Holger Petri, Bad Wildungen*
In der PRISCUS-Liste wird Trospiumchlorid als mögliche Therapiealternative zu Oxybutynin, Tolterodin und Solifenacin empfohlen, die als potenziell inadäquat bei älteren Patienten betrachtet werden [1]. Im Folgenden sollen die pharmakokinetischen Eigenschaften dieser Substanzen aus der PRISCUS-Liste bezüglich Wechselwirkungen auf CYP-Ebene dargestellt werden.
Oxybutynin
Oxybutynin ist ein Substrat von CYP3A4 [2]. Darüber hinaus wird in den AGNP-Leitlinien (AGNP: Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie) Oxybutynin als CYP3A4-Induktor aufgeführt [3]. Bei Substraten, die über dieses Enzym abgebaut werden, könnte der Plasmaspiegel fallen und Dosiserhöhungen somit notwendig werden.
Solifenacin
Solifenacin wird ausschließlich über CYP3A4 metabolisiert. Daher ist bei gleichzeitiger Anwendung von starken CYP3A4-Hemmern die Höchstdosis von Solifenacin auf 5 mg zu begrenzen. Bei Patienten mit stark eingeschränkter Nierenfunktion oder mit mäßig eingeschränkter Leberfunktion ist eine Komedikation mit einem starken CYP3A4-Hemmer sogar kontraindiziert [4]. Wie zu verfahren ist, wenn ein CYP3A4-Induktor gleichzeitig mit Solifenacin verordnet wird, ist der zugehörigen Fachinformation nicht zu entnehmen. Eine klinisch relevante Plasmaspiegelsenkung von Solifenacin ist denkbar mit der Gefahr eines Therapieversagens.
Tolterodin
Der primäre Metabolismus von Tolterodin wird durch das polymorphe Enzym CYP2D6 vermittelt und führt zur Bildung von 5-Hydroxymethyl-Tolterodin (5-HMT) (Abb. 1). Dieser aktive Metabolit ist äquipotent mit der Muttersubstanz. Der Metabolismus bei Personen mit reduzierter CYP2D6 Aktivität (Poor metabolizer; PM) verläuft über CYP3A4 zu N-dealkyliertem Tolterodin, das zur klinischen Wirkung nicht beiträgt [5].
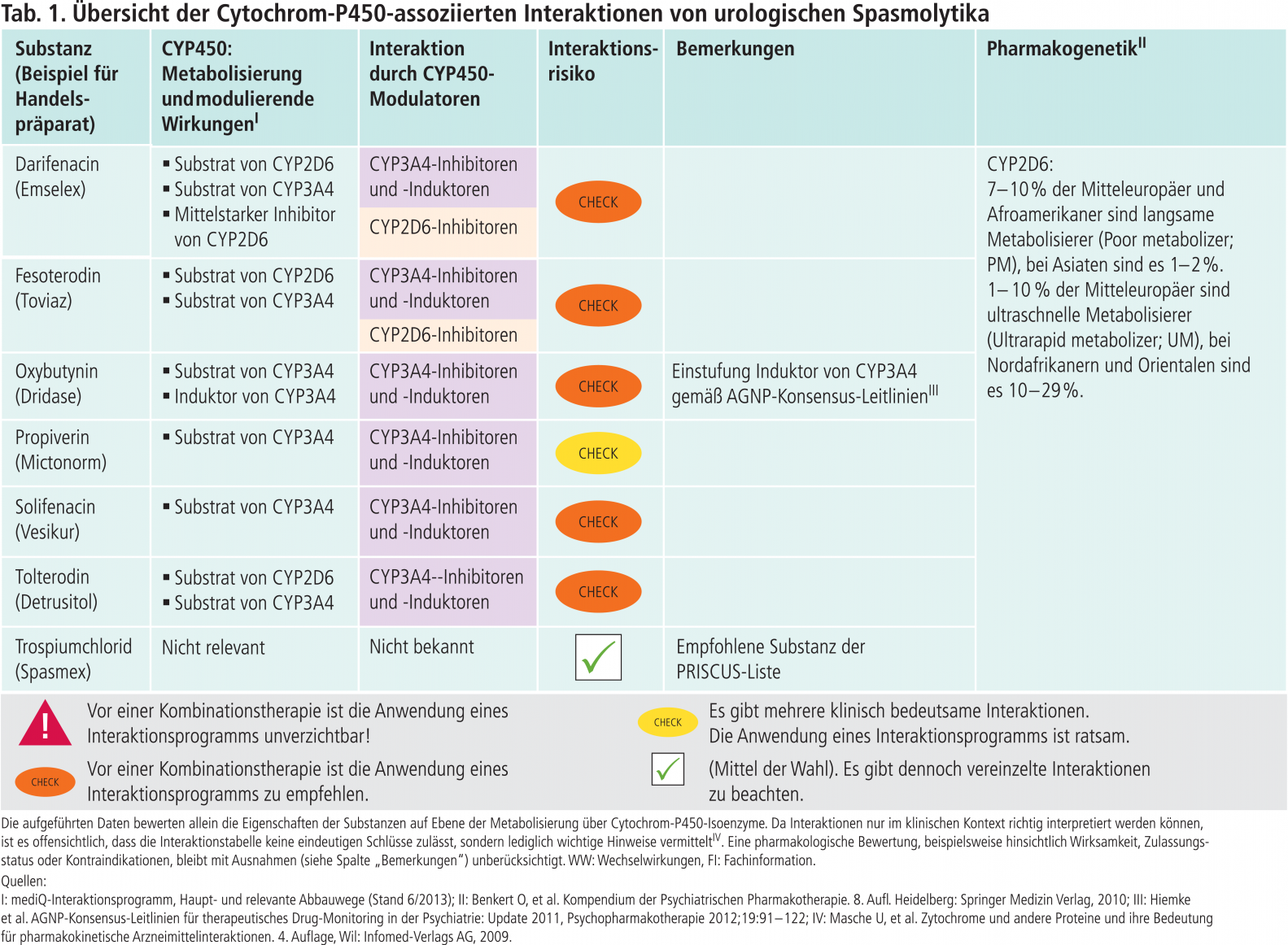
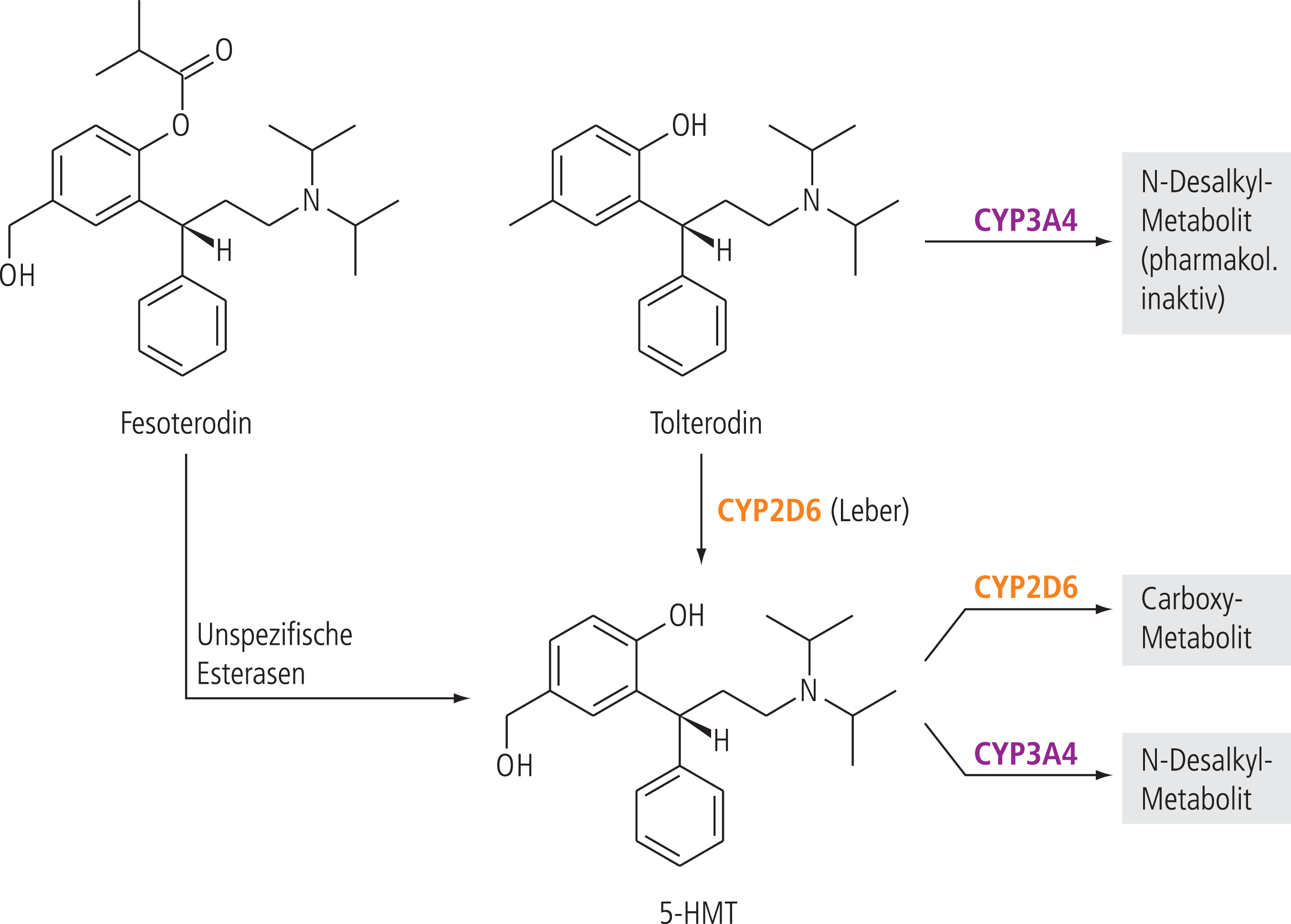
Abb. 1. Metabolismus von Tolterodin und Fesoterodin, 5-HMT =5-Hydroxymethyl-Tolterodin
In der Regel ist der CYP2D6-Status des Patienten nicht bekannt. Der Hersteller rät von der gleichzeitigen Anwendung mit CYP3A4-Hemmern ab [5]. Sollte nämlich gemäß Fachinformation bei einem langsamen CYP2D6-Metabolisierer auch der alternative Metabolisierungsweg blockiert werden, bergen erhöhte Tolterodin-Konzentrationen das Risiko einer Überdosierung. Leider finden sich wie bei Solifenacin in der Fachinformation keine Angaben über die Auswirkungen von CYP3A4-Induktoren auf die Plasmaspiegel von Tolterodin. Zu dieser Fragestellung gibt es jedoch wichtige Hinweise in der Fachinformation von Fesoterodin. Dieses Spasmolytikum ist ein Ester-Prodrug und wird enzymatisch zum aktiven Metaboliten 5-HMT hydrolysiert (Abb. 1). Der weitere Abbau erfolgt über die Isoenzyme CYP2D6 und CYP3A4 [6, 7]. Die gleichzeitige Gabe von Rifampicin, einem starken CYP3A4-Induktor (Abb. 2), senkt die AUC (Fläche unter der Konzentrations-Zeit-Kurve) des aktiven Metaboliten um ungefähr 75% und das führt zu subtherapeutischen Plasmaspiegeln [7]. Da sowohl Tolterodin als auch 5-HMT Substrate von CYP3A4 sind, sollte die gleichzeitige Anwendung von Tolterodin mit CYP3A4-Induktoren in Analogie der Herstellerangabe zu Fesoterodin unterbleiben.
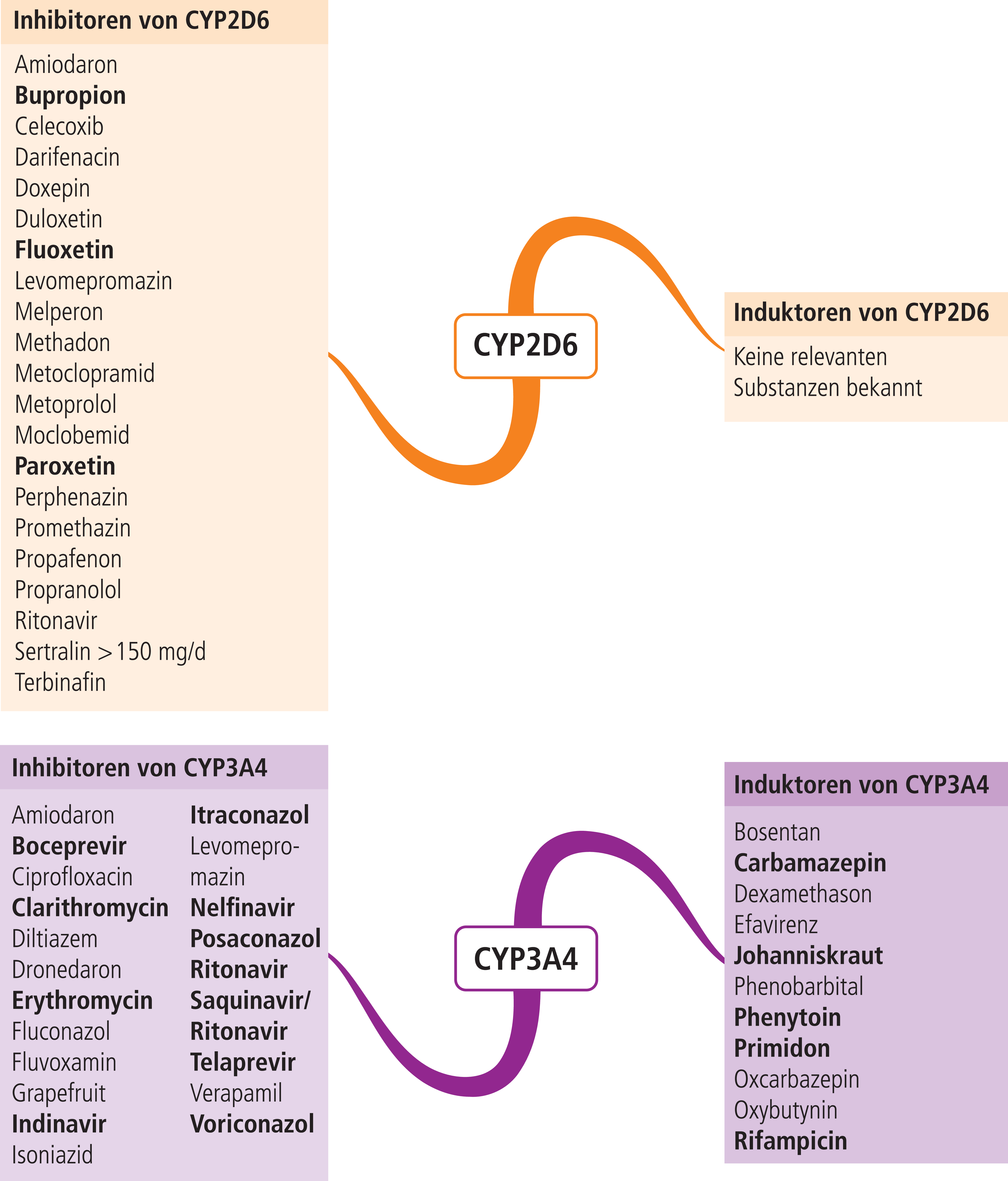
Abb. 2. Auswahl von modulierenden Substanzen (stark wirkende fettgedruckt) mit klinisch relevanter Wirkung auf einzelne CYP450-Isoenzyme (Stand: 05/2013) [Quelle: mediQ-Interaktionsprogramm]
Trospiumchlorid
Trospiumchlorid ist im Gegensatz zu den genannten urologischen Spasmolytika eine quartäre Ammoniumverbindung und überwindet nicht nennenswert die Blut-Hirn-Schranke. Zentralnervöse Nebenwirkungen sind daher unwahrscheinlich, wodurch der besondere Stellenwert in der PRISCUS-Liste erklärbar ist. Aufgrund seiner Struktur werden auch keine stoffwechselbedingten Wechselwirkungen erwartet [8].
Literatur
1. Holt S, Schmiedl S, Thürmann PA. PRISCUS-Liste. Stand 01.02.2011.
2. Fachinformation Dridase®, Stand September 2010.
3. Hiemke C, Baumann P, Bergemann N, Conca A, et al. AGNP-Konsensus-Leitlinien für therapeutisches Drug-Monitoring in der Psychiatrie: Update 2011. Psychopharmakotherapie 2012;19:91–122.
4. Fachinformation Vesikur®, Stand Januar 2013.
5. Fachinformation Detrusitol®, Stand Februar 2010.
6. Malhotra B, Gandelman K, Sachse R, Wood N, et al. The design and development of fesoterodine as a prodrug of 5-hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr Med Chem 2009;16:4481–9.
7. Fachinformation Toviaz®, Stand Oktober 2012.
8. Fachinformation Spasmex®, Stand Oktober 2012.
*Nachdruck aus Krankenhauspharmazie 2013;34:367–70.
Der Artikel wurde unter Einbeziehung von Diskussionsbeiträgen von Dr. Jörg Brüggmann, Berlin, Prof. Dr. Christoph Hiemke, Mainz, und Dr. Jochen Weber, Bad Wildungen, erstellt.
Holger Petri, Zentral-Apotheke der Wicker Kliniken, Im Kreuzfeld 4, 34537 Bad Wildungen, E-Mail: hpetri@werner-wicker-klinik.de
Psychopharmakotherapie 2013; 20(04)