. Von Prodynorphin leiten sich verschiedene Dynorphine ab, die als endogene Liganden des Kappa-Opioid-Rezeptors angesehen werden.
Rolle im Belohnungssystem
Es wird schon lange vermutet, dass Opioidpeptide wie Endorphine und Enkephaline neurochemische Substrate von Belohnungsereignissen darstellen und wichtig für die Vermittlung der damit verbundenen euphorisierenden Wirkung sind. Frühe Studien kamen zu dem Ergebnis, dass sowohl Endorphine als auch Enkephaline intrinsische belohnende Eigenschaften haben. Hierfür spricht, dass Ratten sich diese Opioide im operanten Experiment selbst verabreichen (direkt in die Hirnventrikel [45] und in den NAC [9]). Insbesondere ist das VTA ein „Hot Spot“ für die Induktion einer Verstärkerwirkung (reinforcement) durch Opioide, weil die Verabreichung von Opioid-Rezeptor-Agonisten in das VTA zu einer konditionierten Platzpräferenz führt [14] und außerdem diese Substanzen in das VTA von Ratten selbst appliziert werden [6]. Aus diesen Gründen erscheint es wahrscheinlich, dass die Opioid-Rezeptoren My und Delta, die Zielrezeptoren von Enkephalinen und Endorphinen im VTA und in dem NAC, in wichtiger Weise an den neurobiologischen Mechanismen beteiligt sind, die ein Belohnungsgefühl hervorrufen. Weiter konnte gezeigt werden, dass die tonische Dopaminkonzentration im NAC von endogenen Opioidsystemen reguliert wird [42]. Es blieb allerdings noch viele Jahre ungeklärt, ob Drogen wirklich Belohnungsgefühle durch die Ausschüttung von Endorphinen und Enkephalinen auslösen. Anhand von In-vivo-Mikrodialyseexperimenten zeigten Olive et al. [33] in einer Schlüsselpublikation endgültig, dass Drogen, darunter auch Ethanol, eine Ausschüttung von Beta-Endorphin in den NAC bewirken. Bemerkenswerterweise stellte man anhand der gleichzeitigen Messung der Dopaminkonzentration fest, dass nach der Verabreichung von Alkohol das extrazelluläre Dopamin früher als die Beta-Endorphinkonzentration anstieg, sodass neben der Beta-Endorphin-vermittelten Dopaminausschüttung weitere Mechanismen diskutiert werden, die für eine alkoholinduzierte Stimulation des Belohnungssystems verantwortlich sind (z.B. 5-HT3-Rezeptor-vermittelte Effekte). Allerdings gibt es Studien, in denen die positiven verstärkenden Eigenschaften von My/Delta-Opioid-Rezeptoragonisten bei Injektion in diesen Teil des Hirns nachgewiesen wurden [9]. Daher kann man durchaus annehmen, dass ein Anstieg der extrazellulären Endorphin-Konzentration an der Vermittlung der belohnenden hedonistischen Wirkung von Ethanol und anderen Drogen beteiligt ist. Beta-Endorphin im NAC stammt von POMC-enthaltenden Neuronen im Nucleus arcuatus des Hypothalamus ab [2]. Es ist zum heutigen Zeitpunkt jedoch nicht sicher geklärt, ob der ethanolinduzierte Anstieg der extrazellulären Endorphin-Konzentration im NAC der direkten Aktivierung des Endorphinsystems im Nucleus arcuatus zugrunde liegt.
Wichtig in diesem Zusammenhang ist, dass die von Alkohol induzierte Dopamin-Ausschüttung im NAC durch Opioid-Rezeptorantagonisten wie Naltrexon (z.B. Adepend®) oder Nalmefen (Selincro®) unterdrückt wird und die Suppression von operanter Alkoholselbstverabreichung durch Opioid-Rezeptorantagonisten mit einer Abschwächung des alkoholinduzierten Anstiegs der Dopamin-Konzentration im NAC assoziiert ist [11]. Es konnte auch gezeigt werden, dass Alkohol bevorzugende Ratten (AA-Ratten) eine geringere opioiderge Aktivität in den Regionen aufweisen, die an Alkohol-Reinforcement beteiligt sind [31]; weitere Untersuchungen wiesen bei anderen Alkohol bevorzugenden und Alkohol vermeidenden Tierlinien angeborene Unterschiede im Opioidsystem nach [14, 46]. Schließlich wäre noch darauf hinzuweisen, dass My-Opioidrezeptor-Knockout-Mäuse sich unter unterschiedlichen Testbedingungen keinen Alkohol selbst verabreichen [36]. Diese Ergebnisse belegen, dass die durch Alkohol hervorgerufenen Verstärkungsprozesse nicht nur Dopamin-abhängig, sondern auch von der Funktionsfähigkeit des endogenen Opioidsystems abhängig sind.
Es wird darüber hinaus von einigen Forschern vermutet, dass die auftretende Erhöhung der In-vivo-Synthese sogenannter Tetrahydroisochinoline (TIQ, tetrahydroisoquinolines) nach Alkoholexposition ebenfalls zur Stimulation von Opioid-Rezeptoren und der hedonischen Wirkung von Alkohol beitragen. Diese TIQ sind Kondensationsprodukte aus dem Acetaldehyd des Alkoholabbaus mit Monoaminen. Sie haben eine strukturelle Ähnlichkeit mit einer Vorstufe des Morphins und können an Opioid-Rezeptoren binden. Es gibt Berichte, denen zufolge eine intrakranielle Verabreichung von TIQ sowohl bei Ratten als auch bei Affen eine dosisabhängige Erhöhung der freiwilligen Alkoholeinnahme bewirkt. Somit könnte Alkohol über seine metabolischen Kondensationsprodukte Opioid-Rezeptoren stimulieren. Auf diese Weise ließe sich die nach Alkoholgenuss auftretende Euphorie ebenfalls erklären; eine interessante Hypothese, die allerdings kontrovers beurteilt wird [5].
Insgesamt belegen die Ergebnisse von Tiermodellen, dass endogene Opioide eine entscheidende Rolle bei Belohnungs- und Verstärkungsprozessen durch Alkohol in Wechselwirkung mit dem mesolimbischen Dopaminsystem spielen. Leider ist es im Tiermodell nicht möglich, subjektive Erfahrungen in angemessener Weise zu untersuchen, sodass die Übertragung der Erkenntnisse auf den Menschen limitiert ist. Man mag darüber spekulieren, ob Wohlbefinden und Euphorie nicht weiteren komplexeren Prozessen als nur der zentralen Aktivierung von My/Delta-Opioid-Rezeptoren unterworfen sind, einschließlich eines Gleichgewichts zwischen Stresssystem und den physiologischen Parametern, die vom autonomen Nervensystem kontrolliert werden. In dieser Hinsicht ist es durchaus möglich, dass zukünftige Untersuchungen feststellen werden, dass der Hypothalamus in seiner Rolle als Schnittstelle zwischen Hirn und Körper eine wichtige Rolle in der Suchtentwicklung spielt.
Das mesokortikolimbische System und Suchtentwicklung
Auf die Wechselwirkungen des endogenen Opioidsystems und des mesolimbischen Dopaminsystems wurde ausführlich eingegangen; die genannten Erkenntnisse bilden die Grundlage für den Einsatz von Opioid-Rezeptorantagonisten wie Naltrexon und Nalmefen in der Behandlung von alkoholabhängigen Patienten [26, 39].
Rolle des Opioidsystems bei der Suchtentwicklung
Da im Verlauf einer Suchtentwicklung zunehmend ein negativer emotionaler Status zu Tage tritt, der mit einer Hochregulation des dynorphinergen Systems einhergeht [3], das im Humanhirn direkt Dysphorie bewirkt [35], könnte eine Blockade von Kappa-Opioid-Rezeptoren zu einem zusätzlichen Nutzen in der Behandlung der Alkoholabhängigkeit führen [47]. Eine solche Minderung der über Kappa-Opioid-Rezeptoren vermittelten Dysphorie nach Abklingen der akuten, über My/Delta-Opioid-Rezeptoren vermittelten „belohnenden“ Alkoholeffekte könnte zu einer Minderung des Alkoholverlangens beitragen. Dies wäre ein Grund für eine mögliche Überlegenheit von Nalmefen gegenüber Naltrexon in der Behandlung alkoholkranker Patienten, da diese Substanz als Partialagonist am Kappa-Opioid-Rezeptor wirkt. Ein Partialagonist ist eine Substanz, die mit dem endogenen Liganden am Rezeptor in Wettbewerb tritt, jedoch im Gegensatz zu einem vollen Agonisten nur unvollständig die nachgeschaltete Signaltransduktion aktiviert. Da ein Partialagonist in der Regel in der Lage ist, einen endogenen Liganden konzentrationsabhängig von seinem Rezeptor zu verdrängen, kann Nalmefen die Effekte von Dynorphin funktionell hemmen. Diese besondere pharmakologische Eigenschaft von Nalmefen führt somit zu einer Verminderung dynorphinerger Überaktivität, dabei wird der negative emotionale Status des Patienten vermindert und das damit verbundene Rückfallrisiko; ein Behandlungsprinzip, das durchaus auch bei der Behandlung von depressivem Verhalten als sinnvoll erachtet wird, da präklinische Befunde nahe legen, dass es auch bei der Depression zu einer dynorphinergen Überaktivität kommt.
Rolle von Dopamin bei der Suchtentwicklung
Neben den opioidergen Wirkungen auf das Belohnungssystem haben die Erkenntnisse zum Dopamin bei der Verarbeitung belohnungsankündigender Hinweisreize (z.B. Reize, die mit der Alkoholaufnahme konditioniert werden) für das Verständnis von Suchterkrankungen als „Lernmodell“ eine entscheidende Rolle gespielt. Terry Robinson und Kent Berridge von der Universität Michigan haben in ihrer Incentive Salience Theory of Addiction [37] postuliert, dass es durch wiederholten Substanzkonsum zu einer durch neuroadaptive Prozesse vermittelten Sensibilisierung des dopaminergen mesolimbischen Systems nach wiederholter Drogengabe kommt. Damit einhergehend resultiert zumindest in einigen Individuen auch eine höhere Anreizmotivation für suchtmittelassoziierte Reize [8]. Bei solchen Individuen muss davon ausgegangen werden, dass sie schnell einen Impulskontrollverlust bei der Darbietung bestimmter appetitiver konditionierte Reize erleiden (z.B. Geruchsreize, die mit einer Alkoholaufnahme verbunden sind) und damit auch für eine Suchtentwicklung empfänglich sind.
Dieser Incentive-Salience-Prozess spielt in der Suchtentwicklung sicherlich eine große Rolle; weitere Prozesse sind jedoch ebenfalls von Bedeutung. So kommt es im Verlauf der Suchtentwicklung zu einer zunehmenden Verschiebung der vom NAC abhängigen belohnungsassoziierten zielgerichteten Verhaltensweisen hin zu Reiz-Reaktions-Schemata, die vom dorsalen Striatum als sensomotorische Schnittstelle verarbeitet werden [7]. In der Tat ist mit wiederholtem Alkoholgebrauch eine Verschiebung des phasischen Dopaminsignals vom NAC hin zum dorsalen Striatum zu beobachten [49]. Das dorsale Striatum steuert insbesondere stereotype und automatisierte Verhaltensweisen und so wird es verständlich, dass es mit zunehmend süchtigem Verhalten zu repetitiven eingeengten Verhaltensweisen kommt.
Rolle von Glutamat bei der Suchtentwicklung
Während Dopamin die konditionierten Verstärkungsprozesse und die zunehmend striatal getriebenen Automatismen in Bezug auf chronische Alkoholkonsummuster vermittelt, scheint das fortgeschrittene Stadium von Suchterkrankungen mit zwanghaftem Substanzkonsum und rückfälligem Verhalten wesentlich von der suchtmittelinduzierten Plastizität glutamaterger Neurone im präfrontalen Kortex und dessen Projektionen in den NAC abzuhängen [17]. Kalivas und Volkow schlussfolgern in einer integrativen Betrachtung der vorliegenden Tier- und Bildgebungsstudien, dass dauerhafte synaptische Veränderungen in den glutamatergen Projektionen vom präfrontalen Kortex in den NAC für den zwanghaften Suchtmittelkonsum verantwortlich sind und damit wesentlich zur Aufrechterhaltung von Suchterkrankungen beitragen [17]. Insbesondere Bildgebungsstudien weisen auf eine zentrale Rolle von Neuroadaptionen in den Strukturen des präfrontalen Kortex, als wichtige Regionen für Aufmerksamkeits-, Bewertungs- und Exekutivfunktionen in der Pathogenese von abhängigem Verhalten hin [10, 17]. Vor kurzem konnte eine spezifische, persistierende molekulare Neuroadaption im präfrontalen Kortex nachgewiesen werden [27]. Die Studie zeigt, dass Alkohol speziell einen kleinen Teilbereich des präfrontalen Kortex, das sogenannte infralimbische Areal, schädigt. Bei Ratten kann der daraus resultierende Funktionsverlust mithilfe von molekulargenetischen Werkzeugen repariert werden, wodurch diese wieder die volle Kontrolle über ihr Alkoholsuchverhalten erlangen. Aus der Studie geht weiter hervor, dass eine bestimmte Gruppe von Neuronen im präfrontalen Kortex sehr empfindlich auf Alkohol reagiert, wenn dieser wiederholt in Konzentrationen verabreicht wird, die üblicherweise bei Alkoholikern auftreten (>2,5‰ über mehrere Stunden). Diese Neuronen behalten langfristige Folgeschäden, was sich unter anderem darin ausdrückt, dass sie die Freisetzung des Botenstoffs Glutamat nicht mehr angemessen regulieren können. Zurückzuführen ist dies auf eine mangelnde Autorezeptorfunktion, die normalerweise durch Glutamat-Rezeptoren vom Typ mGluR2 ausgeübt wird. Dieser Funktionsverlust kann direkt mit Suchtverhalten in Verbindung gebracht werden. Es wurde gezeigt, dass die Wiederherstellung des mGluR2-Niveaus im infralimbischen Kortex alkoholabhängiger Ratten ausreichend ist, um deren übermäßiges Bedürfnis nach Alkohol wieder zu normalisieren. Diese Ergebnisse scheinen auch für die Alkoholsucht bei Menschen von Bedeutung zu sein. Im Autopsiematerial von Alkoholikern wurden in der entsprechenden Hirnregion ebenfalls erniedrigte mGluR2-Werte nachgewiesen. Die Untersuchungsergebnisse legen nahe, dass Alkoholabhängigkeit nicht nur zu einer Abnahme von mGluR2-Rezeptoren in neuronalen Netzwerken des präfrontalen Kortex führt, sondern dass der dadurch verursachte Funktionsausfall bei Alkoholsüchtigen auch die Gefahr eines Rückfalls verstärkt. Die im Tiermodell aufgezeigte Möglichkeit einer Reparatur der mGluR2-Autorezeptorfunktion eröffnet neue therapeutische Perspektiven.
Neben den genannten molekularen Veränderungen im präfrontalen Kortex, die für einen zunehmenden Kontrollverlust bei alkoholkranken Patienten stehen, kommt es auch zu pathologischen Veränderungen in der Reiz- und Kontextverarbeitung. Insbesondere für die bevorzugte Enkodierung von Kontext und Alkoholeinnahme ist die reziproke Verbindung zwischen dem VTA und dem Hippokampus von Bedeutung. Während einer abstinenten Phase können dann alkoholassoziierte Umgebungsreize über diese glutamaterge hippokampale VTA-Schleife zu Craving und Rückfall führen [23]. Das Rückfallrisiko hängt somit von der Speicherung impliziter alkoholassoziierter Erinnerungen und der Modulation entsprechender Verhaltensreaktionen ab. Dabei lassen sich Prozesse auf synaptischer Ebene vermuten, wie sie auch charakteristisch für das Langzeitgedächtnis zu sein scheinen. Die Frage, wodurch Erinnerungen persistieren, ist somit hochrelevant für das Verständnis der Alkoholabhängigkeit und bislang auch für das physiologische Gedächtnis noch nicht vollständig beantwortet. In jedem Fall kann von einer physikalischen Reorganisation von Synapsen und den entsprechenden Netzwerken ausgegangen werden. Im Jahr 1973 fanden die Physiologen Bliss und Lomo, dass kurze repetitive Stimulation afferenter Fasern zu einer lang anhaltenden Verstärkung der synaptischen Übertragung führt [1]. Die Autoren bezeichneten diese Phänomene als Langzeitpotenzierung (LTP, long term potentiation) und den umgekehrten Vorgang als Langzeitdepression (LTD, long term depression). Veränderungen in der Ausbildung von LTD, insbesondere einer bestimmten Form von LTD, bei der mGluR2-Rezeptoren im präfrontalen Kortex essenziell sind, konnte im Gehirn von süchtigen Ratten nachgewiesen werden [19]. Dabei zeigte sich, dass süchtige Ratten nicht mehr in der Lage sind, synaptische Plastizität in Form von LTD zu generieren; man spricht in diesem Fall von Anaplastizität [18, 19]. Der Übergang von nichtsüchtigem zu süchtigem Verhalten scheint also auch durch anaplastische Prozesse im mesokortikolimbischen System charakterisiert zu sein [18]. Der Verlust bestimmter Formen von synaptischer Plastizität könnte daher zum pathologischen Prozess der Sucht beitragen, während Drogen-induzierte synaptische Plastizität, wie beispielsweise das Auftreten von LTP nach einer Drogengabe [40] als ein adaptiver physiologische Prozess in unserem ZNS angesehen werden könnte.
Rolle des CRH-Systems bei der Suchtentwicklung
Die basolaterale Amygdala spielt ebenfalls eine wichtige Rolle beim Abruf von Verhaltensreaktionen auf entsprechende spezifische Hinweisreize und bei der Konditionierung von Assoziationen zwischen motivational und emotional relevanten Ereignissen. Im Laufe der Alkoholabhängigkeit stellt sich ein negativer emotionaler Status ein, der neben der über Kappa-Opioid-Rezeptoren vermittelten Dysphorie auch mit einer Hochregulation des Corticotropin-Releasing-Hormone-(CRH-)Systems und insbesondere des CRH-Rezeptors 1 (CRHR1) in der Amygdala in Verbindung gebracht werden kann [13]. CRH ist ein Neuropeptid, das in der hormonalen und extrahormonalen Stressantwort eine entscheidende Rolle spielt. George Koob vom Scripps Institut in La Jolla vertritt seit vielen Jahren die Meinung, dass diese CRH/CRHR1-Veränderungen in der Amygdala zu einer negativen Verstärkung führen, sprich, der Patient trinkt Alkohol, um den durch das CRH-System getriebenen negativen emotionalen Status zu unterdrücken. Demnach würden im Verlauf der Abhängigkeit die durch Alkohol hervorgerufenen positiven Verstärkungsprozesse eine zunehmend untergeordnete Rolle spielen und das CRH-System würde zur Triebfeder eines fortwährenden und sich intensivierenden Alkoholkonsums bzw. Rückfalls [13]. Konsequenterweise müsste demnach eine Blockade des CRHR1 mit einer Trinkmengenreduktion oder Rückfallvermeidung einhergehen, insbesondere dann, wenn Stress einen zusätzlichen negativen emotionalen Zustand bedingt. Am National Institute of Alcohol and Alcoholism (NIAAA) werden zurzeit CRHR1-Antagonisten erstmals in Humanstudien getestet. Erste Ergebnisse sind Ende 2013 zu erwarten.
Zusammenfassung und Ausblick
Der vorliegende Übersichtsartikel vermittelt eine Gesamtsicht der akuten und chronischen zentralen Wirkungen von Alkohol. Die erste Interaktionsebene betrifft die primären Angriffsstellen (Rezeptoren und Ionenkanäle) im Gehirn und anderen Zielorganen. Dabei interagiert Ethanol mit spezifischen Rezeptorsystemen, deren Hemmung (z.B. NMDA-Rezeptoren) oder Aktivitätssteigerung (z.B. GABAA) die akute Alkoholintoxikation bewirkt und dabei subjektive Empfindungen triggert. Nach dem „first hit“ von Ethanol auf diese Rezeptoren und Ionenkanäle setzt eine zweite Welle indirekter Wirkungen auf Monoamine und Opioide ein, die von entscheidender Bedeutung für Reinforcement und die Entwicklung eines Belohnungsgefühls durch Alkohol sind und so das Trinkverhalten beeinflussen.
Im weiteren Verlauf einer Alkoholexposition müssen zudem persistierende Adaptationsprozesse in anderen neurochemischen Funktionssystemen berücksichtigt werden, insbesondere das extrahypothalamische CRH/CRHR1-System und das dynorphinerge/Kappa-Opioid-Rezeptorsystem; über beide können negative Verstärkereffekte vermittelt werden, die zur Aufrechterhaltung des Alkoholkonsums beitragen. Diese Systeme können auch einen negativen emotionalen Status während der Abstinenz bedingen und somit zum Rückfallverhalten beitragen (Abb. 1).
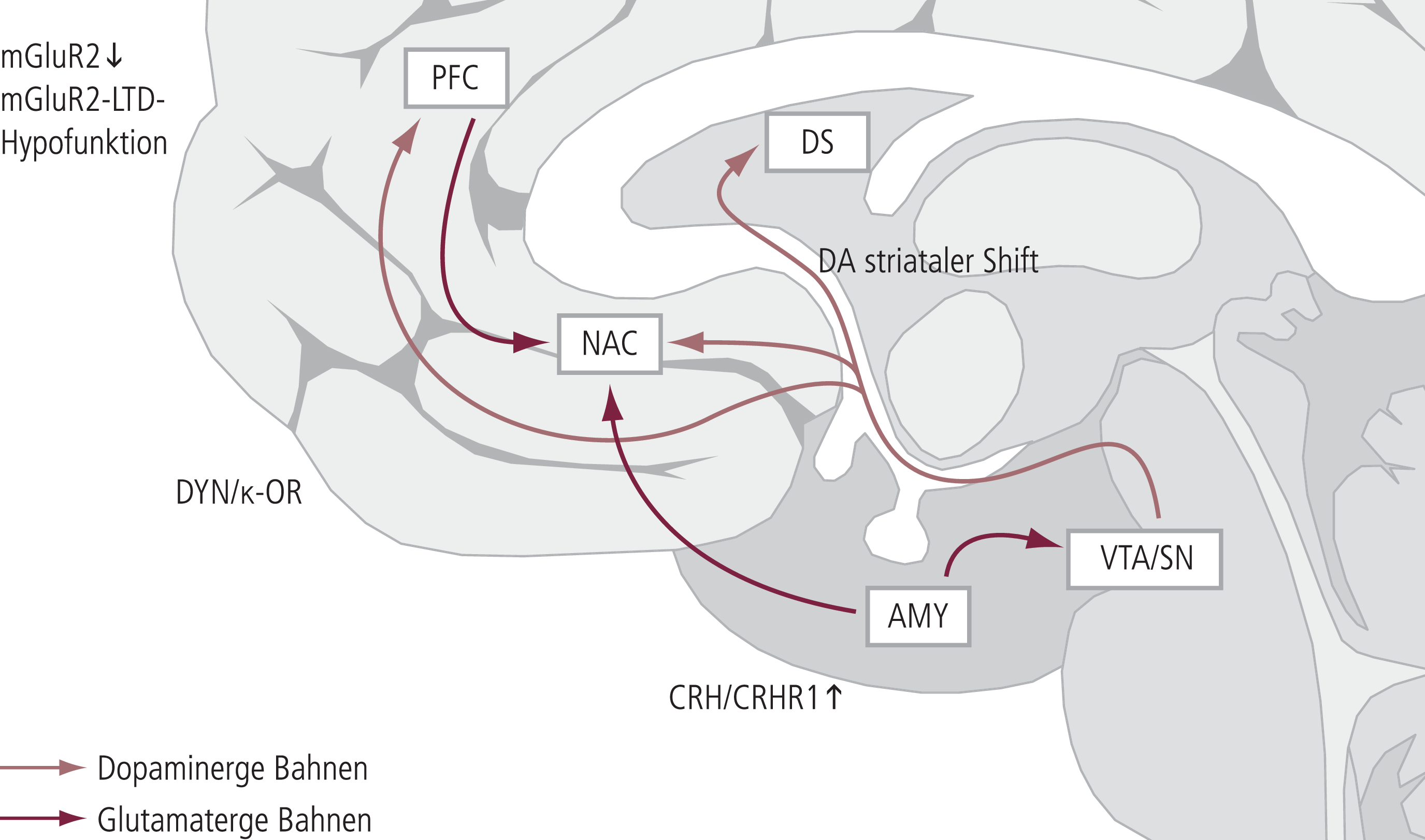
Abb. 1. Die neurobiologischen Charakteristika der Sucht Im Übergang von nichtsüchtigem zu süchtigem Verhalten kommt es zu nachhaltigen Veränderungen im mesokortikolimbischen System. Zentral sind das dorsale Striatum (DS) und der Nucleus accumbens (NAC) dargestellt. Im nichtsüchtigen, jedoch vulnerablen Hirn kann das phasische Dopaminsignal im NAC zu einer zunehmenden Anreizmotivation (incentive salience) führen. Im Verlauf der Suchtentwicklung kommt es zu einer zunehmenden Verschiebung der vom NAC belohnungsassoziierten zielgerichteten Verhaltensweisen hin zu automatisierten Reiz-Reaktions-Schemata, welche von nigrostriatalen Bahnen (Substantia nigra [SN] zum DS) vermittelt werden (striataler Shift). Der präfrontale Kortex (PFC) verliert beim alkoholkranken Patienten seine Impulskontrolle; es kommt zu einer Hypofunktion, getragen durch eine stark verminderte Expression von metabotropen Glutamat(mGluR2)-Autorezeptoren und einem damit verbundenen Verlust von neuronaler Plastizität, d.h., es kann keine mGluR2-vermittelte Long-Term Depression (LTD) mehr auftreten. Ein negativer emotionaler Status bei Entzug und Abstinenz wird durch die Hochregulation des dynorphinergen/Kappa-Opioid-Rezeptorsystems (DYN/κ-OR) im NAC einerseits und durch ein hyperaktives CRH/CRHR1-System in der Amygdala (AMY) anderseits vermittelt. VTA: ventrales Tegmentum
Das Wissen um die molekularen und systemischen Effekte von Alkohol ermöglicht nun, in der Therapie der Alkoholerkrankung spezifische Rezeptor- und Funktionssysteme pharmakologisch zu modulieren [24]. Aktuell stehen hier insbesondere Opioid-Rezeptorantagonisten, Glutamat-Rezeptorantagonisten, GABAerge Substanzen und CRHR1-Antagonisten im Fokus der Forschung. Aufgrund ihrer Wirkansätze lässt sich dabei erwarten, dass die My/Delta-Opioid-Rezeptorantagonisten insbesondere die positiv verstärkenden und motivationalen Aspekte des fortgesetzten Alkoholkonsums (Präferenz, Kontrollverlust) reduzieren, währen die Stress- und Anhedonie-assoziierten negativen Verstärkereffekte eher über das Kappa-opioiderge System, das CRH-System und über antiglutamaterge oder GABAerge Inputs moduliert werden können. Substanzen mit entsprechendem pharmakodynamischem Profil stehen (mit Ausnahme der CRHR1-Antagonisten) klinisch bereits zur Verfügung.
Interessenkonflikterklärung
RS hat Honorare von der Firma Lundbeck erhalten. Eine Einrichtung, für die er tätig ist, hat Gelder von der Firma Lundbeck erhalten.
FK hat Honorare für die Beratung oder Teilnahme an einem Expertenbeirat von den Firmen Lundbeck, Roche und Teva, Honorare für Vorträge und Publikationen von den Firmen Boehringer Ingelheim und Lundbeck, Unterstützung für Kongressbesuche von den Firmen Pfizer und Servier und Forschungsbeihilfen vom Bundesforschungsministerium (BMBF) und der Deutschen Forschungsgemeinschaft (DFG) erhalten.
Literatur
1. Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 1973;232:331–56.
2. Bloom F, Battenberg E, Rossier J, Ling N, et al. Neurons containing ß-endorphin in rat brain exist separately from those containing enkephalin: immunocytochemical studies. Proc Natl Acad Sci USA 1978;75:1591–5.
3. Butelman ER, Yuferov V, Kreek MJ. κ-opioid receptor/dynorphin system: genetic and pharmacotherapeutic implications for addiction. Trends Neurosci 2012;35:587–96.
4. Day JJ, Roitman MF, Wightman RM, et al. Associative learning mediates dynamic shifts in dopamine signaling in the nucleus accumbens. Nat Neurosci 2007;10:1020–8.
5. Deehan GA Jr., Brodie MS, Rodd ZA. What is in that drink: the biological actions of ethanol, acetaldehyde, and salsolinol. Curr Top Behav Neurosci 2013;13:163–84.
6. Devine DP, Wise RA. Self-administration of morphine, DAMGO, and DPDPE into the ventral tegmental area of rats. J Neurosci 1994;14:1978–84.
7. Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 2005;8:1481–9.
8. Flagel SB, Clark JJ, Robinson TE, Mayo L, et al. A selective role for dopamine in stimulus-reward learning. Nature 2011;469:53–7.
9. Goeders NE, Lane JD, Smith JE. Self-administration of methionine enkephalin into the nucleus accumbens. Pharmacol Biochem Behav 1984;20:451–5.
10. Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 2002;159:1642–52.
11. Gonzales RA, Weiss F. Suppression of ethanol-reinforced behavior by naltrexone is associated with attenuation of the ethanol-induced increase in dialysate dopamine levels in the nucleus accumbens. J Neurosci 1998;18:10663–71.
12. Grace AA. The tonic/phase model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction 2000;95:119–28.
13. Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 2007;30:399–406.
14. Herz A. Endogenous opioid systems and alcohol addiction. Psychopharmacology 1997;129:99–111.
15. Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975;258:577–80.
16. Jackson A, Stephens DN, Duka T. A low dose alcohol drug discrimination in social drinkers: relationship with subjective effects. Psychopharmacology 2001;157:411–20.
17. Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 2005;162:1403–13.
18. Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, et al. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 2010;328:1709–12.
19. Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, et al. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry 2013;18:729–37.
20. Kruse SW, Zhao R, Smith DP, Jones DN. Structure of a specific alcohol-binding site defined by the odorant binding protein LUSH from Drosophila melanogaster. Nat Struct Biol 2003;10:694–700.
21. Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 2013, Apr 8 [Epub ahead of print].
22. Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev 2009;89:1379–412.
23. Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron 2005;46:703–13.
24. Litten RZ, Egli M, Heilig M, Cui C, et al. Medications development to treat alcohol dependence: a vision for the next decade. Addict Biol 2012;17:513–27.
25. Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA activated ion current in hippocampal neurons. Science 1989;243:1721–4.
26. Mann K, Bladström A, Torup L, Gual A, et al. Extending the treatment options in alcohol dependence: a randomized controlled study of as-needed nalmefene. Biol Psychiatry 2013;3:706–13.
27. Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, et al. Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. J Neurosci 2013;33:2794–806.
28. Mihic SJ, Ye Q, Wick MJ, Koltchine VV, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature 1997;389:385–9.
29. Moore MS, DeZazzo J, Luk AY, Tully T, et al. Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell 1998;93:997–1007.
30. Noori HR, Spanagel R, Hansson AC. Neurocircuitry for modeling drug effects. Addict Biol 2012;17:827–64.
31. Nylander I, Hyytiä P, Forsander O, Terenius L. Differences between alcohol-preferring (AA) and alcohol-avoiding (ANA) rats in the prodynorphin and proenkephalin systems. Alcohol Clin Exp Res 1994;8:1272–9.
32. Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 1954;47:419–27.
33. Olive MF, Koenig HN, Nannini MA, Hodge CW. Stimulation of endorphin neurotransmission in the nucleus accumbens by ethanol, cocaine, and amphetamine. J Neurosci 2001;21:RC184.
34. Peoples RW, Li C, Weight FF. Lipid vs. protein theories of alcohol action in the nervous system. Annu Rev Pharmacol Toxicol 1996;36:185–201.
35. Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by κ-opiate receptors. Science 1986;233:774–6.
36. Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL et al. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 2000;293:1002–8.
37. Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 1993;18:247–91.
38. Ronald KM, Mirshahi T, Woodward JJ. Ethanol inhibition of N-methyl-D-aspartate receptors is reduced by site-directed mutagenesis of a transmembrane domain phenylalanine residue. J Biol Chem 2001;276: 44729–35.
39. Rösner S, Hackl-Herrwerth A, Leucht S, Vecchi S, et al. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev 2010;12:CD001867.
40. Saal D, Dong Y, Bonci A, et al. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 2003;37:577–82.
41. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science 1997;275:1593–9.
42. Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA 1992;89:2046–50.
43. Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci 1999;22:521–7.
44. Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev 2009;89:649–705.
45. van Ree JM, Smyth DG, Colpaert FC. Dependence creating properties of lipotropin C-fragment (ß-endorphin): evidence for its internal control of behaviour. Life Sci 1979;24:495–502.
46. Vengeliene V, Bilbao A, Molander A, Spanagel R. Neuropharmacology of alcohol addiction. Br J Pharmacol 2008;154:299–315.
47. Walker BM, Zorrilla EP, Koob GF. Systemic κ-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol 2011;16:116–9.
48. Whelan R, Conrod PJ, Poline JB, Lourdusamy A, et al. Adolescent impulsivity phenotypes characterized by distinct brain networks. Nat Neurosci 2012;15:920–5.
49. Willuhn I, Burgeno LM, Everitt BJ, Phillips PE. Hierarchical recruitment of phasic dopamine signaling in the striatum during the progression of cocaine use. Proc Natl Acad Sci USA 2012;109:20703–8.
50. Wise RA. Action of drugs of abuse on brain reward systems. Pharmacol Biochem Behav 1980;13:213–23.
Prof. Dr. Rainer Spanagel, Institut für Psychopharmakologie am Zentralinstitut für Seelische Gesundheit, J 5, 68159 Mannheim, E-Mail: rainer.spanagel@zi-mannheim.de
Prof. Dr. Falk Kiefer, Klinik für Abhängiges Verhalten und Suchtmedizin am Zentralinstitut für Seelische Gesundheit, J 5, 68159 Mannheim
Neurobiology of alcohol addiction
Alcohol produces multiple effects on the central nervous system (CNS). Which molecular mechanisms are primarily involved in mediating those effects? In the past 20 years ethanol binding sites on several membrane receptors and ion channels were identified and it was found that NMDA and GABAA receptors mediate, at least in part, the acute psychotropic effects of alcohol ingestion. This primary action of alcohol which is alcohol intoxication and the hereof resulting subjective effect is followed by a second wave of indirect effects on other neurotransmitter/neuropeptide systems. Especially, the dopamine and opioid systems are recruited and trigger reinforcing and rewarding effects. Molecular interactions between the primary sites of action of ethanol (i.e. NMDA-receptors) and the mesocorticolimbic dopamine system can lead to persistent neuroadaptations that can trigger addictive behavior. Due to reduced prefrontal control and progressively recruitment of a striatal dopaminergic mechanism that facilitates automatized behavior, repetitive and compulsive uncontrolled drug taking behavior is observed. This is paralleled by a negative emotional state during alcohol withdrawal and protracted abstinence – a state that is driven by an enhanced extrahypothalamic corticotropin-releasing hormone-system and an up-regulated dynorphinergic tone and kappa-opioid receptor activation. The knowledge of these neuroanatomical and neurochemical substrates of addictive behavior has greatly facilitated the development of intervention strategies for our patients.
Key words: Addiction, dopamine, glutamate, endogenous opioids, mesocorticolimbic system
Psychopharmakotherapie 2013; 20(05)