Andreas Meid, Mannheim
Die aktuelle Gesetzeslage im deutschen Arzneimittelmarkt fordert die Abgabe von Arzneimitteln nach den so genannten Rabattverträgen [1]. Seit ihrer Einführung im April 2007 sind sie ein ständiger Diskussionsherd im Arzneimittelsektor in Deutschland. Apotheken sind zur Abgabe der Arzneimittel von Herstellern mit (Exklusiv-)Verträgen mit einzelnen gesetzlichen Krankenkassen verpflichtet, was vorrangig zur Kostensenkung im Arzneimittelsektor beitragen soll. Dem stehen Bedenken auf medizinischer Seite gegenüber, etwa durch Therapieumstellungen bei verunsicherten Patienten [2] (siehe Appendix 1). Patientenumfragen zufolge vermag ein solcher Austausch die Compliance zu senken (bei bis zu 65% der Patienten [3]), das Einnahmeverhalten negativ zu beeinflussen (bei bis zu 33% [3]) oder die Verträglichkeit zu verschlechtern (bei 7% bzw. 11% bei über 60-Jährigen [4]). Probleme mit der Verträglichkeit werden in anderen Untersuchungen sogar von rund 22% [5] oder 49% [6] der befragten Patienten nach einer Umstellung berichtet.
Voraussetzung für den Austausch gegen das rabattierte Arzneimittel ist dessen Bioäquivalenz mit dem „Original“, also dem erstzugelassenen Medikament mit in klinischen Studien belegtem Nutzen. Dabei spielt es keine Rolle, ob ein ärztlich verordnetes Generikum gegen ein anderes Generikum, gegen das Original oder umgekehrt ausgetauscht wird. Maßgeblich sind bioäquivalente pharmakokinetische Parameter als Surrogat für die Bioverfügbarkeit von Generikum und Original. Die pharmakokinetischen Messgrößen werden unter enorm standardisierten Bedingungen erhoben, so dass für den klinischen Einsatz wichtige Kovariablen wie Geschlecht, Alter und Gesundheitszustand unberücksichtigt bleiben. Dennoch wird bei nachgewiesener Bioäquivalenz angenommen, dass ein Austausch die klinische Wirksamkeit nicht beeinflusst.
In der Praxis kann aber nicht angenommen werden, dass ein bedenkenloser Austausch uneingeschränkt möglich ist. Erstens gilt die Bioäquivalenz nur für den Vergleich Original und Generikum, wobei die Bioäquivalenz zwischen zwei Generika fraglich bleibt. So können die Abweichungen zwischen zwei Generika deutlich größer sein. Zweitens gibt es pharmakologisch kritische Arzneistoffe, bei denen auch kleine Unterschiede in den Parametern der Bioverfügbarkeit von Bedeutung sind und beim Patienten sowohl maßgeblich die Wirksamkeit als auch die Nebenwirkungen beeinflussen [7]. Drittens können Nebenwirkungen unabhängig von pharmakologischen Grundlagen verstärkt bei verunsicherten Patienten auftreten. Diese unter einem so genannten Nozebo-Effekt leidenden Patienten zeigen zudem eine geringere Compliance und Therapie-Adhärenz (siehe Appendix 1) und gefährden damit den Therapieeffekt [8, 9].
Die Auswirkung der Rabattverträge auf die Therapietreue (Compliance, Adhärenz) gilt als unzureichend untersucht: Neben Patientenbefragungen sind praktisch keine widerspruchsfreien Quellen vorhanden [10]. Auch aus konzeptionellen Gründen sind hierzu nur eingeschränkt klinische Studien durchführbar, so dass Simulationen ein wertvolles Instrument darstellen.
Unter der Voraussetzung eingeschränkter Compliance von Patienten nach einem Arzneimittelaustausch gemäß Rabattvertrag soll daher im vorliegenden Artikel untersucht werden, inwiefern die auszutauschenden Arzneimittel in einer simulierten klinischen Studie tatsächlich auch therapeutische Äquivalenz zeigen könnten. Exemplarisch wurden die kritischen Indikationen Depression und Epilepsie gewählt. In den folgenden simulierten klinischen Studien werden Non-Compliance, Dropouts und Nebenwirkungen für die Äquivalenzuntersuchungen besonders berücksichtigt. Die verschiedenen Endpunkte werden abschließend im Hinblick auf ihre Gleichwertigkeit vor und nach dem Arzneimittelaustausch gemäß Rabattvertrag diskutiert.
Material und Methoden
Wahl der Populationen
In den Patientenkollektiven mit Epilepsie oder psychischen Erkrankungen wurden experimentell deutliche Nozebo-Effekte nachgewiesen [11] (teilweise bis zu 34% [12]). Die Patientencharakteristika bei Depression oder Angstzuständen begünstigen Nozebo-Wirkungen [13, 14], wie sie beispielsweise durch einen Austausch im Rahmen von Rabattverträgen auftreten könnten. Fallberichte und Studien bestätigen, dass durch Arzneimittelaustausch der klinische Erfolg beeinträchtigt wird [15–18], auch wenn eine verallgemeinernde Quantifizierung des Effekts aus dieser Datenlage schwierig ist [19].
Auch bei eingestellten Epileptikern kann durch eine Umstellung die Anfallsfreiheit gefährdet [20], die Anfallshäufigkeit erhöht [21] oder die Zeit bis zum Anfall verkürzt sein [22–24]. Dabei scheint sich der Nozebo-Effekt auch direkt auf die Angabe von Anfällen auszuwirken [25]: Schlafmangel, Stress- und Angstzustände sowie eine negative Erwartungshaltung, etwa durch Verunsicherung nach einem Arzneimittelaustausch gemäß Rabattvertrag, können das Auftreten epileptischer Anfälle ungünstig beeinflussen [26].
Interventionen
Den simulierten Patienten wurden vier Interventionsgruppen zuordnet. Für die Äquivalenzfragestellung relevant sind die Therapiegruppen mit der für die Patienten vertrauten Medikation (Gruppe A, Arzneimittel ohne Umstellung) und der ausgetauschten Medikation (Gruppe B, Arzneimittel nach Umstellung durch Rabattvertrag). Für die Validitätsbeurteilung der Auswertungen wurden zusätzlich als Kontrollen eine Vergleichsgruppe mit der für die Patienten vertrauten Medikation (Gruppe C) und eine Plazebo-Gruppe (Gruppe P) mitgeführt. Eine Verblindung ist bis auf die Plazebo-Gruppe konzeptionell nicht gewünscht, so dass Patienten in den Gruppen A, C und P davon ausgehen, das vertraute Arzneimittel zu erhalten, während es sich bei der Medikation in Gruppe B um einen generischen Austausch mit verändertem Erscheinungsbild handelt. Konkrete Arzneistoffe, Dosierungen, Einnahmeschemata sind für die Fragestellung nicht von Bedeutung.
Simulationsparameter und Endpunkte
Simulationsparameter für das System nach Westfall et al. [27] wurden in Anlehnung an ähnlich konzipierte Studien der Indikationen Depression [28, 29] und Epilepsie [30, 31] gewählt. Als äußerer Rahmen soll eine zwölfwöchige Studie mit sechs Visiten (zweiwöchentlich) für die Veränderung des Scores zum Ausgangswert (Baseline) einer Hamilton-Depressionsskala (HAM-D) unter einer antidepressiven Therapie und für die Zeit bis zum Anfall unter einer antiepileptischen Therapie gelten. Die Annahmen zum Wirksamkeitsparameter sind eine Reduktion zum Baseline-Wert der Hamilton-Depressionsskala um 14 Punkte (Baseline 25, Standardabweichung 1,5) für die Verum-Gruppen A, B und C sowie um 9 Punkte in der Plazebo-Gruppe P. Bei Epilepsie werden die Ereigniswahrscheinlichkeiten von Visite zu Visite über einen Grenzwert für binäre Variablen kontrolliert; in der Simulation betrug dieser Grenzwert 0,8 für die Verum-Gruppen und 0,85 für die Plazebo-Gruppe (für weitere Informationen siehe Appendix 2).
Beide Wirksamkeitsparameter werden durch einen binären Parameter für die Arzneimittelsicherheit ergänzt, welcher den Nozebo-Effekt als neu aufgetretene Nebenwirkungen mit 15% berücksichtigt. Die Compliance ist in der Gruppe nach dem Austausch durch Rabattvertrag um 20% niedriger als ohne Austausch, die Wahrscheinlichkeit für ein Ausscheiden (Dropout) ist um 5% höher. Die Korrelationen zwischen Compliance und Dropout sowie Missbefinden und Dropout betragen jeweils 0,8.
Schließlich wurden 1000 Simulationen durchgeführt, um die Variabilität von simulierten Einzelstudien in der Gesamtzahl auszugleichen.
Statistische Auswertung
Für die Verlaufsdaten im Patientenkollektiv „Depression“ wurde ein gemischtes Modell unter Berücksichtigung wiederholter Messungen gewählt (feste Effekte: Gruppe, Zeit, Gruppe x Zeit). Fehlende Werte wurden nach dem Prinzip „last observation carried forward“ (LOCF) ersetzt. In der Analyse der Ereigniszeiten im Patientenkollektiv „Epilepsie“ wurde ein Cox-Regressionsmodell für die Auswertung der Zeit bis zum epileptischen Anfall herangezogen (Kovariaten: Gruppe, Compliance). Die Fallzahlberechnung für eine TOST(two one-sided tests)-Auswertung nach Schuirmann [32] ergab mit den oben genannten Simulationsparametern bei einer Mindestpower von 90% 51 Patienten pro Gruppe für Verlaufsdaten des Patientenkollektivs „Depression“ und 46 Patienten pro Gruppe für die Ereigniszeiten des Patientenkollektivs „Epilepsie“. Die gewählte Anzahl von 50 Patienten wurde mittels optimaler Tests für den Paarvergleich [34] der Verlaufsdaten sowie für Überlebenszeitanalysen nach Wellek [33] ausgewertet. Als Äquivalenzschranke wurde im Patientenkollektiv „Depression“ der standardisierte Therapieeffekt betrachtet (ε=0,40) und im Patientenkollektiv „Epilepsie“ eine 20%ige Toleranz im Unterschied der Kaplan-Meier-Kurven gewählt (ε=0,73). In Sensitivitätsanalysen wurden die Auswertestrategie der Konfidenzintervallinklusion [35] sowie die multiple Imputation für die Ersetzung fehlender Werte betrachtet.
Alle Simulationen und statistischen Auswertungen wurden mit den SAS®-Versionen 9.2 und 9.3 durchgeführt.
Ergebnisse
Abbildung 1 zeigt beispielhaft jeweils eine ausgewählte simulierte Studie für den zeitlichen Verlauf der Score-Werte auf der Hamilton-Skala (1a) sowie die Kaplan-Meier-Kurven für die Ereigniszeiten epileptischer Anfälle (1b). In der Indikation „Depression“ wurde in Anlehnung an Originalstudien [28, 29] auf Einschlusskriterien zurückgegriffen, wonach Patienten mit mittelschwerer Ausgangslage (Baseline) in die Studie aufgenommen werden (Abb. 1a). Die Ergebnisse können ganz allgemein im Hinblick auf die Auswirkungen negativer Einflüsse auf den Therapieerfolg im zeitlichen Verlauf betrachtet werden. In der hier betrachteten Situation eines Arzneimittelaustauschs in der ambulanten Versorgung sind die Patienten vor dem Austausch idealerweise in stabiler psychischer Verfassung. Somit könnten auch Ereigniszeiten betrachtet werden, etwa die Zeit bis zur Verschlechterung bzw. Rekurrenz einer depressiven Episode. Die im selben Design durchgeführten Simulationen zur Untersuchung der Ereigniszeiten für das Patientenkolletiv „Epilepsie“ (Abb. 1b) lassen ebenso eine Abhängigkeit des Therapieerfolgs von Compliance, gefühlten Nebenwirkungen und Therapie-Adhärenz erkennen. Diese Tendenz zeigte sich konsistent in Sensitivitätsanalysen, in welchen die Auswertungsstrategie „optimaler Tests“ [34] gegen das Prinzip der Konfidenzintervallinklusion [35] getauscht oder die Ersetzung fehlender Werte durch multiple Imputation durchgeführt wurden (Daten nicht gezeigt).
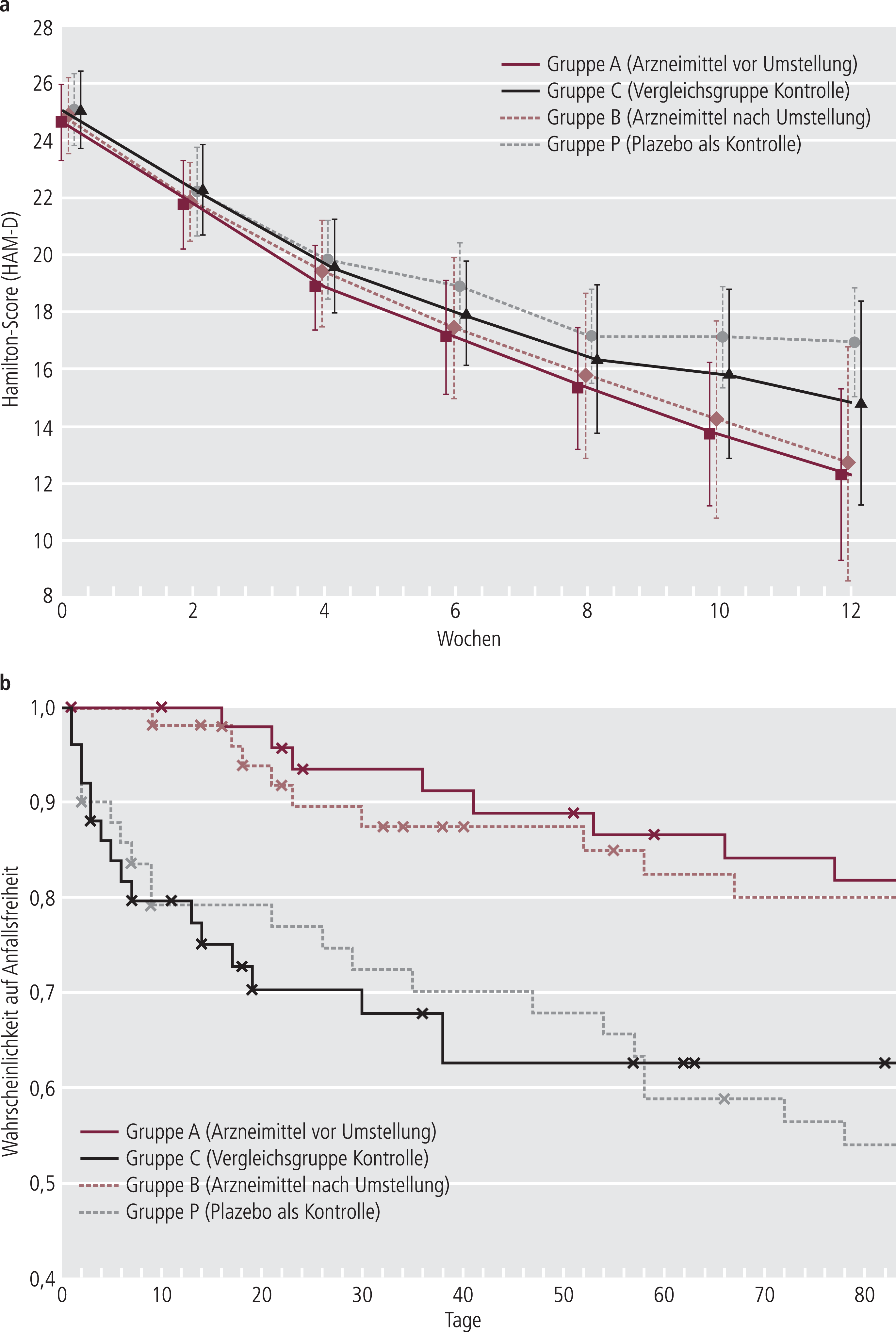
Abb. 1. Exemplarisch eine Verlaufskurve bei Depression (a) und Kaplan-Meier-Kurve bei Epilepsie (b) aus den jeweils 1000 durchgeführten Simulationen
Die Effekte im Mittel über alle 1000 Simulationen sind in Tabelle 1 dargestellt. Die tabellierten Zahlenwerte belegen, dass im Patientenkollektiv „Depression“ eine Population mit erhöhtem Risiko betrachtet wurde, weshalb in den nachfolgenden Äquivalenzuntersuchungen eine strikte Äquivalenzgrenze (ε=0,40) angewendet wurde [34]. Die mittlere ereignisfreie Zeit im Patientenkollektiv „Epilepsie“ weist im Vergleich zu anderen Population in der Indikation „Epilepsie“ (z.B. [30]) auf eine Population mit geringem bis mäßigem Risiko hin. In dieser vergleichsweise stabilen Population wurde in den nachfolgenden Äquivalenzuntersuchungen eine liberalere Äquivalenzgrenze (ε=0,73) gewählt.
Tab. 1. Darstellung der Effekte im Mittel über alle 1000 Simulationen für die Score-Werte der HAMD-Skala für das Patientenkollektiv „Depression“ sowie die mittleren ereignisfreien Tage für das Patientenkollektiv „Epilepsie“ (Zensierungen unberücksichtigt); SD: Standardabweichung
|
Patientenkollektiv „Depression“ |
„Epilepsie“ Ereignisfreie Zeit [Tage] |
||||||||||
|
HAMD-Werte der einzelnen Visiten (Wochen) |
|||||||||||
|
Gruppe |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
||||
|
A |
Mittel |
25,00 |
22,10 |
19,28 |
17,52 |
15,81 |
14,25 |
12,74 |
68,21 |
||
|
SD |
0,20 |
0,22 |
0,24 |
0,26 |
0,31 |
0,37 |
0,44 |
4,20 |
|||
|
C |
Mittel |
25,00 |
22,10 |
19,28 |
17,53 |
15,82 |
14,25 |
12,74 |
68,43 |
||
|
SD |
0,19 |
0,23 |
0,24 |
0,27 |
0,32 |
0,38 |
0,45 |
3,98 |
|||
|
P |
Mittel |
25,00 |
22,55 |
20,18 |
18,85 |
17,55 |
17,22 |
16,89 |
50,63 |
||
|
SD |
0,19 |
0,22 |
0,24 |
0,25 |
0,28 |
0,29 |
0,30 |
5,73 |
|||
|
B |
Mittel |
25,00 |
22,27 |
19,73 |
18,32 |
16,97 |
15,95 |
14,99 |
55,92 |
||
|
SD |
0,19 |
0,23 |
0,27 |
0,32 |
0,39 |
0,49 |
0,59 |
6,63 |
|||
Im Folgenden wurden die für jede Indikation durchgeführten 1000 Studiensimulationen auf Äquivalenz untersucht (Abb. 2). Dazu wurden die Gruppenvergleiche für Gruppe A (ohne Austausch) gegen C (Kontrollgruppe) als Positivkontrolle, A gegen P (Plazebo) als Negativkontrolle sowie A gegen B (nach Austausch durch Rabattvertrag) betrachtet. Die Auswertungen zeigten in den Plazebo-Vergleichen keine oder mehrheitlich keine Äquivalenz auf. Bei Betrachtung der Äquivalenzfragestellung mit der Kontrollgruppe wurde in bis nahezu 95% eine gleichwertige klinische Wirkung nachgewiesen. Der Hauptvergleich ohne und mit einem Austausch durch Rabattverträge zeigte einen klaren Trend: Hier wiesen die ausgewerteten simulierten Studien mehrheitlich Nichtäquivalenz nach (Depression: 629/1000; Epilepsie: 615/1000). Konfirmatorische Auswertungen ergaben immer ein hochsignifikantes Ergebnis (Daten nicht gezeigt). Auch der Einfluss der Compliance auf das Äquivalenzergebnis in einer logistischen Regression zeigte sich hochsignifikant (Daten nicht gezeigt). Abbildung 2 stellt auf einer Sekundärachse die mediane Compliance der Simulationen gegen die Ergebnisse der simulierten Äquivalenzuntersuchungen dar.
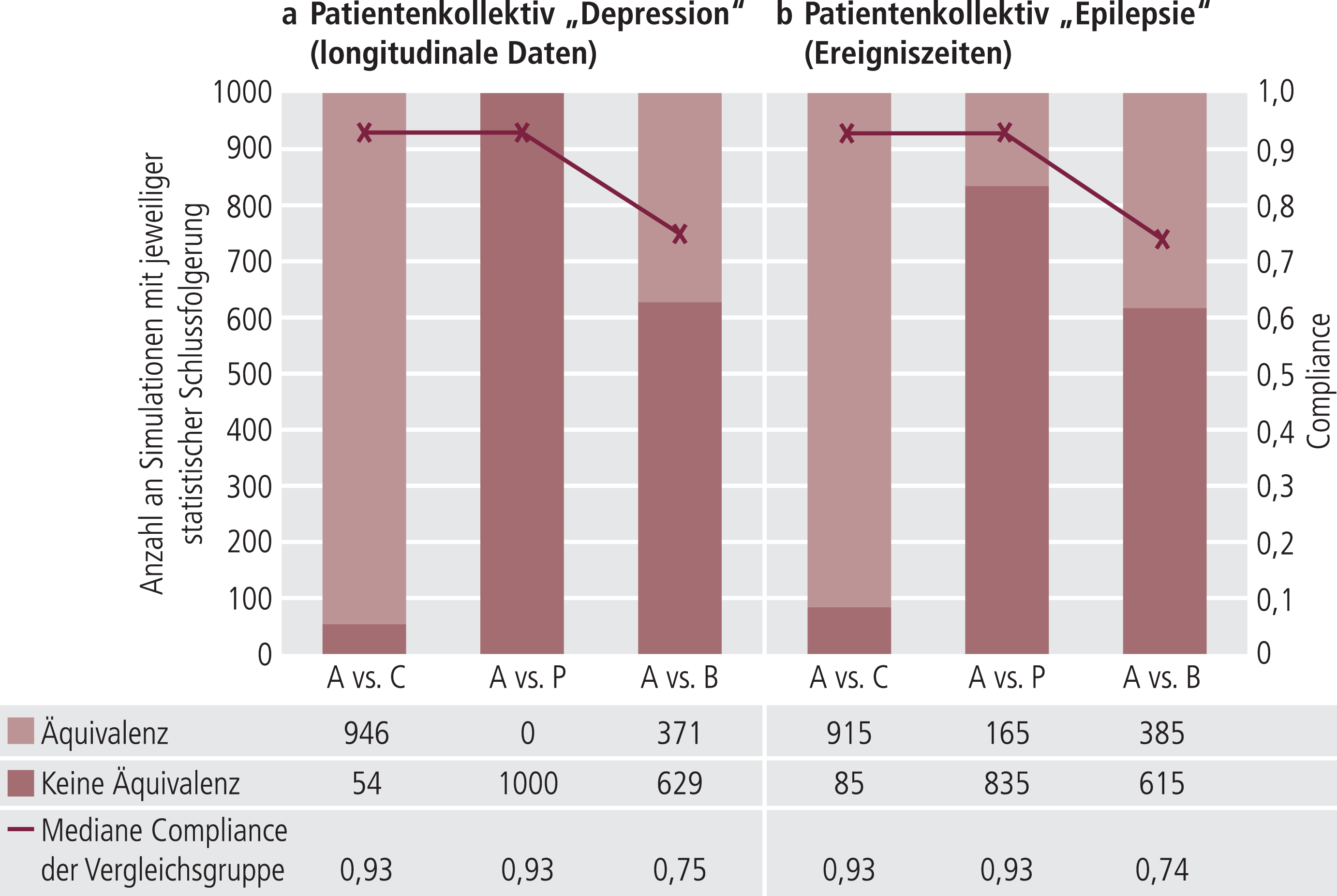
Abb. 2. Ergebnisse der Äquivalenztests in Zahlen simulierter Studien mit äquivalentem (hell) und nicht äquivalentem (dunkel) Ausgang. Dargestellt sind die Therapievergleiche der Gruppen A gegen C (Vergleichsgruppe), A gegen P (Plazebo-Vergleich) und A gegen B (Vergleich ohne und mit einem Arzneimittelaustausch gemäß Rabattvertrag).
Diskussion
Die gewonnenen Ergebnisse sind im Hinblick auf Positiv- und Negativkontrollen als valide anzusehen. In beiden Indikationen und Auswertungsstrategien sind die Therapievergleiche vor und nach einem Austausch gemäß Rabattvertrag mehrheitlich nicht äquivalent, und dies bei relativ liberal gewählten Äquivalenzgrenzen [34]. Diese Ergebnisse entstammen identischen Erwartungen zur Wirksamkeit, aber Unterschieden im Nebenwirkungsprofil, in der Compliance und Dropout-Wahrscheinlichkeit. Letztere beruhen hauptsächlich auf Fallberichten, sind in deren Spannweite aber moderat gewählt [8, 36]. Durch anders gewählte Parameter lässt sich das Simulationsergebnis zwar verändern, die Tendenz bleibt jedoch immer erhalten (Daten nicht gezeigt). Diese Tendenz weist deutlich darauf hin, dass das therapeutische Ziel nach einem Austausch durch Rabattverträge in gewissen Patientenkollektiven gefährdet sein kann.
Modellierungs- und Simulationstechniken sind ein zunehmend etabliertes und auch von Zulassungsbehörden akzeptiertes Instrument in der klinischen Forschung (für eine deutschsprachige Übersichtsarbeit siehe u.a. [37]). In der vorliegenden Arbeit sollen Erkenntnisse mittels Anwendung von Simulationstechniken über eine in der Praxis schwer umzusetzende Fragestellung gewonnen werden. Die klaren Ergebnisse können schon allein aufgrund einer gewissen Unsicherheit in den Annahmen keine Studien ersetzen. Eine solche Studie im Bereich hausärztlicher Versorgung („primary care“) wird aus verschiedenen Gründen jedoch nur schwer realisierbar sein: Zunächst wäre es von Grund auf unethisch, Nozebo-Effekte in einer klinischen Studie zu erzeugen (insbesondere mit einer Plazebo-Gruppe). Daneben kann eine auf das angesprochene Patientenkollektiv bezogene Verzerrung (Bias) durch Zuwendung kaum kontrolliert werden. Die intensive Betreuung innerhalb einer klinischen Studie kann die Annahmen zu Compliance und Adhärenz unter Studienbedingungen verfälschen [38]. Darüber hinaus bestehen unter Feldbedingungen weitere Verzerrungsmöglichkeiten und dadurch hohe Herausforderungen an die interne und externe Validität in der praktischen Durchführung einer solchen Studie.
Als Einflussfaktoren für niedrige Adhärenz und Compliance gelten unter anderem emotionale Gesundheit (Depression), persönliche Erwartungen, ein gestörtes Verhältnis zum behandelnden Personal [39], welche allesamt durch den auf Rabattverträgen beruhenden Austausch beeinflusst werden. Auf den Bereich hausärztlicher Versorgung bezogen kann ein Konditionierungseffekt (siehe Appendix 1) bei Rabattverträgen auch durch den vermittelten Eindruck einer „billigen“ und damit weniger wirksamen oder gar schädlichen Therapie entstehen [40]. Das daraus resultierende Leiden der Patienten ist jedoch real, so dass eine Verbesserung des psychosozialen Umfelds ursächlich hilfreich ist [41]. Daher sind ein vertrauensvolles Verhältnis des Patienten zu Arzt und Apotheker und eine gute Vermittlung von Informationen hervorzuheben, denn ein gut informierter Patient, der sich verstanden fühlt, ist stärker adhärent [42].
Im Artikel wurde in zwei verschiedenen Indikationen mit unterschiedlichen Auswertungsstrategien deutlich gezeigt, dass der Therapieerfolg von Compliance, gefühlten Nebenwirkungen und Therapie-Adhärenz abhängen kann. Auch wenn die simulierten Szenarien nicht auf jedes klinische Setting übertragbar sein mögen, so stehen die Ergebnisse sehr wohl im Einklang mit Umfragen und Kasuistiken und sollten somit weiter zur Sensibilisierung für das Problem beitragen. In der Praxis muss der Therapieerfolg Priorität über vordergründig ökonomische Prinzipien besitzen, gerade weil vermeintliche wirtschaftliche Vorteile kontrovers diskutiert werden [2, 43]. Vor diesem Hintergrund darf das Urteil der in der Betreuung involvierten Ärzte und Apotheker in Einzelfällen nicht durch einen starren bürokratischen Rahmen beeinträchtigt werden. Hierfür belegen die Ergebnisse der Arbeit, dass neben pharmakologisch bedingten Folgen eines Arzneimittelaustauschs bei kritischen Arzneistoffen [7] auch Aspekte der Compliance und Adhärenz sowie des Vertrauens in die Therapie eine Rolle spielen. Diese Aspekte sind ernst zu nehmen, da sie die grundlegende Annahme therapeutischer Gleichwertigkeit zweier Arzneimittel vor und nach einem Austausch gemäß Rabattvertrag in Frage stellen.
Anmerkungen
Appendix 1 zum theoretischen Hintergrund (Compliance und Adhärenz, Plazebo und Nozebo) und Appendix 2 zum methodischen Hintergrund (Clinical trial simulation) sind online zugänglich: Rubrik „Archiv/Download“ auf www.ppt-online.de
Interessenkonflikt
Keiner; Fallbeispiele und angenommene Messwerte wurden aus rein illustrativen Zwecken gewählt und haben keinen Anspruch auf Gültigkeit in realen Studien.
Literatur
Das ausführliche Literaturverzeichnis finden Sie unter "Archiv", "Literatur", Heft 4/2012
1. Nach dem GKV-Wettbewerbsstärkungsgesetz (GKV-WSG) 2007 aktuell verankert in: Sozialgesetzbuch (SGB) Fünftes Buch (V) (gesetzliche Krankenversicherung), §130a, Absatz 8, 2011.
2. May U, Kötting C, Cheraghi T. Non-Compliance als gesundheitspolitische Nebenwirkung – Demoskopie und Problemanalyse am Beispiel der Rabattverträge. Pharm Ztg 2010;6:74–9.
3. Gröber-Grätz D, Gulich M. Die medikamentöse Therapie in der hausärztlichen Versorgung unter dem Aspekt der Rabattverträge der Krankenkassen – Patientensurvey. Z Evid.Fortbild Qual Gesundhwesen (ZEFQ) 2010;104:99–105.
4. Institut für Demoskopie Allensbach. Gesundheits- und Arzneimittelversorgung in der deutschen Bevölkerung: Eine Repräsentativbefragung der Bevölkerung ab 16 Jahre (Umfrage 10042). 2009.
5. Leutgeb R, Mahler C, Laux G, Ärztenetz Weschnetz, Szecsenyi J. Krankenkassen-Rabattverträge: Probleme und Risiken für den Hausarzt bei der Betreuung chronisch kranker Patienten. Dtsch Med Wochenschr 2009;134:181–6.
6. Neises G, Menges A, Palsherm I, Stangl J, et al. Machen Rabattverträge krank? Pharm Ztg 2009;154:74–7.
7. Kojda G, Hafner D. Aut idem – Bedenkenloser Austausch bei Problem-Arzneistoffen und Therapien? Pharm Ztg 2008;25:58–62.
8. Weissenfeld J, Stock S, Lüngen M, Gerber A. The nocebo effect: A reason for patients’ non-adherence to generic substitution? Pharmazie 2010;65:451–6.
9. Liccardi G, et al. Evaluation of the nocebo effect during oral challenge in patients with adverse drug reactions. J Invest Allergol Clin Immunol 2004;14:104–7.
10. arznei-telegramm Korrespondenz. Rabattverträge: Folgen für die Patienten. arznei-telegramm 2012;43:45.
11. Ströhle A. Increased response to a putative panicogenic nocebo administration in female patients with panic disorder. J Psychiatr Res 2000;34:439–42.
12. Uhlenhuth EH, Alexander PE, Dempsey GM, Jones W, et al. Medication side effects in anxious patients: Negative placebo responses? J Affect Disord 1998;47:183–90.
13. Barsky AJ, Saintfort R, Rogers MP, Borus JF. Nonspecific medication side effects and the nocebo phenomenon. JAMA 2002;287:622–7.
14. Drici M-D, Raybaud F, De Lunardo C, Iacono P, et al. Influence of the behaviour pattern on the nocebo response of healthy volunteers. Br J Clin Pharmacol 1995;39:204–6.
15. Van Ameringen M, Mancini C, Patterson B, Bennett M. Symptom relapse following switch from Celexa to generic citalopram: An anxiety disorders case series. J Psychopharmacol 2007;21:472–6.
16. Margolese HC, Wolf Y, Desmarais JE, Beauclair L. Loss of response after switching from brand name to generic formulations: Three cases and a discussion of key clinical considerations when switching. Int Clin Psychopharmacol 2010;25:180–2.
17. Bakish D, Miller C, Hooper C, et al. A double-blind, crossover study comparing generic and brand Fluoxetine in the treatment of major depressive disorder. Int J Neuropsychopharmacol 2000;3(Suppl 1):S234–5.
18. Shields BJ, Nahata MC. Efficacy of brand-name vs. generic fluoxetine. Perspect Psychiatr Care 2003;39:122–35.
19. Borgherini G. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther 2003;25:1578–92.
20. Steinhoff BJ, Runge U, Witte OW, et al. Substitution of anticonvulsant drugs. Ther Clin Risk Manag 2009;5:449–57.
21. Fitzgerald CL, Jacobson MP. Generic substitution of levetiracetam resulting in increased incidence of breakthrough seizures. Ann Pharmacother 2011;45:e27.
22. Bazil CW. Generic substitution: are antiepileptic drugs different? Neurology 2009;5:587–8.
23. Gauthier-Lewis M, Riley TT. Generic substitution of antiepileptic medications in patients with epilepsy: is this a potential problem? Am J Health Syst Pharm 2009;66:1903–4.
24. Lelorier J, Duh MS, Paradis PE, et al. Clinical consequences of generic substitution of lamotrigine for patients with epilepsy. Neurology 2008;70:2179–86.
25. Privitera MD. Generic antiepileptic drugs: current controversies and future directions. Epilepsy Cur 2008;8:113–7.
26. Haut SR, Hall CB, Masur J, Lipton RB. Seizure occurrence: precipitants and prediction. Neurology 2007;69:1905–10.
27. Westfall PH, Tsai K, Ogenstad S, Tomoiaga A, et al. Clinical trials simulation: A statistical approach. J Biopharm Stat 2008;18:611–30.
28. Eli Lilly. A study comparing duloxetine to other antidepressants in the treatment of severe depression (TRY FIRST, Study NCT00666757) [Internet]. 2011. Available on: www.clinicaltrials.gov
29. Forest Laboratories. Escitalopram in adult patients with major depressive disorder (Study NCT00668525) [Internet]. 2011. Available von: www.clinicaltrials.gov
30. Pfizer. A prospective, observational study on the effectiveness of new antiepileptic drugs as first bitherapy in the daily clinical practice (Study NCT00855738) [Internet]. 2011. Available on: www.clinicaltrials.gov
31. Wong ICK, Mawer GE, Sander JWAS, Lhatoo SD. A pharmacoepidemiologic study of factors influencing the outcome of treatment with lamotrigie in chronic epilepsy. Epilepsia 2001;42:1354–8.
32. Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing equivalence of average bioavailability. J Pharmacokinet Biopharm 1987;15:657–80.
33. Wellek S. A log-rank test for equivalence of two survivor functions. Biometrics 1993;49:877–81.
34. Wellek S. Testing statistical hypotheses of equivalence. Chapman & Hall/CRC, 2003.
35. Westlake WJ. Use of confidence intervals in analysis of comparative bioavailability trials. J Pharm Sci 1972;61:1340–1.
36. Himmel W, Simmeroth-Nayda A, Niebling W, Ledig T, et al. What do primary care patients think about generic drugs? Int J Clin Pharmacol Ther 2005;43:472–9.
37. Meid A. Simulationstechniken zur Planung klinischer Studien – Mögliche Anwendungen zur Studienplanung bei Kindern. PZ Prisma 2012;19:116–24.
38. Clarke M, Loudon K. Effects on patients of their healthcare practitioner’s or institution’s participation in clinical trials: a systematic review. Trials 2011;12:16–26.
39. Murdaugh CL, Insel K. Problems with adherence in the elderly. In: Shumaker SA, Ockene JK, Riekert KA (eds.). The Handbook of Health Behavior Change. 3rd edition. New York: Springer Publishing Company, 2009:499–518.
40. Waber RL, Shiv B, Carmon Z, Ariely D. Commercial features of placebo and therapeutic efficacy. JAMA 2008;299:1016–7.
41. Habermann E. Gift und Nocebo. Med Klinik 1998;93:113–8.
42. Osterberg L, Blaschke T. Adherence to medication. NEJM 2005;353:487–97.
43. arznei-telegramm Korrespondenz. Rabattverträge: Segen oder Fluch. arznei-telegramm 2012;43:36–7.
Apotheker Andreas Meid cand. M.Sc. Biometry/Biostatistics, Konstanzer Straße 1–3, 68239 Mannheim, E-Mail: Andreas.Meid@gmx.de
Für die Gesetzliche Krankenversicherung stellen individuelle Arzneimittel-Rabattverträge neben anderen selektiven Vertragsoptionen eine gewichtige Rolle im Bestreben nach Wirtschaftlichkeit der einzelnen Krankenkassen dar. Allein im Jahr 2011 konnten so über 1 Mrd. Euro durch Rabattverträge der einzelnen Krankenkassen mit Arzneimittelherstellern eingespart werden. Eines der zentralen Argumente ist, dass die Substitution von generischen Arzneimitteln nicht mit Nachteilen bei der Wirksamkeiteinhergeht.
Andreas Meid wirft nun in seiner Arbeit erneut die Frage auf, welcher klinische Effekt bei einer Substitution zu erwarten ist. Die von ihm ermittelten Ergebnisse suggerieren einen signifikanten Wirksamkeitsnachteil bei durchgeführter Substitution. Tatsächlich hängt dies im Wesentlichen von den Annahmen ab, die Meid in seiner Modellierung nutzt, insbesondere aber von einem erwarteten Compliance-Nachteil sowie von höheren Abbrecher-Quoten bei erfolgter Substitution.
Meid führt selbst aus, dass nur wenig verfügbare Informationen zur Wirkung von Rabattverträgen oder genauer gesagt zu den Auswirkungen der Umstellung/Substitution auf ein vergleichbares generisches Präparat vorliegen – und die durchgeführten Studien entsprechen nicht den im Allgemeinen hohen Ansprüchen an klinische Studien. Die Frage ist, ob eine Modellierung an dieser Stelle hilfreich ist, die Evidenzlücke zu überbrücken. Sicher ist, dass Meid aufzeigen kann, wie hoch der erwartete Effekt auf die Wirksamkeit ist, wenn die Compliance nach einem Austausch um 20% vermindert ist und gleichzeitig 5% mehr Patienten die Behandlung abbrechen.
Aber sind diese Nachteile realistisch? Ohne konkrete Studiendaten – auch aus Beobachtungsstudien – wird es weiter unklar bleiben. Sicher ist aber etwas anderes: Entsprechend der vereinfachten Zulassung von generischen Arzneimitteln gehen wir von vergleichbaren Wirkprinzipien inklusive positiven und negativen Arzneimittelwirkungen aus. Dies gilt auch für die mit der Nutzung des Arzneimittels verbundene Compliance des Patienten. Die Modellierung von Meid zeigt deshalb nur auf, was sein könnte. Erst ausreichend große pragmatische Studien könnten hier Klarheit bringen. Gänzlich unbetrachtet lässt der Autor übrigens den Einfluss der ärztlichen und pharmazeutischen Beratung, der in einem Studiensetting sicherlich mit beantwortet werden müsste.
Dr. Christoph Vauth, Bremen, hkk – Erste Gesundheit, Martinistraße 26, 28195 Bremen, E-Mail: christoph.vauth@hkk.de
Therapeutic equivalence of drugs substituted under the terms of rebate contracts. Questionable clinical effectiveness after simulated substitution
Introduction: Drug delivery in the German public health sector is carried out in accordance with so-called rebate contracts. Thus, medications containing bioequivalent drug substances have to be substituted for a given statutory health insurance in accordance with its contractual pharmaceutical entrepreneur. This doesn’t account for the present medication at all. Several case studies can be found describing potential complications after such substitutions, in particular for critical dosage forms or patient groups. Since clinical studies are hardly feasible for conceptual reasons, clinical trial simulation provides a useful tool for such investigations. The possible impact of a substitution under the terms of rebate contracts is investigated by means of simulated equivalence studies.
Material and methods: The critical indications of depression and epilepsy have been studied on a patient group resembling original studies. Based on assumptions about efficacy, safety and compliance, 1,000 studies have been simulated, respectively. Analyses have been performed using mixed-effects models for the longitudinal change on the Hamilton depression scale and survival analysis (Cox regression) for the time to epileptic seizures, respectively. In order to estimate the comparison with and without a drug substitution under the terms of rebate contracts, an internal control group (the same parameters for compliance or safety) and a placebo group were carried along.
Results and implications: The internal control group and the placebo group proved validity of the analyses. By the majority of the simulations, the group after a drug substitution under the terms of rebate contracts showed no equivalence to the situation as before, regardless of the analysis method. These results confirm the relevance of case studies and make aware of this problem of primary care of the German public health sector.
Key words: Rebate contracts, generic substitution, bioequivalence, clinical equivalence, clinical trial simulation
Psychopharmakotherapie 2012; 19(04)