Christoph Hiemke, Mainz, Pierre Baumann, Lausanne, Niels Bergemann, Bad Arolsen, Andreas Conca, Bozen, Otto Dietmaier, Weinsberg, Karin Egberts, Würzburg, Mirijam Fric, Wasserburg/Inn, Manfred Gerlach, Würzburg, Christine Greiner, Bonn, Gerhard Gründer, Aachen, Ekkehard Haen, Regensburg, Ursula Havemann-Reinecke, Göttingen, Eveline Jaquenoud Sirot, Königsfelden, Hartmut Kirchherr, Bremen, Gerd Laux, Wasserburg/Inn, Ulrich C. Lutz, Tübingen, Thomas Messer, Pfaffenhofen, Matthias J. Müller, Gießen-Marburg, Bruno Pfuhlmann, Bernhard Rambeck, Bielefeld, Peter Riederer, Würzburg, Bernd Schoppek, München-Haar, Markus J. Schwarz, München, Julia Stingl, Ulm, Manfred Uhr, München, Sven Ulrich, Berlin, Roland Waschgler, Feldkirch, und Gerald Zernig, Innsbruck – TDM-Gruppe der AGNP
Für die Psychopharmakotherapie stehen derzeit etwa 130 Medikamente zur Verfügung [54]. Diese Medikamente sind für die Behandlung vieler psychiatrischer Erkrankungen und deren Symptome wirksam und wichtig. Trotz der medizinisch und wirtschaftlich außerordentlich großen Fortschritte sind die erzielten Therapieerfolge für viele Patienten noch immer nicht zufriedenstellend [5, 6, 396, 661]. Nachdem sich die klinische Forschung während der vergangenen fünf Jahrzehnte auf die Entwicklung neuer Medikamente konzentrierte [521, 522], ist inzwischen erkannt worden, dass eine verbesserte Anwendung der aktuell zugelassenen Medikamente für viele Patienten erhebliche Vorteile bringen kann [45]. Für psychiatrische Erkrankungen wurden in den letzten zehn Jahren evidenzbasierte Leitlinien für die optimale Behandlung veröffentlicht [23, 46, 101, 204, 205, 221, 254, 276, 284, 582, 585, 748].
Ein wertvolles Werkzeug zur Optimierung der Anwendung von Medikamenten unter Berücksichtigung der individuellen Besonderheiten eines Patienten ist das therapeutische Drug-Monitoring (TDM). Der Hauptgrund, TDM für die Steuerung der Psychopharmakotherapie einzusetzen, ist die erhebliche interindividuelle Variabilität der pharmakokinetischen Eigenschaften von Arzneistoffen. Bei gleicher Dosis kann ein mehr als 20-facher Unterschied in der Steady-State-Konzentration auftreten, da sich Patienten in ihrer Aufnahme, Verteilung, Verstoffwechselung und Ausscheidung der Arzneistoffe unterscheiden; Ursachen sind unter anderem Alter, Begleiterkrankungen, Begleitmedikation oder genetische Besonderheiten [61, 310, 311, 334, 335, 374]. Verschiedene galenische Formulierungen des gleichen Arzneistoffs können ebenfalls den Grad und die zeitlichen Muster der Absorption und damit die Arzneistoffkonzentration im Körper beeinflussen. TDM nutzt die Quantifizierung der Arzneistoffkonzentration im Blut (Plasma oder Serum), um die Dosis des einzelnen Patienten so zu titrieren, dass eine Wirkstoffkonzentration aufgebaut wird, bei der mit höchster Wahrscheinlichkeit mit gutem Ansprechen und guter Verträglichkeit bei minimalem Risiko für Toxizität gerechnet werden kann. Darüber hinaus besitzt TDM das bislang ungenutzte Potenzial, die Kosten-Effektivität der Psychopharmakotherapie zu steigern [660]. Für viele Psychopharmaka ist die Quantifizierung der Arzneistoffkonzentration im Blut für die Dosisanpassung klinische Routine geworden. Nachweise für den Nutzen einer TDM-geleiteten Dosiseinstellung existieren für trizyklische Antidepressiva, für eine Reihe von alten und neuen Antipsychotika sowie für konventionelle stimmungsstabilisierende Arzneistoffe [51, 459, 505]. Für Lithium ist aufgrund seines engen therapeutischen Bereichs regelmäßiges TDM obligater Bestandteil der Therapie geworden [133, 395].
Die Vorteile von TDM zur Optimierung der Pharmakotherapie können allerdings nur erreicht werden, wenn die Methode adäquat in die klinische Behandlung integriert ist. Der aktuelle Einsatz von TDM in der psychiatrischen Versorgung ist unbedingt verbesserungsbedürftig [134, 700, 742]. Ähnlich wie in anderen medizinischen Disziplinen zeigten systematische Studien, dass die inadäquate Anwendung von TDM weit verbreitet ist. Sinnlose TDM-Anforderungen vergeuden Labor-Ressourcen und tragen das Risiko, dass irreführende Ergebnisse die klinische Entscheidungsfindung negativ beeinflussen können [122]. Eine Studie über den klinischen Einsatz von TDM zeigte für trizyklische Antidepressiva in einer psychiatrischen Universitätsklinik, dass zwischen 25 und 40% der Anfragen für TDM inadäquat waren, und die Interpretation der Ergebnisse führte bei etwa 20% der Patienten zu nicht angemessenen Dosisanpassungen [700, 742]. Andere typische Fehler sind Nichteinhaltungen von Steady-State-Bedingungen und Übertragungsfehler auf dem Antragsformular [700, 743]. Neuere systematische Studien über die Anwendung von TDM ergaben im Rahmen der Behandlung mit Antidepressiva oder stimmungsstabilisierenden Medikamenten detaillierte Hinweise auf Fehler beim Einsatz von TDM [420, 421].
Vor diesem Hintergrund hat die TDM-Gruppe der Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP) im Jahr 2004 „Best Practice“-Leitlinien für TDM in der Psychiatrie publiziert [51]. Die Leitlinien sind in vielen Laboren und Kliniken in die praktische Arbeit implementiert worden. Sie wurden mehr als 200 Mal in der Literatur zitiert und ins Deutsche [312] und Französische [50] übersetzt. Außerdem gab es spezifische Beschreibungen für TDM von Antidepressiva [52]. Die AGNP-TDM-Konsensus-Leitlinien wurden auch in internationale Leitlinien zur Behandlung von psychischen Erkrankungen integriert [582]. Seit 2004 hat das Wissen über TDM in der Psychiatrie deutlich zugenommen. Es wurden neue Psychopharmaka eingeführt, für die die Anwendung von TDM ebenfalls zu empfehlen ist. Die TDM-Gruppe der AGNP hat daher ihre Leitlinien aus dem Jahre 2004 [52] umfassend aktualisiert. Sie wurden im Oktober 2011 in englischer Sprache publiziert [Hiemke et al. Pharmacopsychiatry 2011;44:195–235] und sind über die Homepage der AGNP (www.agnp.de) frei zugängig. Die vorliegende Publikation ist die deutschsprachige Version der neuen Leitlinien.
Ziele der Konsensus-Leitlinien
Das vorliegende Dokument befasst sich im ersten Teil mit theoretischen Aspekten von TDM. Der zweite Teil beschreibt Indikationen für TDM und definiert therapeutische und dosisbezogene Referenzbereiche für Neuropsychopharmaka. Der dritte Teil beschreibt die praktische Anwendung von TDM, beginnend mit der Anforderung einer Blutspiegelmessung bis hin zur klinischen Entscheidung, eine bestehende Pharmakotherapie entweder unverändert weiterzuführen oder zu ändern.
Mit dem vorrangigen Ziel, die Anwendung von TDM zu verbessern, werden folgende Inhalte behandelt:
- Indikationen für TDM in der Psychiatrie
- Graduierte Empfehlungen zur Anwendung von TDM ohne spezifische Fragestellung
- Definition therapeutischer und dosisabhängiger Referenzbereiche als Orientierungsgrößen für die TDM-gestützte Psychopharmakotherapie
- Festlegung von Warnschwellen für das Labor (laboratory alert levels), wenn Blutspiegel außergewöhnlich hoch sind und der behandelnde Arzt unverzüglich vor potenziellen Gefahren gewarnt werden muss
- Hilfen für die Interpretation der Laborbefunde
- Empfehlungen für die Kombination von TDM mit pharmakogenetischen Tests
Erstellung der Konsensus-Leitlinien
Die aktualisierten Konsensus-Leitlinien wurden von der interdisziplinären TDM-Gruppe der AGNP erarbeitet. Die Gruppe besteht aus klinisch tätigen Psychiatern, Pharmakologen, Biochemikern, Pharmazeuten und Chemikern aus akademischen und nichtakademischen Krankenhäusern und Institutionen in Deutschland, der Schweiz, Österreich und Italien. Sie sind seit vielen Jahren an der Entwicklung und Anwendung von TDM im klinischen Alltag beteiligt. Die Experten sammelten Informationen aus der Literatur und erarbeiteten die vorliegenden „Best Practice“-Leitlinien für TDM in der Psychiatrie. Da TDM für Antidepressiva, Antipsychotika und stimmungsstabilisierende Medikamente weit verbreitet ist, sind diese drei pharmakologischen Klassen umfassend in den vorliegenden Leitlinien vertreten. Anxiolytika, Hypnotika, Antidementiva, Arzneistoffe zur Behandlung von Substanzabhängigkeiten und andere psychotrope Arzneistoffe, für die TDM ebenfalls sinnvoll und nützlich sein kann, wurden mit einbezogen. In besonderen klinischen Situationen kann für jeden Arzneistoff die Messung des Blutspiegels hilfreich sein. Viele Patienten werden gleichzeitig gegen neurologische und psychiatrische Erkrankungen behandelt. Daher haben die aktualisierten Leitlinien auch Informationen über krampflösende Arzneistoffe und Parkinson-Mittel aufgenommen, für die TDM mehr oder weniger etabliert ist [481, 499]. Dies führte zu einer Erweiterung der Leitlinien von ursprünglich 65 Psychopharmaka im Jahr 2004 [51] auf nun 128 Neuropsychopharmaka.
Die im Jahr 2004 und 2005 veröffentlichten AGNP-Konsensus-Leitlinien [51, 312] sowie Leitlinien und Empfehlungen von anderen Experten für TDM in Psychiatrie und Neurologie [317, 400, 488–490, 499, 504, 505] bildeten die Grundlage für das Update. Eine umfangreiche Literaturrecherche, vor allem in MEDLINE, wurde durchgeführt, um TDM-relevante Informationen für die insgesamt 128 ausgewählten neuropsychiatrischen Arzneistoffe zu identifizieren. Die Suche konzentrierte sich auf Begriffe wie „optimal plasma concentrations“, „dose related drug plasma concentrations“, „cytochrome P450 substrate, inducer and inhibitor properties“ und „ratios of concentrations of drug metabolites to parent drugs“. Nach relevanter Literatur wurde auch von Hand in pharmakologischen und klinisch-chemischen Fachzeitschriften gesucht. Mehr als 1000 Artikel wurden identifiziert, beurteilt und ausgewertet. Die extrahierten Daten wurden von sieben Autoren in fünf Tabellen eingetragen. Die Ergebnisse wurden an 20 Mitglieder der TDM-Gruppe gesandt, welche anhand einer Checkliste die extrahierten und analysierten Daten beurteilten. Eine Internet-basierte und Passwort-geschützte Plattform wurde eingerichtet, um den Zugang zur Literatur zu ermöglichen. Die Protokolle und Kommentare der Gutachter wurden an alle Autoren dieser Leitlinien verteilt. Die Abstimmungen über die Inhalte in diesem Dokument erfolgten während zweier Konsensus-Konferenzen und per E-Mail-Kommunikation. Besonders intensiv wurden dabei Definitionen, Referenzwerte, Warnschwellen für das Labor und literaturbasierte, graduierte Empfehlungen für die Anwendung von TDM bearbeitet.
Theoretische Hintergründe
Pharmakokinetik von Psychopharmaka
Die meisten Psychopharmaka zeichnen sich in ihren pharmakokinetischen Eigenschaften aus durch:
- gute Resorption aus dem Gastrointestinaltrakt; der Blutspiegel erreicht nach oraler Einnahme das Maximum innerhalb von 1 bis 6 Stunden
- sehr variabler First-Pass-Metabolismus (systemische Bioverfügbarkeit von 5 bis 90%)
- schnelle Verteilung aus dem Blut in das zentrale Nervensystem mit 2- bis 40-fach höheren Konzentrationen im Gehirn als im Blut
- hohes Verteilungsvolumen im lipophilen Kompartiment (ca. 10 bis 50 l/kg)
- niedrige Talspiegel im Steady-State (ca. 0,1 bis 500 ng/ml für psychiatrische und bis zu 20 µg/ml für neurologische Arzneistoffe)
- langsame, vorwiegend hepatische Elimination aus dem Blut (Halbwertszeit 12 bis 36 Stunden)
- lineare Pharmakokinetik bei therapeutischen Dosen; eine Verdoppelung der täglichen Dosis führt zu einer Verdoppelung des Blutspiegels
- geringe renale Ausscheidung mit entsprechend geringem Einfluss einer Niereninsuffizienz auf die Blutspiegel der Muttersubstanz und ihrer aktiven Metaboliten
- Enzyme der Cytochrom-P450-Familie und UDP-Glucuronosyltransferasen sind die wesentlichen Enzyme für die Metabolisierung
Es gibt jedoch zahlreiche Ausnahmen. Zum Beispiel haben Venlafaxin, Nefazodon, Trazodon, Tranylcypromin, Moclobemid, Quetiapin, Ziprasidon und Rivastigmin kurze Eliminationshalbwertszeiten von 2 bis 10 Stunden, während Aripiprazol und Fluoxetin sehr lange Eliminationshalbwertszeiten aufweisen (72 Stunden für Aripiprazol und 3 bis 15 Tage für Fluoxetin, unter Berücksichtigung seines aktiven Metaboliten Norfluoxetin). Amisulprid, Milnacipran, Memantin, Gabapentin oder Sulpirid werden nicht oder nur unwesentlich hepatisch metabolisiert und hauptsächlich unverändert renal ausgeschieden. Paroxetin weist durch die Hemmung des eigenen Metabolismus eine nichtlineare Pharmakokinetik auf, da ein Stoffwechselprodukt irreversibel an das metabolisierende Enzym bindet und dieses inhibiert [69].
Viele Psychopharmaka kommen als razemische Verbindungen zum Einsatz. Ihre Enantiomere unterscheiden sich allerdings deutlich in ihrer Pharmakodynamik und -kinetik [53, 605]. Bisher ist jedoch nur für Methadon, Methylphenidat und Flupentixol eine TDM-Analytik der Enantiomere etabliert [39, 189]. Die wirksamen Enantiomere von razemischem Methadon und Flupentixol sind (R)-Methadon bzw. cis-(Z)-Flupentixol. Für andere razemische Psychopharmaka, wie Citalopram, Paroxetin, Reboxetin, Venlafaxin, Paliperidon oder Amitriptylin-Metaboliten werden stereoselektive Analysen ausschließlich im Rahmen von Forschungsprojekten und anderen speziellen Anwendungen durchgeführt.
Die meisten Psychopharmaka unterliegen einem Phase-I-Metabolismus durch oxidative (z.B. Hydroxylierung, Dealkylierung, Oxidation zu N-Oxiden, S-Oxidation zu Sulfoxiden oder Sulfonen), reduktive (z.B. Carbonyl-Reduktion zu sekundären Alkoholen) oder hydrolytische Reaktionen. Phase-I-Reaktionen werden überwiegend durch Isoenzyme des Cytochrom-P450(CYP)-Systems katalysiert. Der Mensch besitzt 39 Gene, die für funktionelle CYP-Enzyme kodieren. Die für Psychopharmaka wichtigsten Isoenzyme sind CYP1A2, CYP2B6, CYP2D6, CYP2C9, CYP2C19 und CYP3A4/5 (Tab. 1) [745–747]. In der Regel führen Phase-I-Reaktionen eine polare funktionelle Gruppe ein, was eine Konjugation mit stark polaren Molekülen wie Glucuronsäure oder Schwefelsäure in der Phase-II-Reaktion ermöglicht. Bei den meisten Psychopharmaka ist die Glucuronidierung einer Hydroxyl- (z.B. Oxazepam oder Lorazepam) oder Aminofunktion (z.B. Olanzapin) die wesentliche Phase-II-Reaktion. Darüber hinaus können tertiäre Aminogruppen konjugiert werden, indem quartäre Ammonium-Glucuronide gebildet werden. Phase-II-Enzyme sind im Hinblick auf ihre Substratspezifitäten schlecht charakterisiert. Es scheint bezüglich Substratspezifität eine große Überlappung zwischen den Isoenzymen zu bestehen [143].
Tab. 1. Neuropsychopharmaka und Enzyme der Metabolisierung
|
Arzneistoff (aktive Metaboliten) |
Enzyme, Art der Metabolisierung |
Referenz |
|
Acamprosat |
Keine Metabolisierung |
[578] |
|
Agomelatin |
CYP1A2, CYP2C19 |
[78] |
|
Amantadin |
Keine wesentliche Metaboliserung |
[24] |
|
Alprazolam |
CYP3A4/5 |
[17, 496] |
|
Amisulprid |
Keine wesentliche Metaboliserung |
[566] |
|
Amitriptylin und Amitriptylinoxid (Amitriptylin, Nortriptylin) |
CYP1A2, CYP2C9, CYP2C19, CYP2D6, |
[90, 650, 713] |
|
Aripiprazol (Dehydroaripiprazol) |
CYP2D6, CYP3A4 |
[306, 701] |
|
Asenapin |
Glucuronosyltransferase und CYP1A2 |
[707] |
|
Atomoxetin |
CYP2D6 |
[446] |
|
Benperidol |
Unklar |
[589] |
|
Benserazid |
Hydroxylierung (Isoenzym unbekannt), COMT |
[347] |
|
Biperiden |
Hydroxylierung (Isoenzym unbekannt) |
[628] |
|
Bromocriptin |
CYP3A4 |
[513] |
|
Bromperidol |
CYP3A4 |
[230, 633, 645, 736] |
|
Brotizolam |
CYP3A4 |
[655] |
|
Buprenorphin (Norbuprenorphin) |
CYP2C8, CYP3A4 |
[79, 454] |
|
Bupropion (Hydroxybupropion) |
CYP2B6 |
[309] |
|
Buspiron |
CYP3A4 |
[416] |
|
Cabergolin |
Hydrolyse, CYP3A4 |
[167] |
|
Carbidopa |
Unklarer Metabolismus 1/3 wird unverändert eliminiert |
[575] |
|
Carbamazepin, CBZ |
CYP1A2, CYP2B6, CYP2C8, CYP3A4/5 |
[360, 497] |
|
Chlorpromazin |
CYP1A2, CYP2D6 |
[724] |
|
Citalopram |
CYP2C19, CYP2D6, CYP3A4 |
[97, 227, 739] |
|
Clomipramin (Norclomipramin) |
CYP1A2, CYP2C19, CYP2D6, CYP3A4 |
[244] |
|
Clomethiazol |
CYP2A6, CYP2B6, CYP3A4 |
[116] |
|
Clozapin |
CYP1A2, CYP2C19, CYP3A4 |
[334, 487] |
|
Desipramin |
CYP2D6 |
[244] |
|
Diazepam (Nordazepam, Oxazepam, Temazepam) |
CYP2B6, CYP2C19, CYP3A4 |
[228, 704] |
|
Dihydroergocryptin |
CYP3A4 |
[19, 162] |
|
Diphenhydramin |
CYP2D6 |
[13] |
|
Disulfiram |
CYP1A2, CYP2B6, CYP2E1, CYP3A4 |
[412] |
|
Donepezil |
CYP2D6, CYP3A4 |
[681] |
|
Dothiepin = Dosulepin |
CYP2C19, CYP2D6 |
[740] |
|
Doxepin (Nordoxepin) |
CYP2C9, CYP2C19, CYP2D6 |
[295, 365] |
|
Duloxetin |
CYP1A2, CYP2D6 |
[405] |
|
Entacapon |
Glucuronosyltransferase |
[387] |
|
Escitalopram |
CYP2C19, CYP2D6, CYP3A4 |
[662, 697] |
|
Fluoxetin (Norfluoxetin) |
CYP2B6, CYP2C9, CYP2C19, CYP2D6 |
[404, 588] |
|
Flupentixol |
CYP2D6 |
[148,365] |
|
Fluphenazin |
CYP2D6 |
[746] |
|
Fluvoxamin |
CYP2D6, CYP1A2 |
[354, 450] |
|
Gabapentin |
Wird unverändert renal eliminiert |
[77] |
|
Galantamin |
CYP2D6, CYP3A4 |
[34] |
|
Haloperidol |
CYP2D6, CYP3A4 |
[93, 645] |
|
Iloperidon |
CYP2D6, CYP3A4 |
[106] |
|
Imipramin (Desipramin) |
CYP1A2, CYP2C19, CYP2D6, CYP3A4 |
[244, 413] |
|
Lamotrigin |
Glucuronosyltransferase, CYP2A6 |
[121] |
|
Levodopa |
Dopadecarboxylase, COMT, MAO |
[575] |
|
Levomepromazin |
CYP1A2, CYP2D6 |
[36] |
|
Levomethadon |
CYPC19, CYP2B6, CYP3A4, CYP2D6 |
[145] |
|
Lisurid |
CYP3A4, CYP2D6 |
[539] |
|
Lithium |
Keine Metabolisierung, renale Ausscheidung |
[256, 619] |
|
Lorazepam |
Glucuronosyltransferase |
[164, 196] |
|
Maprotilin (Normaprotilin) |
CYP2D6, CYP1A2 |
[86] |
|
Melatonin |
CYP1A2 |
[296] |
|
Memantin |
Keine wesentliche Metabolisierung |
[251] |
|
Methadon |
CYP2B6, CYP2C19, CYP3A4, CYP2D6 |
[145] |
|
Methylphenidat |
Carboxylesterase 1 |
[468] |
|
Mianserin |
CYP2D6, CYP1A2, CYP3A4 |
[379] |
|
Midazolam |
CYP3A4 |
[220] |
|
Milnacipran |
Kein CYP-abhängiger Metabolismus |
[495, 533] |
|
Mirtazapin |
CYP3A4, CYP1A2, CYP2B6, CYP2D6 |
[397, 630] |
|
Moclobemid |
CYP2C19, CYP2D6 |
[255] |
|
Modafinil |
Amidhydrolyse, CYP3A4 |
[561] |
|
Naltrexon |
Aldoketoreductase AKR1C4 |
[92] |
|
Nortriptylin |
CYP2D6 |
[385, 485, 687] |
|
Olanzapin |
N-Glucuronosyltransferase, Flavinmonooxigenase, CYP1A2, CYP2D6 |
[107] |
|
Opipramol |
CYP2D6 |
Fachinformation |
|
Paliperidon (= 9-Hydroxyrisperidon) |
60% werden unverändert eliminiert, CYP3A4 |
[161] |
|
Paroxetin |
CYP1A2, CYP2D6, CYP3A4 |
[209, 349, 691] |
|
Perazin |
CYP1A2, CYP2C19, CYP3A4, Flavinmonooxigenase |
[629, 725] |
|
Pergolid |
CYP3A4 |
[731] |
|
Perphenazin |
CYP1A2, CYP2C19, CYP2D6, CYP3A4 |
[12, 77, 168, 486] |
|
Pregabalin |
Unverändert renal eliminiert |
[77] |
|
Piribedil |
Demethylierung, p-Hydroxylierung, N-Oxidation |
[168] |
|
Pimozid |
CYP1A2, CYP3A4 |
[171] |
|
Pramipexol |
Keine Metabolisierung |
[62] |
|
Promazin |
CYP1A2, CYP2C19, CYP3A4 |
[726] |
|
Promethazin |
CYP2D6 |
[465] |
|
Quetiapin (Norquetiapin) |
CYP3A4, CYP2D6 |
[38] |
|
Rasagilin |
CYP1A2 |
[277] |
|
Reboxetin |
CYP3A4 |
[307, 716] |
|
Risperidon, RIS (9-hydroxy-RIS) |
CYP2D6, CYP3A4 |
[732] |
|
Ropinirol |
CYP1A2 |
[357] |
|
Rotigotin |
Glucuronosyltransferase, verschiedene andere Abbauwege, Enzyme unbekannt |
[115] |
|
Selegilin |
CYP2B6 |
[60] |
|
Sertindol |
CYP3A4, CYP2D6 |
[729] |
|
Sertralin |
CYP2B6, CYP2C19, CYP2C9, CYP2D6 |
[482, 705] |
|
Thioridazin |
CYP1A2, CYP2C19, CYP2D6, CYP3A4 |
[648, 714] |
|
Tiaprid |
Keine relevante Metabolisierung |
[477] |
|
Tolcapon |
Glucuronyltransferase |
[387] |
|
Trimipramin (Nortrimipramin) |
CYP2C19, CYP2D6, CYP2C9 |
[187] |
|
Tranylcypromin |
Monoaminoxidase, unklar |
[37] |
|
Trazodon |
CYP3A4, CYP2D6 |
[268, 567] |
|
Valproinsäure |
Glucuronosyltransferase, CYP2A6, CYP2B6, CYP2C9, Beta-Oxidation |
[641] |
|
Venlafaxin (O-Desmethylvenlafaxin) |
CYP2C9, CYP2C19, CYP2D6, CYP3A4 |
[217, 434] |
|
Zaleplon |
Aldehydoxidase, CYP3A4 |
[554] |
|
Ziprasidon |
CYP3A4, Aldehydoxidase |
[58, 519] |
|
Zolpidem |
CYP1A2, CYP2C9, CYP3A4 |
[698] |
|
Zopiclon |
CYP2C8, CYP3A4 |
[57, 659] |
|
Zotepin |
CYP1A2, CYP2D6, CYP3A4 |
[596] |
|
Zuclopenthixol |
CYP2D6 |
[330] |
Wenn Enzyme, die fett gedruckt sind, in ihrer Aktivität durch eine Mutation (genetisch) oder durch einen Inhibitor oder Induktor (siehe Tab. 2) vermindert oder gesteigert sind, so kommt es zu einem Anstieg oder Abfall der Plasmakonzentration und daraus folgend zu einer Wirkungsverstärkung, Unverträglichkeit oder Intoxikation bzw. einer Wirkungsabschwächung.
CYP: Cytochrom P450; COMT: Catechol-O-methyltransferase; MAO: Monoaminoxidase
Erstellt durch CH, überprüft und ergänzt durch EJS
Andere enzymatische Systeme wie Aldo-Keto-Reductasen [43], die zur Reduzierung von Ziprasidon zu seinem Dihydro-Derivat [58] oder von Naltrexon zu Naltrexol [92] führen, können ebenfalls beteiligt sein. Ein anderes Beispiel sind die Monoaminoxidasen (MAO) A und B, die Citalopram stereoselektiv zu einem inaktiven sauren Metaboliten deaminieren [562].
Medikamente werden vorwiegend in der Leber und in geringem Maße in extrahepatischen Geweben wie Darmschleimhaut oder Gehirn metabolisiert [59, 238, 444]. Inter- und intra-individuelle Unterschiede in der Plasmakonzentration von Psychopharmaka, das heißt die pharmakokinetische Variabilität, werden durch unterschiedliche Aktivitäten der metabolisierenden Enzyme verursacht. Die Enzymaktivität kann mit zunehmendem Alter sinken und durch Nieren- und Lebererkrankungen verändert sein [374]. Auch wurden geschlechtsspezifische Unterschiede für Psychopharmaka berichtet, aber die Ergebnisse sind widersprüchlich und die klinische Relevanz ist bislang unklar [7–9, 608].
Bei einer Reihe psychoaktiver Arzneistoffe tragen deren Metaboliten zur klinischen Gesamtwirkung des Medikaments bei. Aus diesem Grund muss TDM auch die Quantifizierung der aktiven Metaboliten mit einschließen – beispielsweise im Fall von Clomipramin (Norclomipramin), Doxepin (Nordoxepin), Fluoxetin (Norfluoxetin) oder Risperidon (9-Hydroxyrisperidon). Für Arzneistoffe wie Sertralin oder Clozapin ist die klinische Relevanz ihrer Metaboliten (Norsertralin bzw. Norclozapin) unklar. Doch auch die Analyse pharmakologisch inaktiver Metaboliten liefert nützliche Informationen über den metabolischen Zustand des Patienten oder dessen Compliance [105, 569].
Tabelle 2 zeigt die „normalen“ Verhältnisse der Konzentrationen von Metabolit zur jeweiligen Muttersubstanz. Die berechneten Bereiche beinhalten 68% der unter Standard-Dosierungen zu erwartenden Verhältnisse unter Annahme einer Normalverteilung, das heißt Mittelwert minus eine Standardabweichung bis Mittelwert plus eine Standardabweichung. Ein Verhältnis über oder unter dem „normalen“ Verhältnis Metabolit zu Muttersubstanz (Tab. 2) kann auf eine unregelmäßige Medikamenteneinnahme [546], auf eine Stoffwechselstörung aufgrund einer genetischen Variation [157, 159, 350, 592] oder auf eine Wechselwirkung mit anderen Arzneimitteln hinweisen. Spina und Mitarbeiter haben dies für die Umwandlung von Desipramin zu 2-Hydroxydesipramin gezeigt [618]. Bei Wechselwirkungen zwischen Arzneistoffen erhöht sich das Verhältnis zwischen Metabolit und Muttersubstanz, wenn die enzymatische Metabolisierung durch Einnahme weiterer Medikamente oder pharmakokinetisch relevante Lebensgewohnheiten, wie beispielsweise Rauchen, induziert wird. Arzneistoffe und Substanzen, welche die Stoffwechselenzyme hemmen, führen zu einer Abnahme des Verhältnisses. In Tabelle 3 sind Arzneistoffe angegeben, die in therapeutischen Konzentrationen Inhibitoren und Induktoren von CYP-Enzymen sind.
Tab. 2. Normalbereiche der Verhältnisse der Blutspiegel Metabolit zu Muttersubstanz für Psychopharmaka mit messbaren Metaboliten. Angegeben sind Mittelwertsbereiche±einfache Standardabweichung. Diese Werte sind „normalerweise“ (ohne Begleitmedikamente oder genetisch bedingte pharmakokinetische Besonderheiten) bei 68% der Patienten zu erwarten.
|
Arzneistoff |
Metabolit |
Konzentrationen (MW–SD bis MW+SD) |
Referenz |
|
Amitriptylin |
Nortriptylin* |
0,2–1,8 (n=83) |
[545] |
|
Aripiprazol |
Dehydroaripiprazol (*) |
0,3–0,5 (n=283) PM von CYP2D6: 0,2 (n=8) |
[306, 368, 452] |
|
Bromperidol |
Reduziertes Bromperidol |
0,11–0,51 (n=31) |
[609, 633] |
|
Buprenorphin |
Norbuprenorphin |
0,8–2,0 (n=5) |
[383] |
|
Bupropion |
Hydroxybupropion* |
5–47 (24 h, n=9) 6–30 (12 h, n=9) |
[152, 253] |
|
Buspiron |
6-Hydroxybuspiron |
25–53 (n=20) |
[178] |
|
Carbamazepin |
Carbamazepin-10,11-epoxid |
0,07–0,25 (n=14) |
[338] |
|
Citalopram |
N-Desmethylcitalopram |
0,31–0,60 (n=2 330) |
[549] |
|
Clomipramin |
Norclomipramin* |
0,8–2,6 (n=115) |
[545] |
|
Clozapin |
Norclozapin (*) |
Nichtraucher: 0,5–0,6 (n=98) Raucher: 0,4–0,7 (n=198) |
[308, 500] |
|
Dothiepin |
Nordothiepin |
0–1,4 (n=50) |
[325] |
|
Doxepin |
Nordoxepin |
0,6–1,6 (n=12) PM CYP2C19: 1,8 (n=4) PM CYP2D6: 0,8 (n=6) |
[172, 363] |
|
Escitalopram |
N-Demethylescitalopram |
0,3–1,0 (n=243) |
[548] |
|
Fluoxetin |
Norfluoxetin* |
0,7–1,9 (n=334) |
[545] |
|
Fluvoxamin |
Fluvoxaminsäure |
0–1,2 (n=49) |
[237] |
|
Haloperidol |
Reduziertes Haloperidol |
MW 0,6 |
[673] |
|
Imipramin |
Desipramin* |
0,6–3,2 (n=14) PM CYP2D6 4,1 (n=2) |
[95, 96, 632] |
|
Maprotilin |
N-Desmethylmaprotilin |
1,1–3,7 (n=76) PM CYP2D6 4,9 |
[699] |
|
Mianserin |
N-Desmethylmianserin |
0,5–0,8 (n=182) |
[545] |
|
Mirtazapin |
N-Desmethylmirtazapin |
0,2–1,2 (n=100) |
[591] |
|
Moclobemid |
Moclobemid-N-oxid |
0,8–2,5 (n=6) |
[291] |
|
Olanzapin |
N-Demethylolanzapin |
Nichtraucher: 0,1–0,3 (n=76) Raucher: 0,2–0,4 (n=69) |
[602] |
|
Perazin |
Desmethylperazin |
1,1–3,3 (n=27) |
[91] |
|
Perphenazin |
N-Dealkylperphenazin |
0,6–2,8 (n=54) |
[637] |
|
Quetiapin |
Norquetiapin |
0,1–3,8 (n=25) |
[723] |
|
Reboxetin |
O-Desethylreboxetin |
<0,1 |
[484] |
|
Risperidon |
9-Hydroxyrisperidon* |
EM or IM CYP2D6: 1,5–10,0 PM CYP2D6: ≤1 |
[159, 677] |
|
Risperidon Depot |
9-Hydroxyrisperidon* |
EM: 1,2–4,3 |
[469] |
|
Sertindol |
Dehydrosertindol |
1,1–2,7 (n=6) PM CYP2D6: 1,0 |
[729] |
|
Sertralin |
Norsertralin |
1,7–3,4 (n=348) |
[546] |
|
Trazodon |
m-Chlorophenylpiperazin (mCPP) |
0,04–0,22 (Gesamtbereich) |
[328] |
|
Trimipramin |
Nortrimipramin (*) |
0,26–0,56 (n=25) |
[191] |
|
Venlafaxin |
|
EM or IM CYPD26: 0,3–5,2 PM CYP2D6: ≤0,3 UM CYP2D6: >5,2 |
[592] |
|
0,46–1,48 |
*Pharmakologisch aktiver Metabolit, (*) aktiver Metabolit in vitro, aber unklare Bedeutung in vivo
Wenn Standardabweichungen (SD) für die Verhältnisse (Ratios) der Konzentrationen aus der Literatur nicht verfügbar waren, sondern nur Mittelwerte (MW) und SD für die Einzelsubstanzen, wurden die Bereiche nach dem Gauss‘schen Fehlerfortpflanzungsgesetz berechnet: SD Ratio=[(SD Muttersubstanz x MW Metabolit) + (SD Metabolit x MW Muttersubstanz)]/(MW Metabolit)2
Erstellt durch CH, überprüft durch Sonja Brünen, Christiane Knoth und Elnaz Ostad Haji.
Tab. 3. Inhibitoren und Induktoren von Enzymen, die am Abbau von Psychopharmaka beteiligt sind
|
Inhibierende Arzneistoffe |
Inhibierte Enzyme |
Induzierende Arzneistoffe |
Induzierte Enzyme |
|
Amiodaron |
CYP2C9, CYP2D6, CYP3A4 |
Carbamazepin |
CYP1A2, CYP2B6, CYP2C9, CYP3A4 |
|
Bupropion |
CYP2D6 |
Dexamethason |
CYP2C9, CYP3A4 |
|
Bromocriptin |
CYP3A4 |
Efavirenz |
CYP2B6, CYP3A4 |
|
Chinidin |
CYP2D6 |
Ethanol |
CYP2E1 |
|
Cimetidin |
CYP1A2, CYP2D6, CYP3A4 |
Ginkgo biloba |
CYP2E1 |
|
Ciprofloxacin |
CYP1A2 |
Isoniazid |
CYP2C19 |
|
Clarithromycin |
CYP3A4 |
Johanniskraut |
CYP2C19, CYP3A4 |
|
Clopidogrel |
CYP2B6 |
Oxybutynin |
CYP3A4 |
|
Disulfiram |
CYP2E1 |
Phenobarbital |
CYP2C9, CYP2C19, CYP3A4 |
|
Duloxetin |
CYP2D6 |
Phenytoin |
CYP2B6, CYP2C9, CYP2C19, CYP3A4 |
|
Enoxacin |
CYP1A2 |
Primidon |
CYP2C9, CYP2C19, CYP3A4 |
|
Erythromycin |
CYP3A4 |
Rauch |
CYP1A2 |
|
Felbamat |
CYP2C19 |
Rifabutin |
CYP3A4 |
|
Fluconazol |
CYP2C19, CYP2C9, CYP3A4 |
Rifampicin |
CYP1A2, CYP2B6, CYP2C9, CYP2C19 |
|
Fluoxetin und Norfluoxetin |
CYP2D6, CYP2C19 |
Ritonavir |
CYP2B6, CYP2C9, CYP3A4 |
|
Fluvoxamin |
CYP1A2, CYP2C9, CYP2C19, CYP3A4 |
||
|
Indinavir |
CYP3A4 |
||
|
Isoniazid |
CYP1A2, CYP2A6, CYP2C19, CYP3A4 |
||
|
Itraconazol |
CYP2B6, CYP3A4 |
||
|
Ketoconazol |
CYP3A4 |
||
|
Levomepromazin |
CYP2D6 |
||
|
Melperon |
CYP2D6 |
||
|
Metoclopramid |
CYP2D6 |
||
|
Metoprolol |
CYP2D6 |
||
|
Miconazol |
CYP2C9, CYP2C19 |
||
|
Mifepriston |
CYP3A4 |
||
|
Moclobemid |
CYP2C19, CYP2D6 |
||
|
Nelfinavir |
CYP3A4 |
||
|
Norfloxacin |
CYP1A2 |
||
|
Omeprazol |
CYP2C19 |
||
|
Paroxetin |
CYP2D6 |
||
|
Perazin |
CYP1A2 |
||
|
Pergolid |
CYP2D6 |
||
|
Perphenazin |
CYP2D6 |
||
|
Propafenon |
CYP1A2, CYP2D6 |
||
|
Propranolol |
CYP2D6 |
||
|
Ritonavir |
CYP2D6, CYP3A4 |
||
|
Saquinavir |
CYP3A4, CYP2C9 |
||
|
Ticlopidin |
CYP2B6 |
||
|
Troleandomycin |
CYP3A4 |
||
|
Valproinsäure |
CYP2C9 |
||
|
Verapamil |
CYP3A4 |
||
|
Voriconazol |
CYP2C9, CYP3A4 |
Bei Kombination der oben angegebenen Inhibitoren oder Induktoren kann es bei Einnahme von Psychopharmaka, die bevorzugt über das inhibierte bzw. induzierte Enzym abgebaut werden (siehe Tab. 1) zu klinisch relevanten pharmakokinetischen Arzneimittelwechselwirkungen kommen. Letzteres wurde geprüft mit den Interaktionsdatenbanken www.mediq.ch und www.psiac.de. Erstellt durch CH, geprüft durch EJS
Pharmakogenetik von Neuropsychopharmaka
Pharmakogenetische Faktoren sind in den vergangenen Jahren als wesentlich für die Pharmakokinetik und Pharmakodynamik von Neuropsychopharmaka erkannt worden [199, 457]. Einige CYP-Isoenzyme weisen eine hohe genetische Variabilität auf [745–747]. Wenn eine Abweichung in der Allelfrequenz in mindestens 1% der Bevölkerung auftritt, wird dies als genetischer Polymorphismus bezeichnet.
Die Anzahl der aktiven Allele in einem Gen bestimmt, wie viel Enzymprotein exprimiert wird. Langsame Metabolisierer (poor metaboliser, PM) weisen eine mangelnde funktionelle Allelexpression auf; intermediäre Metabolisierer (IM) sind entweder genetisch heterozygot, also Träger eines aktiven und eines inaktiven Allels (oder eines Allels mit reduzierter Aktivität) oder zweier Allele mit reduzierter Aktivität. Extensive Metabolisierer (EM) besitzen den Wildtyp mit zwei aktiven Allelen; bei ultra-schnellen Metabolisierern (UM) findet man verstärkte enzymatische Aktivität durch Amplifikation der funktionellen Allele [66]. Die Genpolymorphismen der metabolisierenden Enzyme können klinisch äußerst relevant sein. So können bei PM unerwartete Nebenwirkungen und Toxizität durch überhöhte Blutspiegel und bei UM fehlendes Ansprechen aufgrund subtherapeutischer Blutspiegel auftreten.
Sogenannte Prodrugs sind inaktive Vorstufen eines Arzneistoffs, die erst durch die Verstoffwechselung aktiviert werden. Beispiele sind Codein oder Clopidogrel, welche durch CYP2D6 bzw. CYP2C19 aktiviert werden. PM-Patienten sind nicht in der Lage solche Prodrugs zu pharmakologisch aktiven Metaboliten umzubauen.
Andere Enzymsysteme, wie UDP-Glucuronosyltransferasen, zeigen ebenfalls genetische Polymorphismen [155], deren klinische Relevanz in der Pharmakopsychiatrie bislang allerdings unklar ist.
Die Verfügbarkeit von Methoden zur CYP-Genotypisierung nimmt stetig zu, und es wurden bereits Leitlinien für ihren Einsatz in der klinischen Praxis veröffentlicht [675]. Die funktionelle Bedeutung vieler Genotypen ist jedoch nach wie vor unklar. Für einige Enzyme, wie CYP3A4, ist trotz großer interindividueller Variabilität des Phänotyps kein genetischer Polymorphismus zuzuordnen. Daher kann es vorteilhaft sein, Methoden zur Phänotypisierung mit Modellsubstanzen wie Coffein für CYP1A2, Omeprazol für CYP2C19, Dextromethorphan für CYP2D6 oder Midazolam für CYP3A4/5 zu verwenden [403, 643]. Eine solche Phänotypisierung analysiert die aktuelle metabolische Situation eines Patienten. Der Phänotyp kann durch Umweltfaktoren wie Rauchen oder Begleitmedikamente beeinflusst werden [201, 601, 749]. Mit der Genotypisierung wird ein „trait marker“ identifiziert, der nicht durch Umweltfaktoren beeinflusst wird. Die Genotypisierung kann mit und ohne Medikamenteneinnahme durchgeführt werden und ihr Ergebnis ist lebenslang gültig.
Neuere Untersuchungen zeigen, dass auch der Efflux-Transporter P-Glycoprotein (P-gp) in der Darmschleimhaut und Blut-Hirn-Schranke für die pharmakokinetische Variabilität von Psychopharmaka relevant ist [1]. Dieses Transportprotein, ein Mitglied der Transporter-Familie mit einer ATP-bindenden Cassette (ABC), wird vom sogenannten Multidrug-Resistenz-Gen (ABCB1 oder MDR1) kodiert. Es existiert ein Genpolymorphismus, der aktuell ausschließlich durch Genotypisierung identifizierbar ist. Eine Phänotypisierung (z.B. mit Digoxin) ist in der Praxis nicht anwendbar [129, 183, 210, 389]. Für den Genpolymorphismus von P-gp gibt es Hinweise, dass die klinische Relevanz ähnlich hoch sein könnte wie die von CYP-Enzymen. Für Antidepressiva, die Substrate von P-gp sind, wurde eine Abhängigkeit der Arzneimittelwirkung vom P-gp-Genotyp nachgewiesen [668]. Auch für Quetiapin gibt es Hinweise, dass sowohl der Blutspiegel von Quetiapin als auch die antipsychotische Wirksamkeit vom P-gp-Genotyp des behandelten Patienten abhängen [470]. Im Hinblick auf das Auftreten erwünschter und unerwünschter klinischer Effekte von Psychopharmaka weisen erste Berichte auf einen Einfluss des Genpolymorphismus von P-gp hin [279, 560].
Dosis und Wirkstoffkonzentration im Blut
In aller Regel wird für die TDM-geleitete Dosis-Optimierung eine Blutspiegelmessung unter Steady-State-Bedingungen vorgenommen. Steady State ist erreicht, wenn die Arzneistoffaufnahme gleich der Arzneistoffausscheidung ist. Nach wiederholter Medikamenteneinnahme ist das Steady State nach vier Eliminationshalbwertszeiten zu 94% erreicht und nach fünf Eliminationshalbwertszeiten zu 97%. Für mehr als 90% aller Psychopharmaka ist bei regelmäßig eingenommener Erhaltungsdosis das Steady State innerhalb von einer Woche erreicht.
Die Dosis, die erforderlich ist, um eine gewünschte Steady-State-Konzentration eines Arzneimittels zu erreichen, kann berechnet werden, wenn das Dosierungsintervall (t), die Clearance (CL) und die Bioverfügbarkeit (F) für das Medikament bekannt sind. Die Berechnung basiert darauf, dass die Konzentration im Blut (c) proportional zur Erhaltungsdosis (De) im Steady State ist. Wenn die totale Clearance des Wirkstoffs (CLt) bekannt ist, gilt folgende Gleichung:
De = D×F/t = c×CLt
Basierend auf diesem Zusammenhang ist es auch möglich, die dosisabhängige Wirkstoffkonzentration im Blut zu berechnen, die in Blutproben von Patienten unter Medikation mit einer bestimmten Dosis zu erwarten ist [285]:
c = De/CLt
Für jedes Arzneimittel stehen solche Daten zur Verfügung, da sie bei der Beantragung der Zulassung vorgelegt werden müssen. Sie stammen meist aus pharmakokinetischen Studien an gesunden Probanden oder Patienten, die mit fixen Dosen behandelt wurden. Wenn die Clearance des Medikaments als arithmetischer Mittelwert ± einfache Standardabweichung ermittelt wurde, kann daraus ein dosisbezogener Referenzbereich für ein Normalkollektiv berechnet werden [285].
Definition des dosisbezogenen Referenzbereichs
Der „dosisbezogene Referenzbereich“ wird in den vorliegenden Leitlinien als Konzentrationsbereich angegeben, der für ein Arzneimittel bei einer bestimmten Dosis im Blut zu erwarten ist. In diesem „dosisbezogenen Referenzbereich“ sollten 68% aller Wirkstoffkonzentrationen enthalten sein, mit denen unter Standardbedingungen im Blut eines „normalen“ Patienten zu rechnen ist. Dabei ist „normal“ durch die Population der jeweiligen klinischen Studie definiert. Das Studienkollektiv besteht üblicherweise aus 18 bis 65 Jahre alten Personen ohne relevante Komorbidität, Komedikation oder genetische Anomalien im Arzneimittel-Metabolismus.
In Tabelle 4 sind Faktoren für die Berechnung der dosisbezogenen Referenzbereiche für die wichtigsten Psychopharmaka dargestellt. Die zu erwartenden dosisbezogenen Referenzbereiche werden durch Multiplikation der Faktoren „C/Dlow“ bzw. „C/Dhigh“ mit der täglichen Dosis berechnet. Man muss sich dabei allerdings bewusst sein, dass viele Patienten im klinischen Kontext nicht alle oben genannten Bedingungen erfüllen.
Tab. 4. Gesamtclearance (CLt), Bioverfügbarkeit (F), Dosierungsintervall (τ) und Faktoren (C/Dlow and C/Dhigh) zur Berechnung von dosisbezogenen Plasmakonzentrationen (C/D) von Psychopharmaka
|
Arzneistoff |
N |
CLt–SD bis CLt+SD [ml/min] |
F |
τ [h] |
C/Dlow [ng/ml/mg] |
C/Dhigh [ng/ml/mg] |
Literatur |
|
Antidepressiva |
|||||||
|
Amitriptylin |
8 |
198–373 |
0,5 |
24 |
0,93 |
1,75 |
[165] |
|
Amitriptylinoxid |
12 |
331–539 |
0,8 |
24 |
1,03 |
1,68 |
[384] |
|
Bupropion |
17 |
2500–11300 |
1,0 |
24 |
0,06 |
0,28 |
[665] |
|
Citalopram |
8 |
367–545 |
0,8 |
24 |
1,02 |
1,51 |
[616] |
|
Clomipramin |
9 |
583–933 |
0,5 |
24 |
0,37 |
0,60 |
[198] |
|
Desipramin |
12 |
1633–2333 |
0,5 |
24 |
0,15 |
0,21 |
[2] |
|
Desvenlafaxin |
7 |
233–396 |
1,0 |
24 |
1,75 |
2,98 |
[520] |
|
Dothiepin = Dosulepin |
22 |
674–3960 |
0,3 |
24 |
0,05 |
0,31 |
[740] |
|
Doxepin |
85 |
769–2644 |
1,0 |
24 |
0,18 |
0,27 |
[100] |
|
Duloxetin |
12 |
610–1733 |
0,5 |
24 |
0,20 |
0,57 |
[600] |
|
Escitalopram |
24 |
360–960 |
0,8 |
24 |
0,58 |
1,54 |
[607] |
|
Fluoxetin |
n.b. |
600–833 |
0,7 |
24 |
0,60 |
0,83 |
[18] |
|
Fluvoxamin |
6 |
807–1960 |
1,0 |
24 |
0,35 |
0,86 |
[163] |
|
Imipramin |
n.b. |
791–1029 |
0,4 |
24 |
0,28 |
0,37 |
[100] |
|
Maprotilin |
6 |
503–1747 |
0,8 |
24 |
0,32 |
1,10 |
[415] |
|
Mianserin |
n.b. |
843–1 948 |
0,3 |
24 |
0,11 |
0,25 |
[137] |
|
Mirtazapin |
10 |
455–945 |
0,5 |
24 |
0,37 |
0,85 |
[651] |
|
Nordoxepin |
85 |
504–2738 |
1,0 |
24 |
0,25 |
1,38 |
[445] |
|
Nortriptylin |
n.b. |
300–1117 |
0,5 |
24 |
0,31 |
1,16 |
[664] |
|
Paroxetin |
30 |
1561–10856 |
1,0 |
24 |
0,06 |
0,44 |
[213] |
|
Reboxetin |
n.b. |
22–51 |
1,0 |
24 |
12,55 |
31,10 |
[141] |
|
Sertralin |
|
1313–2213 (m) |
1,0 |
24 |
0,31 |
0,53 |
[565] |
|
793–2357 (f) |
1,0 |
24 |
0,29 |
0,88 |
||
|
Trazodon |
8 |
73–103 |
1,0 |
24 |
6,72 |
9,47 |
[473] |
|
Trimipramin |
12 |
898–1215 |
0,40 |
24 |
0,23 |
0,31 |
[165, 364] |
|
Venlafaxin O-Desmethylvenlafaxin |
18 |
747–1540 |
1,0 |
24 |
0,45 |
0,93 |
[372] |
|
315–618 |
1,0 |
24 |
1,12 |
2,2 |
|||
|
Antipsychotika |
|||||||
|
Amisulprid |
78 |
520–693 |
0,5 |
24 |
0,50 |
0,67 |
[566] |
|
Asenapin |
n.b. |
867–1190 |
0,35 |
24 |
0,18 |
0,28 |
[707] |
|
Aripiprazol |
6 |
47–70 |
0,9 |
24 |
8,63 |
12,85 |
[417] |
|
Benperidol |
14 |
1073–2240 |
0,5 |
24 |
0,15 |
0,31 |
[589] |
|
Bromperidol |
14 |
3570–7938 |
1,0 |
24 |
0,09 |
0,19 |
[390] |
|
Chlorpromazin |
11 |
1043–1510 |
0,1 |
24 |
0,05 |
0,07 |
[738] |
|
Chlorprothixen |
3 |
918–1448 |
0,2 |
24 |
0,10 |
0,15 |
[534] |
|
Clozapin |
16 |
258–728 |
0,5 |
24 |
0,48 |
1,35 |
[128, 176, 332] |
|
Flupentixol |
3 |
440–490 |
0,6 |
24 |
0,85 |
0,95 |
[348] |
|
Fluphenazindecanoat |
12 |
2380–3940 |
1,0 |
24 |
0,18 |
0,29 |
[197] |
|
Haloperidol |
6 |
420–680 |
0,6 |
24 |
0,61 |
0,99 |
[123] |
|
Haloperidoldecanoat |
420–680 |
1,0 |
336 672 |
0,073 0,036 |
0,118 0,059 |
[123] |
|
|
Melperon |
6 |
1484–2898 |
0,6 |
24 |
0,14 |
0,28 |
[83] |
|
Levomepromazin |
8 |
913–4737 |
0,5 |
24 |
0,07 |
0,38 |
[149] |
|
Olanzapin |
491 |
233–637 |
0,8 |
24 |
0,87 |
2,38 |
[67] |
|
Paliperidon |
n.b |
31–98 |
0,3 |
24 |
1,99 |
6,31 |
[161] |
|
Perphenazin |
8 |
1009–2566 |
0,4 |
24 |
0,11 |
0,28 |
[195] |
|
Pimozid |
7 |
21–553 |
0,5 |
24 |
0,64 |
16,53 |
[581] |
|
Quetiapin |
10 |
1146–2421 |
1,0 |
24 |
0,13 |
0,21 |
[7, 435] |
|
Risperidon, oral |
8 |
91–171 |
0,7 |
24 |
3,50 aktive Fraktion |
14,00 aktive Fraktion |
[159] |
|
Risperidon Depot |
n.b. |
91–171 |
1,0 |
336 |
0,29 aktive Fraktion |
0,55 aktive Fraktion |
[606] |
|
Sertindol |
6 |
133–600 |
1,0 |
24 |
1,16 |
5,22 |
[728] |
|
Sulpirid |
6 |
331–499 |
0,25 |
24 |
0,35 |
0,52 |
[717] |
|
Thioridazin |
11 |
404–982 |
0,60 |
24 |
0,42 |
1,03 |
[117] |
|
Zotepin |
14 |
467–10267 |
1,0 |
24 |
0,07 |
1,49 |
[642] |
|
Ziprasidon |
12 |
303–397 |
0,6 |
24 |
1,05 |
1,36 |
FI |
|
Zuclopenthixol |
8 |
867–2300 |
0,4 |
24 |
0,13 |
0,35 |
[337] |
|
Antikonvulsiva, Stimmungsstabilisierer |
|||||||
|
Carbamazepin |
n.b. |
58–74 |
1,0 |
24 |
9,40 |
11,93 |
FI |
|
Felbamat |
10 |
29,1–33,3 |
1,0 |
24 |
20,85 |
23,86 |
[556] |
|
Lamotrigin |
129 |
22–49 |
1,0 |
24 |
14,09 |
31,28 |
[118] |
|
Levetiracetam |
216 |
52–72 |
1,0 |
24 |
9,65 |
13,35 |
[535] |
|
Lithium |
n.b. |
10–40 |
1,0 |
24 |
17,36 |
69,44 |
[706] |
|
Oxcarbazepin |
7 |
1703–5063 |
1,0 |
24 |
0,14 |
0,41 |
[319, 694] |
|
Primidon |
8 |
30–47 |
1,0 |
24 |
14,78 |
23,15 |
[423] |
|
Topiramat |
6 |
21–31 |
1,0 |
24 |
22,47 |
33,55 |
[179] |
|
Valproinsäure |
9 |
4,5–9,8 |
1,0 |
24 |
71,23 |
154,32 |
[682] |
|
Anxiolytika und Hypnotika |
|||||||
|
Alprazolam |
6 |
34–83 |
0,8 |
24 |
6,73 |
16,53 |
[496, 604] |
|
Bromazepam |
10 |
50–91 |
1,0 |
24 |
7,67 |
13,95 |
[352] |
|
Brotizolam |
8 |
85–141 |
0,7 |
24 |
4,93 |
8,17 |
[341] |
|
Buspiron |
41 |
1260–2702 |
0,04 |
24 |
0,01 |
0,02 |
[41] |
|
Clonazepam |
9 |
63–90 |
0,8 |
24 |
6,17 |
8,82 |
[259] |
|
Diazepam |
48 |
10–43 |
0,9 |
24 |
14,53 |
62,50 |
[264] |
|
Lorazepam |
15 |
36–109 |
0,8 |
24 |
5,10 |
15,43 |
[266] |
|
Oxazepam |
|
36–167 |
0,8 |
24 |
3,33 |
15,22 |
[260] |
|
29–109 |
0,8 |
24 |
5,12 |
18,90 |
||
|
Triazolam |
13 |
326–584 |
0,9 |
24 |
1,01 |
1,81 |
[263] |
|
Zaleplon |
10 |
868–1330 |
0,3 |
24 |
0,16 |
0,25 |
[265] |
|
Zolpidem |
10 |
266–364 |
0,67 |
24 |
1,28 |
1,75 |
[265] |
|
Zopiclon |
10 |
250–883 |
1 |
24 |
0,79 |
2,78 |
[411] |
|
Antidementiva |
|||||||
|
Donepezil |
14 |
112–217 |
1,0 |
24 |
3,20 |
6,20 |
[463] |
|
Galantamin |
8 |
268–400 |
1,0 |
24 |
1,74 |
2,59 |
[744] |
|
Rivastigmin |
20 |
29–64 (Pflaster, TTS) |
0,5 |
24 |
5,43 |
11,97 |
[391] |
|
Arzneimittel zur Behandlung Substanz-bezogener Abhängigkeitserkrankungen |
|||||||
|
Acamprosat |
24 |
1741–4221 |
1,0 |
24 |
0,16 |
0,40 |
[287] |
|
Buprenorphin |
6 |
356–947 |
0,4 |
24 |
0,29 |
0,78 |
FI |
|
Bupropion |
17 |
2500–11300 |
1,0 |
24 |
0,06 |
0,28 |
[665] |
|
Methadon |
12 |
75–148 |
0,95 |
24 |
4,46 |
8,80 |
[474, 727] |
|
Naltrexon 6β-Naltrexol |
453 |
2077–2590 |
1,0 |
24 |
0,27 |
0,33 |
[182] |
|
928–1242 |
0,56 |
0,75 |
|||||
|
Vareniclin |
1878 |
170–176 |
1,0 |
24 |
3,95 |
4,08 |
[540] |
FI: Fachinformation; n.b.: nicht berichtet
Dosisbezogene Referenzbereiche werden erhalten durch Multiplikation von C/Dlow mit der Dosis (unterer Wert) bzw. C/Dhigh mit der Dosis (oberer Wert). Arzneimittel der Tabelle 5 wurden in diese Tabelle nicht aufgenommen, wenn keine Clearance-Daten aus der Literatur verfügbar waren.
Erstellt durch EH und CG, überprüft und ergänzt durch CH.
Arzneistoffspiegel im Blut und im Gehirn
Die pharmakologische Aktivität eines Psychopharmakons hängt von der Verfügbarkeit im Zielorgan Gehirn ab. Allerdings ist das Gehirn vom Blut durch zwei Barrieren, die Blut-Hirn-Schranke (BHS) und die Blut-Liquor-Schranke getrennt, die vom Arzneistoff überwunden werden müssen [154]. Die meisten Psychopharmaka gelangen aufgrund ihrer hohen Fettlöslichkeit durch passive Diffusion ins Gehirn. Die BHS ist eine physikalische Barriere, die das zirkulierende Blut vom zentralen Nervensystem trennt. Sie besteht aus kapillären Endothelzellen, die miteinander durch tight junctions verbunden sind [154]. Sie begrenzt effizient den Austausch von gelösten Stoffen zwischen Blut und extrazellulärer Flüssigkeit im Gehirn und schützt dadurch das Gehirn vor potenziell schädlichen Chemikalien. Wie bereits erwähnt, sind eine Reihe von psychoaktiven Arzneistoffen wie Risperidon, Aripiprazol oder Venlafaxin Substrate von P-gp [180, 370, 668]. Als Konsequenz können die Verhältnisse der Gehirn- zu Blutkonzentrationen von Psychopharmakon zu Psychopharmakon ganz unterschiedlich sein, obwohl sie ähnliche physikochemische Eigenschaften (Octanol-Wasser-Verteilung) besitzen. In Tierstudien fanden sich Verhältnisse von 0,22 für Risperidon [29] bis 34 für Fluphenazin [27]. Trotz der sehr unterschiedlichen Verhältnisse von Gehirn- zu Blutspiegel der verschiedenen Psychopharmaka haben Tierversuche gezeigt, dass Steady-State-Blutspiegel von Psychopharmaka sehr gut mit den Konzentrationen im Gehirn korrelieren. Diese Korrelationen sind besser als die zwischen Dosis und Gehirnspiegel. Dies wurde für trizyklische Antidepressiva [249], Trazodon [173] und Olanzapin [28] gezeigt. Der Blutspiegel eines Arzneistoffs kann daher als ein zuverlässiger Surrogat-Marker für Steady-State-Konzentrationen im Gehirn betrachtet werden.
Arzneistoffkonzentration im Blut und die Besetzung von Zielstrukturen im Gehirn
Die Positronen-Emissions-Tomographie (PET) ermöglicht die Analyse der Rezeptor-Besetzung des zentralen Nervensystems in vivo [207, 274]. Antipsychotika wirken vorwiegend über die Blockade von D2-artigen Dopamin-Rezeptoren. Die Blockade der D2-Rezeptoren durch Antipsychotika reduziert die Bindung von radioaktiven PET-Liganden [207, 272]. Mit diesem Ansatz und in Verbindung mit der Quantifizierung der Verdrängung von Dopamin-Rezeptor-Radioliganden hat sich gezeigt, dass die Blutspiegel von Psychopharmaka sehr gut mit der Rezeptor-Besetzung im Gehirn korrelieren. In Übereinstimmung mit der hohen Variabilität der Arzneistoffkonzentrationen im Blut unter gleichen Dosen wurde festgestellt, dass die Rezeptor-Besetzung besser mit Blutspiegeln als mit der Tagesdosis korreliert [313]. Eine optimale klinische Wirksamkeit wurde bei 70 bis 80% Rezeptor-Besetzung gesehen, wobei ab einer Rezeptor-Besetzung von 80% die Schwelle für das Auftreten von extrapyramidalen Nebenwirkungen definiert wurde [207, 480].
PET wurde auch verwendet, um in vivo die Serotonin-Transporter-Besetzung mit SSRI zu analysieren [442, 443]. Mit Hilfe eines Serotonin-Transporter-Radioliganden wurde gezeigt, dass die Blutspiegel von Citalopram, Paroxetin, Fluoxetin und Sertralin gut mit der Serotonin-Transporter-Besetzung korrelieren. Dabei zeigte sich, dass für eine optimale klinische Wirkung eine Belegung von mindestens 80% erreicht werden sollte [442, 443]. PET-Untersuchungen haben somit für zahlreiche Antipsychotika und Antidepressiva äußerst relevante Informationen für die Festlegung therapeutisch wirksamer Blutspiegel geliefert [274].
„Therapeutisches Fenster“ – therapeutischer Referenzbereich
TDM basiert auf der Annahme, dass es einen Zusammenhang zwischen dem Blutspiegel und der klinischen Wirkung gibt (Besserung, unerwünschte Wirkungen). Es wird außerdem angenommen, dass es einen Plasmakonzentrationsbereich des Wirkstoffs gibt, das sogenannte „therapeutische Fenster“, der sich durch maximale Effektivität bei guter Verträglichkeit und Sicherheit auszeichnet. Studien über die Beziehungen zwischen Plasmakonzentration und klinischer Besserung haben dieses Konzept seit den 60er-Jahren für Lithium, trizyklische Antidepressiva und klassische Antipsychotika bestätigt. Systematische Übersichtsarbeiten und Metaanalysen haben überzeugend nachgewiesen, dass es eine signifikante Beziehung zwischen Plasmakonzentration und klinischer Wirkung für Nortriptylin, Imipramin und Desipramin gibt [51]. Für Amitriptylin hat eine Metaanalyse von 45 Studien gezeigt, dass verschiedene statistische Ansätze zu nahezu identischen Ergebnissen führen [672, 674]. Für neue Antipsychotika wie Aripiprazol [612], Olanzapin [509] oder Risperidon [737] wurde ebenfalls eine enge Beziehung zwischen Plasmakonzentration und klinischer Wirksamkeit berichtet.
Für den anschaulichen Begriff „therapeutisches Fenster“ gibt es viele Synonyme, wie beispielsweise „therapeutischer Referenzbereich“, „therapeutischer Bereich“, „optimale Plasmakonzentration“, „wirksamer Blutspiegel“, „Zielkorridor“, „Zielspiegel“ oder „orientierender therapeutischer Bereich“ [51]. Der vorliegende Konsens verwendet den Begriff „therapeutischer Referenzbereich“ in Übereinstimmung mit Leitlinien für TDM für Antiepileptika [499]. Der „therapeutische Referenzbereich“ wurde in dieser Konsensus-Leitlinie für neuropsychiatrische Arzneistoffe wie folgt definiert:
Definition des therapeutischen Referenzbereichs
Die „therapeutischen Referenzbereiche“, die in dieser Leitlinie angegeben werden (Tab. 5), definieren Bereiche von Wirkstoffkonzentrationen im Blut mit einer unteren Grenze, unterhalb derer eine durch das Medikament induzierte therapeutische Reaktion relativ unwahrscheinlich ist, und einer oberen Grenze, ab der die Verträglichkeit abnimmt oder oberhalb derer es relativ unwahrscheinlich ist, dass eine therapeutische Verbesserung erreicht werden kann.
Tab. 5. Empfohlene Referenzbereiche, Warnschwellen für das Labor und literaturbasierte Empfehlungen zur Anwendung von TDM
|
Arzneistoffe und aktive Metaboliten |
Therapeutischer Referenz-bereich (Konsensus) |
t1/2 |
Warnschwelle für das Labor (Konsensus) |
Empfehlung zur Anwendung von TDM* (Konsensus) |
CF (nmol/l = CF x ng/ml) |
Literatur |
Kommentar |
|
Antidepressiva |
|||||||
|
Agomelatin |
7–300 ng/ml 1–2 h nach Einnahme von 50 mg |
1–2 h |
600 ng/ml |
4 |
4,11 |
[78] |
Wegen der raschen Elimination sind keine messbaren Talspiegel unter Dauereinnahme zu erwarten; Messungen, bevorzugt von Cmax, sollten auf spezifische Fragestellungen begrenzt sein |
|
Amitriptylin plus Nortriptylin |
80–200 ng/ml |
10–28 h 30 h |
300 ng/ml |
1 |
3,41 3,61 |
[282, 502, 672] |
|
|
Bupropion plus Hydroxbupropion |
225–1500 ng/ml |
8–26 h 17–47 h |
2000 ng/ml |
3 |
4,17 3,91 |
[151, 152, 636, 529, 636] |
Bupropion, und in gewissem Umfang auch sein Metabolit, sind bei Raumtemperatur instabil; Plasma oder Serum müssen gefroren aufbewahrt werden (mindestens bei –20°C) |
|
Citalopram |
50–110 ng/ml |
33 h |
220 ng/ml |
2 |
3,08 |
[42, 73, 111, 339, 388, 442, 471, 491, 549, 598] |
N-demethylierte Metaboliten scheinen nicht zur pharmakologischen Wirkung beizutragen; veränderte Elimination in PM oder schnellen Metabolisierern (CYP2C19) |
|
Clomipramin plus Norclomipramin |
230–450 ng/ml |
16–60 h 36 h |
450 ng/ml |
1 |
3,18 3,32 |
[239] |
Hydroxylierte Metaboliten |
|
Desipramin |
100–300 ng/ml |
15–18 h |
300 ng/ml |
2 |
3,75 |
[502] |
Veränderte Elimination in PM oder UM (CYP2D6) |
|
Desvenlafaxin |
100–400 ng/ml |
11 h |
800 ng/ml |
2 |
3,80 |
[520] |
|
|
Dosulepin = Dothiepin |
45–100 ng/ml |
18–21 h |
200 ng/ml |
2 |
3,39 |
[102, 325, 414, 541] |
|
|
Doxepin plus Nordoxepin |
50–150 ng/ml |
15–20 h |
300 ng/ml |
2 |
3,58 3,77 |
[172, 321, 393, 445] |
Veränderte Elimination in PM oder UM |
|
Duloxetin |
30–120 ng/ml |
9–19 h |
240 ng/ml |
2 |
3,36 |
[21, 640, 703] |
Keine aktiven Metaboliten, beschleunigte Elimination bei Rauchern [222] |
|
Escitalopram |
15–80 ng/ml |
30 h |
160 ng/ml |
2 |
3,08 |
[409, 679] |
N-demethylierte Metaboliten scheinen nicht zur pharmakologischen Wirkung beizutragen; veränderte Elimination in PM oder schnellen Metabolisierern (CYP2C19) |
|
Fluoxetin plus Norfluoxetin |
120–500 ng/ml |
4–6 Tage 4–16 Tage |
1000 ng/ml |
2 |
3,23 3,39 |
[84, 187, 410, 442, 545] |
Lange Eliminationshalbwertszeit von Norfluoxetin und lang anhaltende Hemmung von CYP2D6 beachten |
|
Fluvoxamin |
60–230 ng/ml |
20 h |
>500 ng/ml |
2 |
3,14 |
[353, 587, 631, 634, 639] |
Hemmung von CYP1A2, CYP2C19 |
|
Imipramin plus Desipramin |
175–300 ng/ml |
11–25 h 15–18 h |
300 ng/ml |
1 |
3,57 3,75 |
[72, 229, 245, 510, 538] |
Hydroxylierte Metaboliten |
|
Maprotilin |
75–130 ng/ml |
20–58 h |
220 ng/ml |
2 |
3,60 |
[231, 321, 384] |
Aktiver Metabolit |
|
Mianserin |
15–70 ng/ml |
14–33 h |
140 ng/ml |
3 |
3,78 |
[191, 192, 453] |
|
|
Milnacipran |
50–110 ng/ml |
5–8 h |
220 ng/ml |
2 |
2,24 |
[206, 315] |
|
|
Mirtazapin |
30–80 ng/ml |
20–40 h |
160 ng/ml |
2 |
3,77 |
[257, 367, 397, 440, 552, 591] |
Der N-demethylierte Metabolit scheint nicht zur pharmakologischen Wirkung beizutragen |
|
Moclobemid |
300–1000 ng/ml |
2–7 h |
2000 ng/ml |
3 |
3,72 |
[225, 291, 327] |
Die Metaboliten tragen nicht zur pharmakologischen Wirkung bei |
|
Nortriptylin |
70–170 ng/ml |
30 h |
300 ng/ml |
1 |
3,80 |
[30, 31, 504, 506, 510] |
Hydroxylierte Metaboliten |
|
Paroxetin |
30–60 ng/ml |
12–44 h |
120 ng/ml |
3 |
3,04 |
[242, 243, 410, 443] |
|
|
Reboxetin |
60–350 ng/ml |
13–30 h |
700 ng/ml |
3 |
3,19 |
[483, 484] |
Der Metabolit trägt nicht zur pharmakologischen Wirkung bei |
|
Sertralin |
10–150 ng/ml |
26 h |
300 ng/ml |
2 |
3,27 |
[15, 49, 258, 281, |
Der N-demethylierte Metabolit hat eine zweifach längere Eliminationshalbwertszeit als Sertralin und 1/20 der pharmakologischen Aktivität von Sertralin |
|
Tranylcypromin |
≤ 50 ng/ml |
1–3 h |
100 ng/ml |
4 |
7,51 |
[103, 329] |
Wegen der irreversiblen Hemmung von Monoaminoxidasen gibt es keine Korrelation zwischen Plasmakonzentrationen und pharmakologischen Effekten |
|
Trazodon |
700–1000 ng/ml |
4–11 h |
1200 ng/ml |
2 |
2,69 |
[250, 262, 268, 447, 590] |
|
|
Trimipramin |
150–300 ng/ml |
23 h |
600 ng/ml |
2 |
3,40 |
[142, 187, 223, 326] |
Metaboliten mit ähnlicher pharmakologischer Aktivität wie Muttersubstanz |
|
Venlafaxin plus O-Desmethylvenlafaxin |
100–400 ng/ml |
5 h 11 h |
800 ng/ml |
2 |
3,61 3,80 |
[85, 241, 316, 443, 545, 550, 592, 684, 696] |
Für die meisten Patienten ist in vivo O-Desmethylvenlafaxin das aktive Prinzip, N-demethyliertes Venlafaxin scheint nicht zur pharmakologischen Wirkung beizutragen. Bei niedrigen Konzentrationen wirkt Venlafaxin als SSRI |
|
Antipsychotika |
|||||||
|
Amisulprid |
100–320 ng/ml |
12–20 h |
640 ng/ml |
1 |
2,71 |
[64, 89, 441, 461, |
Keine Metabolisierung |
|
Aripiprazol |
150–500 ng/ml |
60–80 h |
1000 ng/ml |
2 |
2,23 |
[33, 273, 306, 368, 452, 612] |
Der Metabolit Dehydroaripiprazol ist in vitro aktiv, es ist unklar, in welchem Umfang er zur klinischen Wirkung beiträgt |
|
Asenapin |
2–5 ng/ml |
24 h |
10 ng/ml |
4 |
3,50 |
[707] |
Absorption über die Mundschleimhaut |
|
Benperidol |
1–10 ng/ml |
5 h |
20 ng/ml |
3 |
2,62 |
[472, 589] |
Unter Langzeitbehandlung mit hohen Dosen werden möglicherweise höhere Spiegel toleriert wegen adaptiver Veränderungen |
|
Bromperidol |
12–15 ng/ml |
20–36 h |
30 ng/ml |
2 |
4,38 |
[609, 656, 735] |
|
|
Chlorpromazin |
30–300 ng/ml |
15–30 h |
800 ng/ml |
2 |
3,14 |
[127, 559] |
|
|
Chlorprothixen |
20–300 ng/ml |
8–12 h |
400 ng/ml |
3 |
3,17 |
[542] |
|
|
Clozapin |
350–600 ng/ml |
12–16 h |
1000 ng/ml |
1 |
3,06 |
[175, 507, 493, |
Hauptmetabolit N-Desmethylclozapin: ungeklärte antipsychotische Aktivität |
|
Flupentixol |
0,5–5 ng/ml (cis-Isomer) |
20–40 h |
15 ng/ml |
2 |
2,30 |
[40, 543, 564] |
Messung des aktiven cis-Isomers notwendig |
|
Fluphenazin |
1–10 ng/ml |
16 h |
15 ng/ml |
1 |
2,29 |
[564, 680] |
|
|
Fluspirilen |
0,1–2,2 ng/ml |
7–14 Tage |
4,4 ng/ml |
2 |
2,10 |
[611] |
|
|
Haloperidol |
1–10 ng/ml |
12–36 h |
15 ng/ml |
1 |
2,66 |
[74, 214, 480, 494, 508, 674, 680] |
Unter Langzeitbehandlung mit hohen Dosen werden durch adaptive Rezeptorveränderungen möglicherweise höhere Spiegel toleriert |
|
Iloperidon |
5–10 ng/ml |
18–33 h |
20 ng/ml |
3 |
2,34 |
[476, 576] |
|
|
Levomepromazin |
30–160 ng/ml |
16–78 h |
320 ng/ml |
3 |
3,04 |
[656] |
Inhibitor von CYP2D6 |
|
Melperon |
30–100 ng/ml |
4–6 h |
200 ng/ml |
3 |
3,80 |
[83, 324] |
Inhibitor von CYP2D6 |
|
Olanzapin |
20–80 ng/ml |
30–60 h |
150 ng/ml |
1 |
3,20 |
[32, 56, 63, 132, 208, 240, 418, 478, 509, 602, 711] |
Unter Olanzapinpamoat ist mit einem Postinjektionssyndrom zu rechnen, wenn die Plasmakonzentration auf über 150 ng/ml steigt |
|
Paliperidon |
20–60 ng/ml |
23 h |
120 ng/ml |
2 |
2,35 |
[26, 70, 131, 466] |
Paliperidon = 9-Hydroxyrisperidon, aktiver Metabolit von Risperidon |
|
Perazin |
100–230 ng/ml |
8–16 h |
460 ng/ml |
1 |
2,95 |
[91] |
Inhibitor von CYP1A2 |
|
Perphenazin |
0,6–2,4 ng/ml |
8–12 h |
5 ng/ml |
1 |
2,48 |
[564, 637, 680] |
|
|
Pimozid |
15–20 ng/ml |
23–43 h |
20 ng/ml |
3 |
2,17 |
[649] |
|
|
Pipamperon |
100–400 ng/ml |
17–22 h |
500 ng/ml |
3 |
2,66 |
[82, 517] |
|
|
Prothipendyl |
5–10 ng/ml |
2–3 h |
20 ng/ml |
4 |
3,35 |
[436] FI |
|
|
Quetiapin |
100–500 ng/ml |
7 h |
1000 ng/ml |
2 |
2,61 |
[112, 212, 236, 299, 498, 603, 627, 689, 723] |
Aktiver Metabolit Norquetiapin, hemmt die Wiederaufnahme von Noradrenalin. Wenn die retardierte Formulierung am Vorabend eingenommen wurde und die Blutentnahme am Morgen danach erfolgt, sind die zu erwartenden Plasmakonzentrationen doppelt so hoch wie die Talspiegel vor der abendlichen Einnahme [212]. |
|
Risperidon plus 9-Hydroxyrisperidon |
20–60 ng/ml |
3 h 24 h |
120 ng/ml |
2 |
2,44 2,35 |
[150, 406, 426, 437, 469, 475, 553, 557, 617, 729, 737] |
Aus dem Verhältnis der Konzentrationen 9-Hydroxyrisperidon zu Risperidon lässt sich auf den CYP2D6-Genotyp schließen (siehe Tab. 2) |
|
Sertindol |
50–100 ng/ml |
55–90 h |
200 ng/ml |
2 |
2,27 |
[71, 109, 110, 653, 728, 729] |
Aktiver Metabolit Dehydrosertindol (Konzentration bei therapeutischen Dosen 40–60 ng/ml); konzentrationsabhängige Zunahme des QT-Intervalls durch Blockade von kardialen Kaliumkanälen |
|
Sulpirid |
200–1000 ng/ml |
8–14 h |
1000 ng/ml |
2 |
2,93 |
[460, 656] |
Keine Metaboliten, renale Elimination |
|
Thioridazin |
100–200 ng/ml |
30 h |
400 ng/ml |
1 |
2,70 |
[190, 656] |
Kontraindiziert bei PM (CYP2D6) |
|
Ziprasidon |
50–200 ng/ml |
6 h |
400 ng/ml |
2 |
2,55 |
[126, 419, 427, 688, 695] |
Das Arzneimittel sollte mit einer Mahlzeit eingenommen werden, ansonsten ist mit niedrigeren Plasmakonzentrationen zu rechnen |
|
Zotepin |
10–150 ng/ml |
13–16 h |
300 ng/ml |
3 |
3,01 |
[376, 642] |
|
|
Zuclopentixol |
4–50 ng/ml |
15–25 h |
100 ng/ml |
3 |
2,49 |
[330, 371, 692] |
|
|
Stimmungsstabilisierer |
|||||||
|
Carbamazepin |
4–10 µg/ml |
10–20 h |
20 µg/ml |
2 |
4,23 |
[512] |
Der aktive Metabolit Carbamazepin-10,11-Epoxid trägt zur klinischen Wirkung bei |
|
Lamotrigin |
3–14 µg/ml |
7–23 h |
30 µg/ml |
2 |
3,90 |
[455, 558] |
Bisher kein definierter Bereich für den stimmungsstabilisierenden Effekt; Valproinsäure verlängert die Eliminationshalbwertszeit auf 48–70 h |
|
Lithium |
0,5–1,2 mmol/l (4–8 µg/ml) |
24 h |
1,2 mmol/l (8 µg/ml) |
1 |
125,8 |
[593, 721] |
Altersabhänige Zunahme der Eliminationshalbwertszeit |
|
Valproinsäure |
50–100 µg/ml |
18 h |
120 µg/ml |
2 |
6,93 |
[16, 216, 301, 683] |
Manche Patienten benötigen 120 µg/ml in der akuten manischen Phase |
|
Antikonvulsiva |
|||||||
|
Carbamazepin |
4–12 µg/ml |
10–20 h |
20 µg/ml |
2 |
4,25 |
[87, 338, 499] |
Der aktive Metabolit Carbamazepin-10,11-Epoxid trägt zur klinischen Wirkung bei |
|
Clobazam plus N-Desmethylclobazam |
30–300 ng/ml 300–3 000 ng/ml |
18–42 h |
500 ng/ml 5000 ng/ml |
2 |
3,33 3,49 |
[278, 499] |
Der aktive N-demethylierte Metabolit trägt zur klinischen Wirkung bei |
|
Clonazepam |
20–70 ng/ml |
40 h |
80 ng/ml |
2 |
3,17 |
[44, 464, 499] |
7-Aminometabolit mit pharmakologischer Aktivität |
|
Ethosuximid |
40–100 µg/ml |
33–55 h |
120 µg/ml |
2 |
7,08 |
[88,499] |
|
|
Felbamat |
30–60 µg/ml |
15–23 h |
100 µg/ml |
2 |
4,20 |
[290, 343, 499] |
|
|
Gabapentin |
2–20 µg/ml |
6 h |
25 µg/ml |
3 |
5,84 |
[75–77, 343, 398, 499] |
|
|
Lacosamid |
1–10 µg/ml |
13 h |
20 µg/ml |
2,66 |
[47] |
||
|
Lamotrigin |
3–14 µg/ml |
7–23 h |
20 µg/ml |
2 |
3,90 |
[88, 343, 455, 456, 499, 610] |
Valproinsäure verlängert die Eliminationshalbwertszeit auf 48–70 h |
|
Levetiracetam |
10–40 µg/ml |
6–8 h |
100 µg/ml (Morgenspiegel) |
2 |
3,87 |
[88, 343, 430, 499] |
|
|
Mesuximid plus N-Desmethylmesuximid |
10–40 µg/ml |
1–3 h 36–45 h |
45 µg/ml |
2 |
4,92 und 5,29 |
[88] |
Der Metabolit ist in vivo das wirksame Prinzip |
|
Oxcarbazepin plus 10-Hydroxycarbazepin |
10–35 µg/ml |
5 h 10–20 h |
40 µg/ml |
2 |
3,96 und 3,73 |
[88, 343, 428, 499] |
|
|
Phenobarbital |
10–40 µg/ml |
80–120 h |
50 µg/ml |
1 |
4,31 |
[88, 499] |
|
|
Phenytoin |
10–20 µg/ml |
20–60 h |
25 µg/ml |
1 |
3,96 |
[88, 380, 499] |
|
|
Pregabalin |
2–5 µg/ml |
6 h |
10 µg/ml |
3 |
6,28 |
[68, 77, 88, 343, |
|
|
Primidon |
5–10 µg/ml |
14–15 h |
25 µg/ml |
2 |
4,58 |
[88, 499] |
Für den aktiven Metaboliten Phenobarbital ist der Referenzbereich 10–40 µg/ml |
|
Rufinamid |
5–30 µg/ml |
7 h |
40 µg/ml |
2 |
4,20 |
[511] |
|
|
Stiripentol |
1–10 µg/ml |
4–13 h |
15 µg/ml |
2 |
4,27 |
[503] |
|
|
Sultiam |
2–8 µg/ml |
3–30 h |
12 µg/ml |
2 |
3,46 |
[88, 375, 429] |
|
|
Tiagabin |
20–200 ng/ml |
7–9 h |
300 ng/ml |
2 |
2,66 |
[88, 235, 343, 499] |
|
|
Topiramat |
2–8 µg/ml (Morgenspiegel) |
21 h |
16 µg/ml |
3 |
2,95 |
[88, 226, 343, 431, 499] |
|
|
Valproinsäure |
50–100 µg/ml |
18 h |
120 µg/ml |
2 |
6,93 |
[16, 88, 216, 301, 499, 682, 683] |
|
|
Vigabatrin |
2–10 µg/ml |
5–8 h |
20 µg/ml |
4 |
7,74 |
[88, 342, 398, |
|
|
Zonisamid |
10–40 µg/ml |
60 h |
40 µg/ml |
2 |
4,71 |
[247, 448, 449] |
|
|
Anxiolytika/Hypnotika |
|||||||
|
Alprazolam |
5–50 ng/ml |
12–15 h |
100 ng/ml |
4 |
3,22 |
[586, 686] |
Bei chronischer Einnahme von Benzodiazepinen können effektive Plasmakonzentrationen deutlicher höher liegen (mögliche Toleranzentwicklung) |
|
Bromazepam |
50–200 ng/ml |
15–35 h |
300 ng/ml |
4 |
3,16 |
[218, 286, 586] |
|
|
Brotizolam |
4–10 ng/ml (Cmax) |
3–6 h |
20 ng/ml |
4 |
2,53 |
[341, 669] |
|
|
Buspiron |
1–4 ng/ml |
2–3 h |
8 ng/ml |
3 |
2,59 2,49 |
[178, 580, 586] |
Aktiver Metabolit 6-Hydroxybuspiron |
|
Chlordiazepoxid |
400–3 000 ng/ml |
5–30 h |
3500 ng/ml |
4 |
3,48 |
[408, 586] |
|
|
Clonazepam |
4–80 ng/ml |
19–30 h |
100 ng/ml |
4 |
3,17 |
[181, 467, 586] |
|
|
Diazepam und Metaboliten |
200–2 500 ng/ml |
24–48 h |
3000 ng/ml§ |
4 |
3,51 |
[224, 261, 264, 586] |
Aktive Metaboliten sind Nordazepam, Oxazepam und Temazepam |
|
Flunitrazepam |
5–15 ng/ml |
10–30 h |
50 ng/ml |
4 |
3,20 |
[80, 425] |
|
|
Lorazepam |
10–15 ng/ml |
12–16 h |
30 ng/ml |
4 |
3,20 |
[164, 196, 218, 267] |
|
|
Lormetazepam |
2–10 ng/ml |
8–14 h |
100 ng/ml |
4 |
2,98 |
[3, 515] |
|
|
Midazolam |
6–15 ng/ml Cmax: 60–80 ng/ml |
1–3 h |
1000 ng/ml |
4 |
3,06 |
[35, 261, 323] |
|
|
Nitrazepam |
30–100 ng/ml |
18–30 h |
200 ng/ml |
4 |
3,56 |
[467, 586] |
|
|
Nordazepam |
20–800 ng/ml |
50–90 h |
1500 ng/ml |
4 |
3,69 |
[586] |
|
|
Opipramol |
50–500 ng/ml |
11 h |
1000 ng/ml |
3 |
2,87 |
[386] |
|
|
Oxazepam |
200–1 500 ng/ml |
4–15 h |
2000 ng/ml |
4 |
3,49 |
[586] |
|
|
Pregabalin |
2–5 µg/ml |
6 h |
10 µg/ml |
3 |
6,28 |
[76, 77] |
|
|
Temazepam |
20–900 ng/ml |
5–13 h |
1000 ng/ml |
4 |
3,51 |
[586] |
|
|
Triazolam |
2–20 ng/ml |
1–5 h |
40 ng/ml |
4 |
4,12 |
[586] |
|
|
Zolpidem |
80–150 ng/ml |
1–4 h |
300 ng/ml |
4 |
3,23 |
[586] |
|
|
Zopiclon |
10–50 ng/ml |
5 h |
150 ng/ml |
4 |
3,48 |
[586] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C) |
|
Antidementiva |
|||||||
|
Donepezil |
30–75 ng/ml |
70–80 h |
75 ng/ml |
2 |
2,64 |
[492, 563, 652] |
|
|
Galantamin |
30–60 ng/ml |
8 h |
90 ng/ml |
3 |
3,48 |
[322, 333, 734] |
|
|
Memantin |
90–150 ng/ml |
60–100 h |
300 ng/ml |
3 |
5,58 |
[251, 378] |
|
|
Rivastigmin |
|
1–2 h |
40 ng/ml |
3 |
4,00 |
[597] |
|
|
[147, 391] |
||||||
|
Arzneimittel zur Behandlung Substanz-bezogener Störungen |
|||||||
|
Acamprosat |
250–700 ng/ml |
13 h |
1000 ng/ml |
3 |
8,68 |
[287, 288, 424] |
|
|
Buprenorphin |
0,7–1,6 ng/ml Cmax: <9 ng/ml nach 24 mg |
2–5 h |
10 ng/ml (Cmax) |
2 |
2,38 |
[120, 130, 383] |
|
|
Bupropion plus Hydroxybupropion |
550–1500 ng/ml |
20 h 20 h |
2000 ng/ml |
2 |
4,17 3,91 |
[345] |
Bupropion ist instabil; Plasma oder Serum nach der Blutenentnahme eingefroren aufbewahren (–20o C) |
|
Clomethiazol |
100–5000 ng/ml |
2–5 h |
300 ng/ml§ |
4 |
6,19 |
[672] |
Patienten mit Alkoholabhängigkeit tolerieren deutlich höhere Plasmaspiegel als gesunde Kontrollpersonen |
|
Disulfiram |
50–400 ng/ml |
7 h |
500 ng/ml |
3 |
3,37 |
[203, 344, 586] |
Disulfiram (DSF) ist eine Vorstufe, der aktive Metabolit Diethylthiomethylcarbamat (DDTC-Me) wurde für die TDM-geleitete Dosisfindung von Disulfiram vorgeschlagen [344]. Unter 300 mg DSF wurden Steady-State- Konzentrationen (MW ± SD) von 170±10 ng/ml für DSF und 290 ± 20 ng/ml für DDTC-Me gefunden. |
|
Levomethadon |
250–400 ng/ml |
14–55 h |
400 ng/ml 100 ng/ml§ |
2 |
3,23 |
[146] |
In Patienten ohne Opiat-Abhängigkeit sind effektive oder toxische Plasmakonzentrationen deutlich niedriger; bei chronischem Gebrauch können im Einzelfall „toxische“ Plasmakonzentrationen notwendig sein, um das Auftreten von Entzugssymptomen zu verhindern |
|
Methadon |
400–600 ng/ml |
24–48 h |
600 ng/ml 300 ng/ml§ |
2 |
3,23 |
[146, 188, 595] |
|
|
Naltrexon plus 6β-Naltrexol |
25–100 ng/ml |
4 h 13 h |
200 ng/ml |
2 |
3,06 3,04 |
[99, 211, 252, 424] |
Unter In-vivo-Bedingungen ist im Wesentlichen der weniger potente Metabolit Naltrexol wirksam |
|
Vareniclin |
4–5 ng/ml |
24 h |
10 ng/ml |
3 |
4,73 |
[202, 532] |
|
|
Antiparkinson-Medikamente |
|||||||
|
Amantadin |
0,3–0,6 µg/ml |
10–14 h |
1,2 µg/ml |
3 |
5,98 |
[320] |
|
|
Biperiden |
Cmax: 1–6,5 ng/ml 0,5–2 h nach 4 mg |
18–24 h |
13 ng/ml |
3 |
3,21 |
[270] |
|
|
Bornaprin |
Cmax: 0,7–7,2 ng/ml 1–2 h nach 4 mg |
30 h |
14 ng/ml |
3 |
3,04 |
[433] |
|
|
Bromocriptin |
Niedrigdosis (2,5 mg): max. Dosis (25 mg): |
38 h |
8 ng/ml |
3 |
1,53 |
[168] |
|
|
Cabergolin |
Cmax: 58–144 pg/ml, 0,5–4 h nach Einnahme seit mindestens 4 Wochen |
63–68 h |
390 pg/ml |
3 |
2,21 |
[168] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C) |
|
Carbidopa |
Cmax: 20–200 ng/ml 2 h nach Einnahme |
2 h |
400 ng/ml |
3 |
4,42 |
[574] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C) |
|
Levodopa |
Cmax: 0,9–2,0 µg/ml 0,6–0,9 h nach 250 mg kombiniert mit 25 mg Carbidopa |
1–3 h |
5 µg/ml |
3 |
5,07 |
[4, 135, 479, 574] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C) Eliminationshalbwertszeit und Plasmakonzentrationen steigen unter Komedikation mit Carbidopa oder Benserazid |
|
0,7–10,9 µg/ml |
||||||
|
Entacapon |
Cmax: 0,4–1,0 µg/ml |
0,5 h |
2 µg/ml |
3 |
3,28 |
[304, 570] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C) |
|
Pramipexol |
0,39–7,17 ng/ml |
8–12 h |
15 ng/ml |
3 |
4,73 |
[730] |
|
|
Ropinirol |
0,4–6,0 ng/ml |
3–10 h |
12 ng/ml |
3 |
3,84 |
[657] |
|
|
Tiaprid |
Cmax: 1–2 µg/ml |
3–4 h |
4 µg/ml |
3 |
3,05 |
[108] |
|
|
Tolcapon |
Cmax: 3–6 µg/ml |
2 h |
12 µg/ml |
3 |
3,66 |
[177, 346] |
|
|
Andere |
|||||||
|
Atomoxetin |
200–1000 ng/ml 60–90 min nach Einnahme von 1,2 mg/kg/d |
4 h |
2000 ng/ml |
3 |
3,91 |
[233, 302, 446, 583] |
Referenzbereich gilt für Cmax, welches bei Remittern gemessen wurde; die Eliminationshalbwertszeit beträgt 21 h in PM (CYP2D6) |
|
Dexmethylphenidat |
13–23 ng/ml 4 h nach 20 mg |
2 h |
44 ng/ml |
2 |
4,29 |
[663] |
5,2–5,5 ng/ml sind mit 50% Dopamintransporterblockade assoziiert [614] |
|
Methylphenidat |
13–22 ng/ml d-Methylphenidat 2 h nach 20 mg „immediate release” oder 6–8 h nach 40 mg „extended release” |
2 h |
44 ng/ml |
2 |
4,29 |
[331, 422, 614] |
Instabil bei Raumtemperatur; Plasma oder Serum gefroren aufbewahren (<–20°C). Der Referenzbereich bezieht sich auf Cmax. |
|
Modafinil |
1000–1700 ng/ml nach 200 mg/d |
10–12 h |
3400 ng/ml |
3 |
4,21 |
[733] |
|
Die Plasmakonzentrationen sind in Masseneinheiten angegeben. Eine Umrechnung in molare Einheiten erfolgt durch Multiplikation mit dem Umrechnungsfaktor (conversion factor, CF) nmol/l=ng/mlxCF.
*Empfehlungsgrade siehe Kasten auf Seite 111.
§ Der aktive Metabolit trägt zu erwünschten und unerwünschten Wirkungen bei. Die angegebenen Referenzbereiche und die Warnschwellen für das Labor beziehen sich nur auf die Muttersubstanz.
Für Bupropion, Carbamazepin, Lamotrigin und Valproinsäure sind zwei Referenzbereiche angegeben entsprechend den zwei unterschiedlichen Indikationen.
Erstellt durch CH, PB, SU, BR und HK, überprüft durch AC, OD, KE, MF, MG, CG, GG, EH, UH-R, CH, EJS, HK, GL, UL, TM, BP, BS, MU, SU, GZ
Der therapeutische Referenzbereich ist ein orientierender, populationsbezogener Wert, der nicht unbedingt für alle Patienten gültig sein muss. Einzelne Patienten können ein optimales therapeutisches Ansprechen bei einer Wirkstoffkonzentration zeigen, die außerhalb des therapeutischen Referenzbereichs liegt. Letztlich sollte für jeden Patienten die „individuelle therapeutische Konzentration“ identifiziert werden, bei der das Ansprechen auf die Psychopharmakotherapie am besten ist.
Die von der TDM-Gruppe der AGNP empfohlenen therapeutischen Referenzbereiche sind in Tabelle 5 angegeben. Sie wurden aus der Literatur durch die oben beschriebene strukturierte Überprüfung abgeleitet. Für nur 15 neuropsychiatrische Arzneistoffe wurden in der Literatur therapeutische Referenzbereiche aus randomisierten klinischen Studien gefunden. Für die meisten Arzneistoffe wurden die Referenzbereiche aus Studien mit therapeutisch wirksamen Dosierungen abgeleitet. Daher besteht ein Bedarf an weiteren Studien, um therapeutische Referenzbereiche für Neuropsychopharmaka zu definieren.
Die in Tabelle 5 aufgeführten Referenzbereiche beziehen sich in der Regel auf die primäre Indikation. Einige Arzneistoffe sind jedoch für verschiedene Indikationen zugelassen. Zum Beispiel werden Antidepressiva auch für die Behandlung von Angstzuständen eingesetzt, und Antipsychotika werden zunehmend genutzt, um manische Symptome zu behandeln. Für diese Anwendungsbereiche existieren bislang so gut wie keine Informationen zu einer optimalen Plasmakonzentration. Ausnahmen sind Carbamazepin, Lamotrigin und Valproinsäure, die deshalb zweifach in Tabelle 5 aufgeführt sind. Darüber hinaus sei erwähnt, dass therapeutische Referenzbereiche für Kinder oder Jugendliche sowie für ältere Patienten noch nicht ausreichend durch Studien belegt sind.
Bestimmung der unteren Grenze des therapeutischen Referenzbereichs
Die Einschätzung eines therapeutischen Referenzbereichs beruht auf der Annahme einer unteren und einer oberen Grenze der Konzentrationen im Blut, bei denen der Arzneistoff wirksam ist. Eine allgemein akzeptierte Methode zur Berechnung dieser Grenzen gibt es nicht. Wenn immer möglich, sollte die untere Grenze des therapeutischen Bereichs eines Arzneistoffs durch Studien über die Beziehung von Plasmakonzentrationen und klinischer Wirksamkeit belegt sein. Unterhalb dieser Grenze werden im Vergleich zu einer Plazebo-Behandlung keine therapeutischen Effekte erwartet. Das optimale Studiendesign für die Auswertung der unteren Grenze des therapeutischen Bereichs ist eine randomisierte, prospektive Doppelblind-Studie, in der Patienten mit Dosierungen behandelt werden, die zu einem definierten Plasmakonzentrationsbereich des Arzneistoffs führen. Eine solche Studie wurde von Van der Zwaag und Mitarbeitern an mit Clozapin behandelten Patienten durchgeführt [678]. Die Patienten wurden auf drei verschiedene Blutspiegel des antipsychotischen Arzneistoffs eingestellt. Verglichen mit der Gruppe, die auf niedrige Blutspiegel eingestellt war, wurde bei Patienten mit mittleren und hohen Blutspiegeln eine deutliche Überlegenheit der Wirksamkeit von Clozapin festgestellt. Ähnliche Untersuchungen wurden mit Imipramin und Mirtazapin durchgeführt [98]. Solche Studien sind jedoch eine beträchtliche logistische Herausforderung. Wesentlich einfacher durchzuführen sind Studien mit fester Dosis. Sie werden daher für die Bestimmung der unteren Grenze des therapeutischen Referenzbereichs bevorzugt eingesetzt [672, 674].
Für die Abschätzung der Schwellenwerte der therapeutischen Referenzbereiche hat sich die „Receiver Operating Characteristics“ (ROC)-Analyse als hilfreich erwiesen [289]. Ein ROC-Plot ermöglicht die Identifizierung eines Cut-off-Werts, der Responder von Nonrespondern unterscheidet und dabei die Sensitivität und Spezifität des Parameters „Arzneistoffblutspiegel“ schätzt. Die Nützlichkeit der ROC-Analyse wurde für eine Reihe antipsychotischer und antidepressiver Arzneistoffe nachgewiesen [461, 505, 510, 703].
Bestimmung der oberen Grenze des therapeutischen Referenzbereichs
In der ersten Studie über TDM in der Psychiatrie wurde für Nortriptylin eine U-förmige Beziehung zwischen Plasmakonzentration und klinischer Wirkung berichtet [31]. Dem Fehlen einer Wirkung bei hohen Konzentrationen wurde der Wirkungsmechanismus des trizyklischen Antidepressivums auf monoaminerge Neurone zugeschrieben. Gemäß aktuellem Wissen scheint es jedoch wahrscheinlicher, dass die reduzierte Besserung bei hohen Konzentrationen auf der erhöhten Wahrscheinlichkeit für Nebenwirkungen beruht. Die Obergrenze des therapeutischen Bereichs ist daher auch in dieser Leitlinie durch das Auftreten von Nebenwirkungen definiert. Für die meisten Nebenwirkungen (Typ-A-Nebenwirkungen) wird angenommen, dass sie eine Funktion der Dosis und der Wirkstoffkonzentration im Körper sind [335]. Diese Annahme ist für motorische Nebenwirkungen der Neuroleptika [536] und für unerwünschte Arzneimittelwirkungen der trizyklischen Antidepressiva [153, 282] bestätigt worden. Für Paroxetin wurde eine positive Korrelation zwischen Wirkstoffkonzentration im Blut und dem Auftreten von Symptomen eines Serotonin-Syndroms gefunden [303]. Wenn solche Daten verfügbar sind, ist es möglich, die Obergrenze des therapeutischen Bereichs zu berechnen [461].
Für viele der in Tabelle 5 aufgelisteten Psychopharmaka fehlen jedoch verwertbare Daten über den Zusammenhang zwischen Plasmakonzentration und der Häufigkeit von Nebenwirkungen. Fallberichte über Probleme bezüglich Verträglichkeit oder über Vergiftungen können oft nicht verwendet werden, da Messungen der Wirkstoffkonzentration im Blut fehlen. Sporadische Berichte über Todesfälle und Vergiftungen sind von begrenztem Wert. Die meisten Konzentrationen im Blut, von denen berichtet wird, dass sie letal waren, liegen weit über den Arzneistoffkonzentrationen, die mit einer maximalen therapeutischen Wirkung verbunden sind [544, 622]. Post-mortem-Umverteilung von Arzneistoffen aus dem bzw. in das Blut kann außerdem zu dramatischen Veränderungen der Plasmakonzentrationen führen [382, 518], wobei die Richtung der Veränderung nicht allgemein vorhersagbar ist [359].
Die Abschätzung eines oberen Schwellenwerts, oberhalb dessen die Verträglichkeit abnimmt oder das Risiko einer Vergiftung erhöht ist, ist daher schwieriger als die Einschätzung der unteren Schwelle, vor allem für Arzneistoffe mit einem breiten therapeutischen Index wie SSRIs.
Einführung einer Warnschwelle für das Labor
Wie oben erläutert, sind für die meisten Psychopharmaka die Plasmakonzentrationen mit einem erhöhten Risiko für Toxizität sehr viel höher als die in Tabelle 5 dargestellten oberen Grenzwerte des therapeutischen Referenzbereichs. Für die vorliegenden Leitlinien haben wir daher Plasmakonzentrationen definiert, oberhalb derer es unwahrscheinlich ist, dass die therapeutische Wirkung gesteigert werden kann, und darüber hinaus eine „Warnschwelle für das Labor“ neu eingeführt (Tab. 5).
Definition von Warnschwellen für das Labor
Die in dieser Leitlinie aufgeführten „Warnschwellen für das Labor“ (Tab. 5) entsprechen Arzneistoffkonzentrationen oberhalb des empfohlenen Referenzbereichs, ab denen das Labor den behandelnden Arzt unverzüglich zu verständigen hat.
Die Warnschwellen beruhen idealerweise auf Berichten über Intoleranz oder Vergiftungen, bei denen Wirkstoffkonzentrationen im Blut gemessen worden waren. Ohne Datenbasis wurde eine Plasmakonzentration festgelegt, die 2-fach höher als die obere Grenze des therapeutischen Referenzbereichs liegt.
Die Meldung des Labors sollte zu einer Dosisreduktion führen, wenn der Patient Anzeichen von Intoleranz oder Toxizität zeigt. Wenn die hohe Wirkstoffkonzentration durch den Patienten toleriert wird und gleichzeitig bei Dosisreduktion die Gefahr der Symptomverschlechterung besteht, sollte die Dosis unverändert bleiben. Die klinische Entscheidung, insbesondere wenn die Dosis nicht geändert wird, muss in der Krankenakte dokumentiert werden.
Von populationsbezogenen zu individuellen Referenzwerten
Alle in Tabelle 5 aufgeführten therapeutischen Referenzbereiche sind populationsbezogen ermittelt. Sie sind eine erste Orientierung und nicht zwangsläufig für jeden Patienten passend. Einzelne Patienten können optimal auf eine Wirkstoffkonzentration ansprechen, die außerhalb des therapeutischen Referenzbereichs liegt. Man sollte daher versuchen, für den Patienten eine optimale „individuelle therapeutische Konzentration“ zu finden [61, 523]. Für Lithium hat sich gezeigt, dass der empfohlene Plasmakonzentrationsbereich davon abhängt, ob sich der Patient in einer akuten manischen Episode oder in der Erhaltungsphase befindet [593]. Gaertner und Kollegen ermittelten in einer Studie zur Rückfallprävention für jeden einzelnen Patienten in der Phase der Erhaltungstherapie die optimale Plasmakonzentration von Clozapin für eine stabile Remission [232].
Graduierte Empfehlungen für die Messung der Blutspiegel von Neuropsychopharmaka
Der Nutzen von TDM hängt von der klinischen Situation und dem jeweiligen Medikament ab. Bei Verdacht auf Non-Compliance oder Intoxikation ist die Messung von Blutspiegeln generell für jedes Medikament und jeden Patienten sinnvoll. Es ist jedoch immer noch umstritten, bei welchen Psychopharmaka TDM in der klinischen Routine eingesetzt werden sollte. Basierend auf empirischen Daten wurden in den Leitlinien von 2004 fünf Empfehlungsgrade zum Einsatz von TDM für 65 Psychopharmaka definiert. Diese Definitionen wurden überarbeitet und auf vier Empfehlungsgrade reduziert, die nunmehr von „dringend empfohlen“ bis „potenziell nützlich“ wie folgt definiert sind (siehe Kasten).
Graduierte Empfehlungen zur Anwendung von TDM ohne spezifische Fragestellung
1=TDM dringend empfohlen
Evidenz: Die berichteten therapeutischen Referenzbereiche sind durch Studien validiert. Kontrollierte klinische Studien haben positive Effekte von TDM nachgewiesen. Es gibt Berichte über Unverträglichkeit oder Vergiftungen bei hohen Plasmakonzentrationen.
Empfehlung: TDM ist für die Dosisfindung und für spezielle Indikationen dringend empfohlen. Für eine Behandlung mit Lithium ist TDM obligat.
Klinische Konsequenzen: Bei therapeutischen Plasmakonzentrationen besteht die höchste Wahrscheinlichkeit des Ansprechens bzw. einer Remission; bei „subtherapeutischen“ Plasmakonzentrationen ist in der Akutbehandlung die Ansprechrate mit Plazebo vergleichbar, unter chronischer Behandlung besteht ein erhöhtes Risiko für einen Rückfall. Bei „supratherapeutischen“ Plasmakonzentrationen besteht ein erhöhtes Risiko für eine Unverträglichkeit oder Vergiftung.
2=TDM empfohlen
Evidenz: Die berichteten therapeutischen Referenzbereiche wurden aus Untersuchungen gewonnen, bei denen Plasmakonzentrationen unter therapeutisch wirksamen Dosierungen mit den klinischen Wirkungen korreliert wurden. Es gibt Berichte über verminderte Verträglichkeit oder Vergiftungen bei „supratherapeutischen“ Plasmakonzentrationen.
Empfehlung: TDM ist für die Dosisfindung und für spezielle Indikationen oder Problemlösungen empfohlen.
Klinische Konsequenzen: TDM erhöht die Wahrscheinlichkeit des Ansprechens bei Therapieversagern. Bei „subtherapeutischen“ Plasmakonzentrationen besteht ein erhöhtes Risiko für schlechtes Ansprechen; bei „supratherapeutischen“ Plasmakonzentrationen besteht ein erhöhtes Risiko für Unverträglichkeit oder Vergiftung.
3=TDM nützlich
Evidenz: Die berichteten therapeutischen Referenzbereiche wurden aus Plasmakonzentrationen errechnet, die unter wirksamer Dosierung in pharmakokinetischen Studien gefunden wurden. Untersuchungen über Plasmakonzentrationen und davon abhängige pharmakodynamische Wirkungen fehlen bisher, oder sie beruhen auf retrospektiven Analysen von TDM-Daten, einzelnen Fallberichten oder klinischer Erfahrung.
Empfehlung: TDM ist nützlich für spezielle Indikationen oder bei spezifischen Problemen.
Klinische Konsequenzen: Durch TDM kann man einschätzen, ob Plasmakonzentrationen für eine bestimmte Dosis plausibel sind oder ob bei Nonrespondern, die zu niedrige Plasmakonzentrationen aufweisen, durch Dosissteigerung eine klinische Besserung erwartet werden kann.
4=TDM potenziell nützlich
Evidenz: Plasmakonzentrationen korrelieren aufgrund besonderer pharmakologischer Eigenschaften des Arzneimittels nicht mit klinischen Effekten; beispielsweise bei irreversibler Blockade eines Enzyms, oder die Dosierung kann leicht anhand klinischer Symptome eingestellt werden, wie zum Beispiel Schlafinduktion durch Hypnotika.
Empfehlung: TDM wird nicht für die Dosisfindung empfohlen, kann aber für spezielle Indikationen oder besondere Probleme potenziell nützlich sein.
Klinische Konsequenzen: TDM sollte auf spezifische Fragestellungen beschränkt werden.
Nach der Literatur-basierten Analyse wurde TDM für 15 der 128 untersuchten Neuropsychopharmaka als „dringend empfohlen“ eingestuft und für 52 als „empfohlen“. Für 44 Neuropsychopharmaka wurde TDM als „nützlich“ und für 19 als „potenziell nützlich“ (Tab. 5) eingestuft.
TDM wird nach Tabelle 5 für die meisten trizyklischen Antidepressiva „dringend empfohlen“. Denn es reduziert das Risiko von Vergiftungen [103, 459, 510, 718]. Für viele trizyklische Antidepressiva konnte ein Zusammenhang zwischen Plasmakonzentration und klinischer Wirksamkeit gezeigt werden. Für SSRI ist TDM in der klinischen Praxis von geringer klinischer Bedeutung [6, 537, 644], da die Toxizität dieser Antidepressiva gering ist [166, 314, 646, 715]. Daten aus Schweden zeigen allerdings, dass TDM von SSRI bei älteren Patienten zu einer kostengünstigeren Behandlung führt, indem man mit TDM die minimale effektive Dosis ermittelt [410]. Für Citalopram ergab eine aktuelle Beobachtungsstudie, dass niedrige Plasmakonzentrationen, die eine Woche nach Behandlungsbeginn gemessen worden waren, prädiktiv für späteres Therapieversagen waren [491]. Patienten, die Citalopram-Plasmakonzentrationen über 50 ng/ml aufwiesen, zeigten signifikant besseres Ansprechen auf der Hamilton-Rating-Skala für Depression. Einen Nachweis für eine statistisch signifikante Beziehung zwischen Wirkstoffkonzentration und Therapieeffekt gibt es bisher weder für die tetrazyklischen Antidepressiva Maprotilin, Mianserin und Mirtazapin noch für Trazodon und Reboxetin und die MAO-Hemmer Moclobemid und Tranylcypromin.
Für die typischen Antipsychotika Haloperidol, Perphenazin und Fluphenazin und für die atypischen Antipsychotika Amisulprid, Clozapin, Olanzapin und Risperidon wird TDM dringend empfohlen (Tab. 5). Eine Überdosierung kann zu extrapyramidalen Nebenwirkungen führen. Die Vermeidung einer Überdosierung von typischen Antipsychotika durch TDM wird daher eher als Verbesserung der Lebensqualität und weniger als eine Steigerung der Arzneimittelsicherheit angesehen [136]. Für Clozapin steigt mit den Blutspiegeln das Risiko für Krampfanfälle [507]. TDM von Antipsychotika ist auch nützlich, wenn von einer oralen auf eine Depot-Form oder umgekehrt umgestellt wird.
Für die stimmungsstabilisierenden und/oder antimanischen Medikamente Lithium, Valproinsäure und Carbamazepin sind die therapeutischen Referenzbereiche und toxische Konzentrationen gut validiert. Deshalb wird TDM für diese Arzneistoffe dringend empfohlen (Tab. 5). Für Lithium gilt TDM als Standard guter klinischer Praxis [170, 185, 280, 593, 706, 721]. Bei langfristiger Nutzung werden Plasmakonzentrationen von 0,5 bis 0,8 mmol/l empfohlen. Für eine akute Behandlung mit Lithium kann es gerechtfertigt sein, die Konzentration auf bis zu 1,2 mmol/l zu erhöhen.
Arzneimittel mit antidementiven Eigenschaften sind Donepezil, Rivastigmin, Galantamin und Memantin. TDM wird nur selten bei der medikamentösen Behandlung von Demenzen eingesetzt, obwohl es Hinweise für einen Nutzen gibt. Für Donepezil wurde gezeigt, dass bei Plasmakonzentrationen über 50 ng/ml der therapeutische Effekt deutlich besser ist als bei niedrigeren Wirkstoffkonzentrationen [563].
Die meisten Anxiolytika und Hypnotika gehören zur Klasse der Benzodiazepine. Anxiolytische und hypnotische Effekte treten rasch ein. Die Behandlung kann daher durch den unmittelbaren klinischen Eindruck gesteuert werden. Bei Ausbleiben einer therapeutischen Wirkung unter üblicher Dosierung kann TDM jedoch klären, ob dies durch pharmakokinetische Besonderheiten oder durch Toleranzausbildung aufgrund von Medikamentenmissbrauch bedingt ist. Bei der Behandlung von Panikattacken kann TDM von Alprazolam nützlich sein [722].
Zur Behandlung der Opiat-Abhängigkeit werden die Opiatagonisten Methadon, R-Methadon (Levomethadon), Buprenorphin, l-a-Acetylmethadol (LAAM) und Slow-Release-Formulierungen von Morphin verwendet. TDM ist für Patienten unter Behandlung mit Methadon oder R-Methadon indiziert. Die Nützlichkeit von TDM für die Überwachung der Behandlung mit den „Anti-Craving“-Medikamenten Acamprosat und Naltrexon, die zur Behandlung der Alkoholabhängigkeit eingesetzt werden, wurde vor kurzem untersucht [99]. TDM wurde empfohlen, um die moderate Wirksamkeit dieser Arzneistoffe zu steigern.
Für Antikonvulsiva ist TDM gut etabliert, vor allem für alte Wirkstoffe, die toxischer sind als die neuen [499].
Für Parkinson-Medikamente ist TDM bisher nicht etabliert. Die Datenlage für Dopaminagonisten ist zu schwach, um für die Festlegung auf Referenzbereiche eingesetzt werden zu können. Bezüglich Levodopa ist eine Korrelation zwischen Plasmakonzentration und kurzfristiger klinischer Reaktion beschrieben [479]. Die neurologischen Arzneistoffe wurden in diese Leitlinie eingeschlossen (Tab. 1 und 5), da psychiatrische Patienten gegebenenfalls Parkinson-Medikamente erhalten können, um unerwünschte motorische Wirkungen der Psychopharmaka zu reduzieren. Für die meisten dieser Arzneistoffe sind Cmax-Werte angegeben.
Indikationen für die Messung der Plasmakonzentrationen von Psychopharmaka
Tabelle 6 ist eine Liste von Indikationen für TDM in der Psychiatrie. Welche Indikation zutreffend ist, muss im Einzelfall individuell geprüft und bewertet werden. Ähnlich wie bei allen diagnostischen Tests sollte TDM nur angefordert werden, wenn das Ergebnis eine Antwort auf eine klar definierte Fragestellung liefern kann.
Tab. 6. Typische Indikationen zur Anforderung einer Blutspiegelmessung in der Psychiatrie
|
Dosisfindung nach Eindosierung oder Dosisänderung |
|
Obligatorisches TDM aus Gründen der Arzneimittelsicherheit (z.B. Lithium) |
|
Verdacht auf unzuverlässige Einnahme der Medikamente, unzureichende Adhärenz |
|
Kein/ungenügendes Therapieansprechen bei empfohlener Dosis |
|
Unerwünschte Arzneimittelwirkung bei empfohlener Dosis |
|
Kombination von Medikamenten mit Wechselwirkungspotenzial |
|
TDM in Pharmakovigilanzprogrammen |
|
Rückfallprävention unter Erhaltungstherapie |
|
Rezidiv unter Erhaltungstherapie bei empfohlener Dosis |
|
Genetische Besonderheit im Arzneimittelmetabolismus (Defektmutante, Genmultiplikation) |
|
Schwangere oder stillende Patientin |
|
Patient im Kindes- oder Jugendalter (bis 18 Jahre) |
|
Alterspatient (>65 Jahre) |
|
Patient mit verminderter Intelligenz |
|
Patient mit pharmakokinetisch relevanter Komorbidität (z.B. Leberfunktionsstörung oder Niereninsuffizienz) |
|
Forensischer Patient |
|
Arzneimittelumstellung auf Generikum |
Für Arzneimittel mit gut definierten therapeutischen Referenzbereichen und mit einem engen therapeutischen Index sollten die Plasmakonzentrationen auch ohne spezifische Fragestellung für die Dosisfindung nach Eindosierung oder Dosisänderung gemessen werden. Für Lithium und 14 Antidepressiva, Antipsychotika und Antikonvulsiva sind positive Effekte von TDM nachgewiesen worden (Tab. 5). Für Lithium ist TDM aus Gründen der Sicherheit zwingend geboten.
Bei Verdacht auf Non-Compliance oder bei mangelhafter klinischer Besserung unter der empfohlenen Dosierung ist TDM ein wertvolles Hilfsmittel für die Behandlung mit allen in dieser Leitlinie aufgeführten Arzneistoffen. Compliance ist ein Problem während der Dauermedikation [10, 55, 401]. Bei Patienten mit Schizophrenie [55, 351] und bei Patienten mit unipolaren oder bipolaren Störungen reicht die Häufigkeit von Non-Compliance von 10 bis 69% [401, 439]. Um die Compliance zu überprüfen, werden verschiedene mehr oder weniger unsichere Methoden wie Tabletten-Zählen, Hinweise in den täglichen Kadex-Aufzeichnungen, direktes Befragen des Patienten oder das klinische Urteil des behandelnden Arztes verwendet [11, 355, 685, 708]. Studien haben gezeigt, dass Ärzte die Compliance ihrer Patienten nicht verlässlich vorhersagen können [104, 579]. TDM ist eine große Hilfe, da es sich um eine objektive Methode handelt, die dem behandelnden Arzt anzeigt, ob das Medikament in einer Konzentration vorliegt, die für die Besserung der Symptome wahrscheinlich ausreicht. Abweichungen vom erwarteten dosisbezogenen Referenzbereich (Tab. 4) geben Hinweise, ob der Patient seine Medikamente genommen hat, wobei die gleichzeitige Bestimmung der Metaboliten zusätzlich klären kann, ob das Medikament auch kontinuierlich genommen wurde. Zur Interpretation müssen jedoch mögliche Wechselwirkungen mit Begleitmedikamenten, die zu pharmakokinetischen Interaktionen führen können, berücksichtigt werden (Tab. 3). Reis und Mitarbeiter [546] analysierten durch wiederholte Messung der Blutspiegel die Compliance von Patienten, die mit Sertralin behandelt wurden. Aus der Variation des Verhältnisses der Konzentrationen von Norsertralin zu Sertralin konnte auf versteckte und partielle Non-Adhärenz geschlossen werden. Um diese Vorgehensweise praktisch nutzen zu können, wurden die vorliegenden Leitlinien mit Daten über zu erwartende Konzentrationsverhältnisse Metabolit zu Muttersubstanz für 32 Psychopharmaka aus Daten der Literatur berechnet (Tab. 2). Darüber können Hinweise erhalten werden, ob eine gemessene niedrige Plasmakonzentration durch reduzierte Bioverfügbarkeit, beschleunigten Abbau oder mangelnde Compliance bedingt ist. Für weitergehende Analysen ist zu empfehlen, mehrere Blutproben pro Tag abzunehmen. Die gemessenen Plasmakonzentrationen können dann mit errechneten Plasmakonzentrationen verglichen werden, die anhand bekannter pharmakokinetischer Daten zu erwarten sind [4, 78, 340, 626, 654]. Dann kann man relativ sicher erschließen, ob reduzierte Bioverfügbarkeit, beschleunigter Abbau oder mangelnde Compliance ursächlich für die ungewöhnlich niedrigen Plasmaspiegel waren.
Wenn eine klinische Verbesserung unter empfohlenen Dosen nicht ausreichend ist und das Medikament gut vertragen wird, kann TDM klären, ob das Medikament zu niedrig dosiert ist und ob es sinnvoll ist, die Dosis zu erhöhen.
Wenn unter zugelassenen Dosen unerwünschte Arzneimittelwirkungen auftreten, kann die Messung der Plasmakonzentration klären, ob die Nebenwirkungen durch zu hohe Wirkstoffkonzentrationen verursacht wurden. Dies ist dann besonders relevant, wenn eine klinische Besserung erzielt wurde. So lässt es sich prüfen, ob die Dosis ohne Wirkverlust verringert werden kann. Dies ist wahrscheinlich, wenn die Plasmakonzentrationen im oberen therapeutischen Referenzbereich (Tab. 5) oder darüber liegen.
Bei der Kombination mit Arzneistoffen, die Inhibitoren oder Induktoren Arzneimittel-metabolisierender Enzyme sind (Tab. 3), werden pharmakokinetische Wechselwirkungen auftreten, wenn die Komedikation ein Substrat des gehemmten oder induzierten Enzyms ist (Tab. 1). Bei Kombinationen mit einem Induktor oder Inhibitor (Tab. 3) sollte die Anpassung der Dosis mit Hilfe von TDM gesteuert werden, um den Verlust der Wirksamkeit, eine schlechte Verträglichkeit oder eine Intoxikation durch Wechselwirkung mit anderen Arzneimitteln zu vermeiden [215, 244, 594]. In Bezug auf Lebensgewohnheiten ist Rauchen von hoher klinischer Relevanz für Arzneimittel, die Substrate von CYP1A2 sind (Tab. 1). Dieses Isoenzym wird dosisabhängig durch Bestandteile von Zigarettenrauch (polyzyklische aromatische Kohlenwasserstoffe, nicht Nicotin) induziert. Seine Aktivität steigt bei 1 bis 5, 6 bis 10 bzw. >10 Zigaretten pro Tag um das 1,2-, 1,5- bzw. 1,7-fache [201]. Auf der anderen Seite nimmt die CYP1A2-Aktivität bis zum vierten Tag nach abrupter Beendigung starken Rauchens deutlich ab [200]. Die Effekte des Rauchens bzw. Rauchstopps sollten daher berücksichtigt werden, wenn Patienten unter Therapie mit einem CYP1A2-Substrat (Tab. 1) wie Clozapin [81, 676], Duloxetin [222] oder Olanzapin [749] behandelt werden. Es sollte auch erwähnt werden, dass viele pharmakokinetische Arzneimittelinteraktionen entweder zufällig durch die Messung der Blutspiegel oder durch eine retrospektive Analyse von TDM-Datenbanken gefunden werden [112, 537].
In Pharmakovigilanz-Programmen wird die Sicherheit der Medikamenteneinnahme unter Alltagsbedingungen untersucht [271, 285]. Im Falle beobachteter Nebenwirkungen ist die Messung der Plasmakonzentration höchst hilfreich, um die Ursachen für eine unerwünschte Arzneimittelwirkung zu klären [335].
Rückfallprävention ist ein wesentliches Ziel der Erhaltungstherapie. Regelmäßiges TDM ist eine relativ kostengünstige Methode, um Rückfälle, die zur Hospitalisierung führen können, zu verhindern [377]. Bei schizophrenen Patienten hat sich gezeigt, dass Schwankungen der Clozapin-Blutspiegel prädiktiv für Rückfälle [232, 670] bzw. Hospitalisierungen [627] sind. Die regelmäßige Überprüfung der Arzneistoffspiegel sorgt erfahrungsgemäß für eine zuverlässigere Einnahme der Medikation und senkt damit das Risiko eines Rückfalls durch Absetzen der Medikamente. Ein Tag im Krankenhaus ist etwa 4- bis 16-mal teurer als eine Blutspiegelmessung eines Arzneistoffs im Labor.
Empfehlung
Obwohl die klinische Evidenz begrenzt ist, empfehlen wir aus eigener Erfahrung eine regelmäßige Messung der Blutspiegel unter Erhaltungstherapie, mindestens alle 3 bis 6 Monate, um Rückfälle und Hospitalisierungen zu verhindern. Die Häufigkeit von Blutspiegeluntersuchungen sollte erhöht werden, wenn der Patient bekanntermaßen zu Non-Compliance neigt. Ebenso sollte TDM bei pharmakokinetisch relevanten Änderungen der Komedikation oder des Rauchverhaltens eingesetzt werden.
Bei Patienten mit genetischen Besonderheiten von Enzymen des Arzneimittelabbaus müssen die Dosierungen angepasst werden. Kirchheiner und Mitarbeiter [362, 365] berechneten aufgrund pharmakokinetischer und pharmakodynamischer Erkenntnisse notwendige Dosisanpassungen für PM oder UM von CYP2D6. Doch auch im Fall eines bestätigten abnormen CYP-Genotyps wird TDM empfohlen, da die Genotypisierung nur grob vorhersagen kann, in welchem Umfang sich Blutspiegel beim individuellen Patienten ändern [496, 497, 625].
Für spezielle Patientengruppen, wie Schwangere oder stillende Mütter, Kinder oder Jugendliche [373, 194], Menschen mit geistigen Behinderungen oder ältere Patienten, insbesondere Patienten über 75 Jahren [374], wird TDM unbedingt zur Risikominimierung empfohlen.
Bei jeder Psychopharmakotherapie von schwangeren oder stillenden Frauen sollte sichergestellt werden, dass die Plasmakonzentration des Arzneistoffs im therapeutischen Referenzbereich ist, um seitens der Mutter das Risiko eines Rückfalls und seitens des Fötus bzw. Kindes die Arzneistoffexposition [169, 174] zu minimieren. Die renale Clearance und die Aktivität der Isoenzyme CYP3A4, CYP2D6 und CYP2C9, sowie von Uridin-5‘-diphosphat-Glucuronosyltransferase sind während der Schwangerschaft erhöht, während die Aktivitäten von CYP1A2 und CYP2C19 verringert sind [21]. TDM sollte bei Schwangeren und/oder Müttern mindestens einmal pro Trimester und innerhalb von 24 Stunden nach der Geburt durchgeführt werden [65].
Viele Psychopharmaka sind für den Einsatz bei Kindern und Jugendlichen nicht zugelassen [248]. Während der Entwicklung ändern sich Pharmakokinetik und Pharmakodynamik [194, 438, 514, 516]. Bei Jugendlichen, die unter psychotischen Störungen leiden, ist komorbider Arzneimittelmissbrauch sehr verbreitet, und die Zuverlässigkeit der Einnahme von Antipsychotika ist in der Regel marginal [318]. Daher ist TDM bei diesen Patienten dringend empfohlen. Zur Verbesserung der Datenlage über die Wirksamkeit und Verträglichkeit von Psychopharmaka unter Alltagsbedingungen wurde ein TDM-Netzwerk für Patienten der Kinder- und Jugendpsychiatrie eingerichtet (siehe www.tdm-kjp.de/eng/contact.html).
Bei älteren Patienten, die häufig überempfindlich auf Arzneistoffe reagieren, ist TDM sinnvoll, um zu klären, ob pharmakokinetische oder -dynamische Ursachen vorliegen, wenn unerwünschte Wirkungen auftreten [666]. Altern ist mit stetig zunehmenden Beeinträchtigungen der funktionalen Reserven verschiedener Organe verbunden [407], insbesondere die Niere ist betroffen. Auch die Körperzusammensetzung verändert sich signifikant [361, 374]. Die hepatische Clearance kann bis zu 30% reduziert sein. Dabei sind eher Phase-I-Reaktionen als Phase-II-Reaktionen beeinträchtigt. Auf der anderen Seite gibt es keine generellen altersabhängigen Veränderungen in der CYP-Isoenzym-Aktivität [374]. Altersbedingte Veränderungen in physiologischen und pharmakokinetischen Funktionen sowie die häufige Komorbidität und Polypharmazie erschweren die Pharmakotherapie bei älteren Patienten [125]. Daher steigt mit zunehmendem Alter vor allem die Variabilität der Pharmakokinetik und -dynamik. Das Ansprechen auf Psychopharmaka ist bei Alterspatienten erfahrungsgemäß schwächer als bei jungen Patienten. TDM sollte daher für diese Patienten eingesetzt werden, insbesondere um die Sicherheit und Verträglichkeit zu verbessern.
Bei Personen mit geistiger Behinderung werden bevorzugt Antipsychotika der neuen Generation verwendet. Kürzlich veröffentlichte Leitlinien empfehlen TDM für diese Patienten zumindest dann, wenn sie Risperidon oder Olanzapin erhalten [158]. Aus ethischen und rechtlichen Gründen sind die Patienten mit geistiger Behinderung aus klinischen Studien ausgeschlossen. Auf der anderen Seite müssen viele dieser Patienten Medikamente einnehmen. Bei diesen Personen kann es schwierig sein, zwischen krankheits- und medikamentenbedingten Gründen einer Symptomverschlechterung zu unterscheiden. Zur Abklärung wird TDM empfohlen [158].
In der forensischen Psychiatrie, in der vor allem Antipsychotika zum Einsatz kommen, ist das primäre Behandlungsziel die Unterdrückung von gefährlichem Verhalten [458, 462]. Um konsequent das Risiko für Gewalt und Aggression zu reduzieren, ist die Einnahme der verordneten Medikation von wesentlicher Bedeutung [658]. Daher wird TDM für diese Patientengruppe unbedingt empfohlen. Es ist jedoch nicht nachgewiesen, ob Plasmakonzentrationen, die bei Patienten der Allgemeinpsychiatrie wirksam sind, auch bei forensisch-psychiatrischen Patienten wirksam sind. Castberg und Spigset [113] analysierten Daten einer forensischen Hochsicherheits-Einheit und stellten fest, dass bei forensischen Patienten höhere Dosen als bei einer Kontrollgruppe verabreicht wurden. Die dosisabhängigen Plasmakonzentrationen von Olanzapin waren in der Gruppe der forensischen Patienten im Vergleich zur Kontrollgruppe signifikant niedriger, die von Quetiapin aber höher.
Hinweise auf ein auftretendes Problem nach dem Wechsel von einem Originalpräparat auf ein Generikum (und umgekehrt) sind derzeit noch Gegenstand der Forschung und Daten hierzu sind bislang rar [124, 139].
Eine weitere mögliche Indikation für TDM, welche nicht in Tabelle 6 aufgeführt ist, ergibt sich aus der zunehmenden Verfügbarkeit von gefälschten Arzneimitteln über das Internet [599]. Die WHO hat im Jahr 2006 ein Programm gestartet, um diesen illegalen Handel zu bekämpfen. Es liegen keine Daten über den illegalen Markt für Psychopharmaka vor, aber Patienten können zusätzliche Medikamente (meist zur Selbstmedikation) aus dieser Quelle beziehen. Die Hersteller gefälschter Medikamente halten sich oft nicht an die Vorschriften zu Reinheit und Stabilität und erhöhen somit das Risiko für Wechselwirkungen und unerwünschte Arzneimittelwirkungen.
Praktische Hinweise für TDM in der Psychiatrie
Voraussetzung für einen effektiven TDM-Service ist die Verfügbarkeit von geeigneten analytischen Methoden. Die Ergebnisse sollten dem behandelnden Arzt innert 48 Stunden vorliegen. Zur Ergebnismitteilung gehört auch eine Befundkommentierung durch einen Experten, der über pharmakokinetisches und pharmakotherapeutisches Wissen verfügt [184]. Wie in Abbildung 1 dargestellt, beginnt TDM mit der Anforderung einer Blutspiegelmessung durch den Arzt und endet mit der Entscheidung des Arztes, wie die Psychopharmakotherapie des einzelnen Patienten durch den behandelnden Arzt angepasst werden soll.
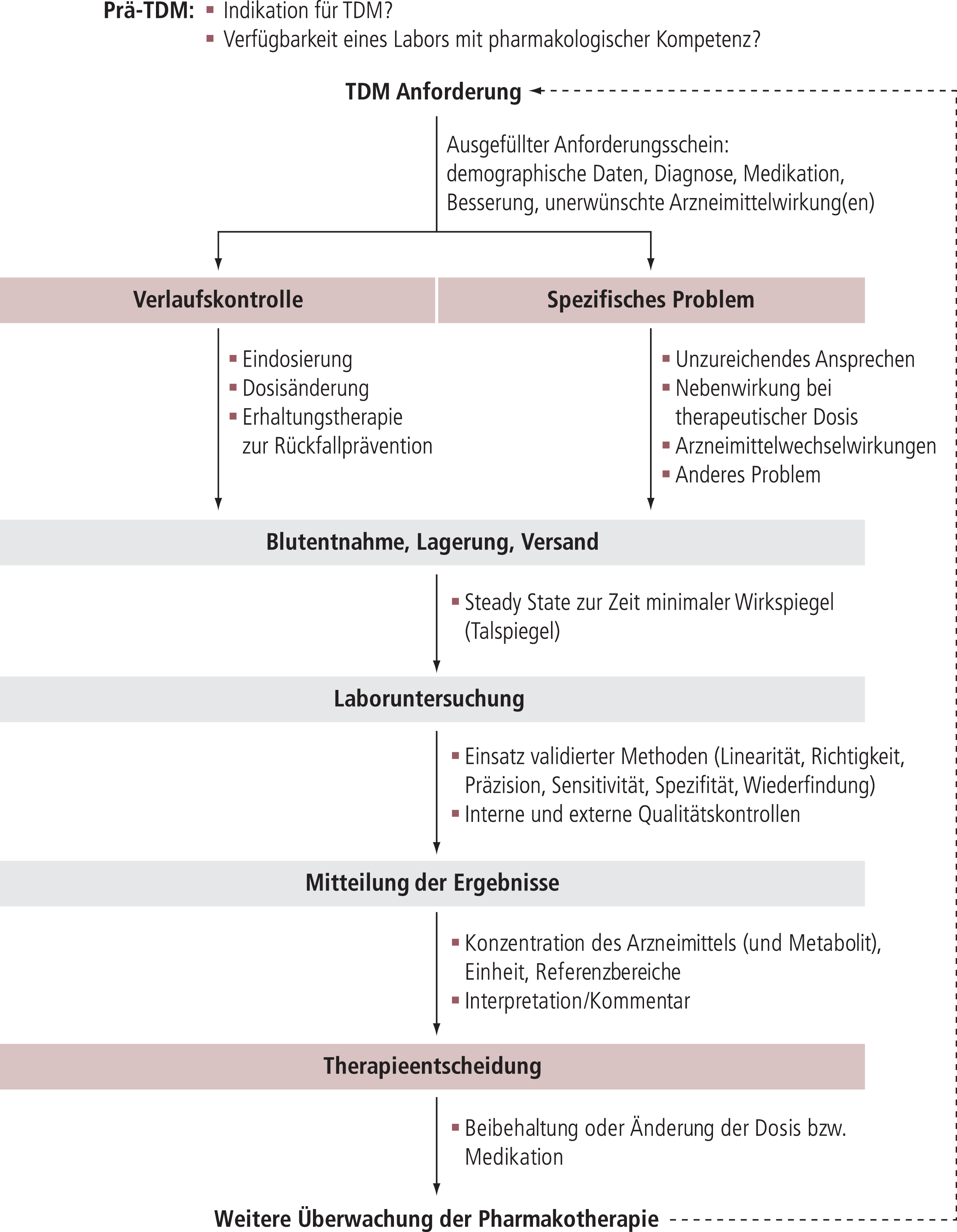
Abb. 1. Schematische Darstellung der Vorgehensweise bei Anwendung von TDM für die Psychopharmakotherapie. TDM ohne spezifische Fragestellung (Verlaufskontrolle) wird in erster Linie bei Arzneistoffen mit engem therapeutischem Index und gut definiertem therapeutischem Referenzbereich angewendet. TDM ist im Einzelfall für jeden Arzneistoff sinnvoll, wenn es um spezielle Probleme geht, wie fehlendes Ansprechen oder Auftreten von Nebenwirkungen unter empfohlener Dosierung (siehe Tab. 6).
Anforderung einer Blutspiegelmessung
Wie bereits erwähnt, sollte eine Blutspiegelmessung nur angefordert werden, wenn es klare Hinweise gibt, dass das Ergebnis eine Antwort auf eine spezielle Frage geben kann. Wer sich über die Fragestellung nicht im Klaren ist, wird von dem Ergebnis wenig Hilfe erwarten können. Typische Indikationen sind in Tabelle 6 aufgelistet. Zur Problemlösung reicht eine einzelne Messung oft nicht aus. Zum Beispiel kann eine Reihe von Messungen in angemessenen Zeitabständen erforderlich sein, um zu klären, ob ein niedriger Blutspiegel entweder durch schlechte Compliance, reduzierte Bioverfügbarkeit oder ungewöhnlich rasche Verstoffwechselung verursacht wird.
Eine TDM-Anforderung erfordert ein ausgefülltes Antragsformular (Abb. 2). Dies ist bedeutsam für eine effektive Laboranalyse und eine angemessene Interpretation der Ergebnisse [501, 635]. Das Formular sollte den Patienten per Namen oder Code identifizieren, sowie demographische Daten, Diagnose, Medikation, Grund für die Anforderung, den Wirkstoffnamen und möglichst auch den Handelsnamen des Medikaments sowie Dosis, Galenik, Zeitpunkt der letzten Änderung der Dosis und der Blutentnahme enthalten. Ein kurzer Kommentar zur klinischen Situation ist notwendig, wenn mit dem Befund Empfehlungen gegeben werden sollen. Wir empfehlen die Verwendung von validerten Ratingskalen zur objektiven Symptomerfassung, wie beispielsweise die Clinical-Global-Impression-(CGI-)Skala [283], um die Schwere der Erkrankung und die therapeutische Verbesserung zu quantifizieren. Um auch die Häufigkeit und Schwere von Nebenwirkungen zu bewerten, ist die Verwendung der UKU-Skala sinnvoll [402]. Die Erfahrung zeigt allerdings, dass es viele behandelnde Ärzte ablehnen oder für überflüssig halten, detaillierte Informationen zu dokumentieren. Die Angaben auf dem Anforderungsschein sind oft unvollständig. Alternativ kann interessierten Ärzten vom Labor eine telefonische Beratung angeboten werden.
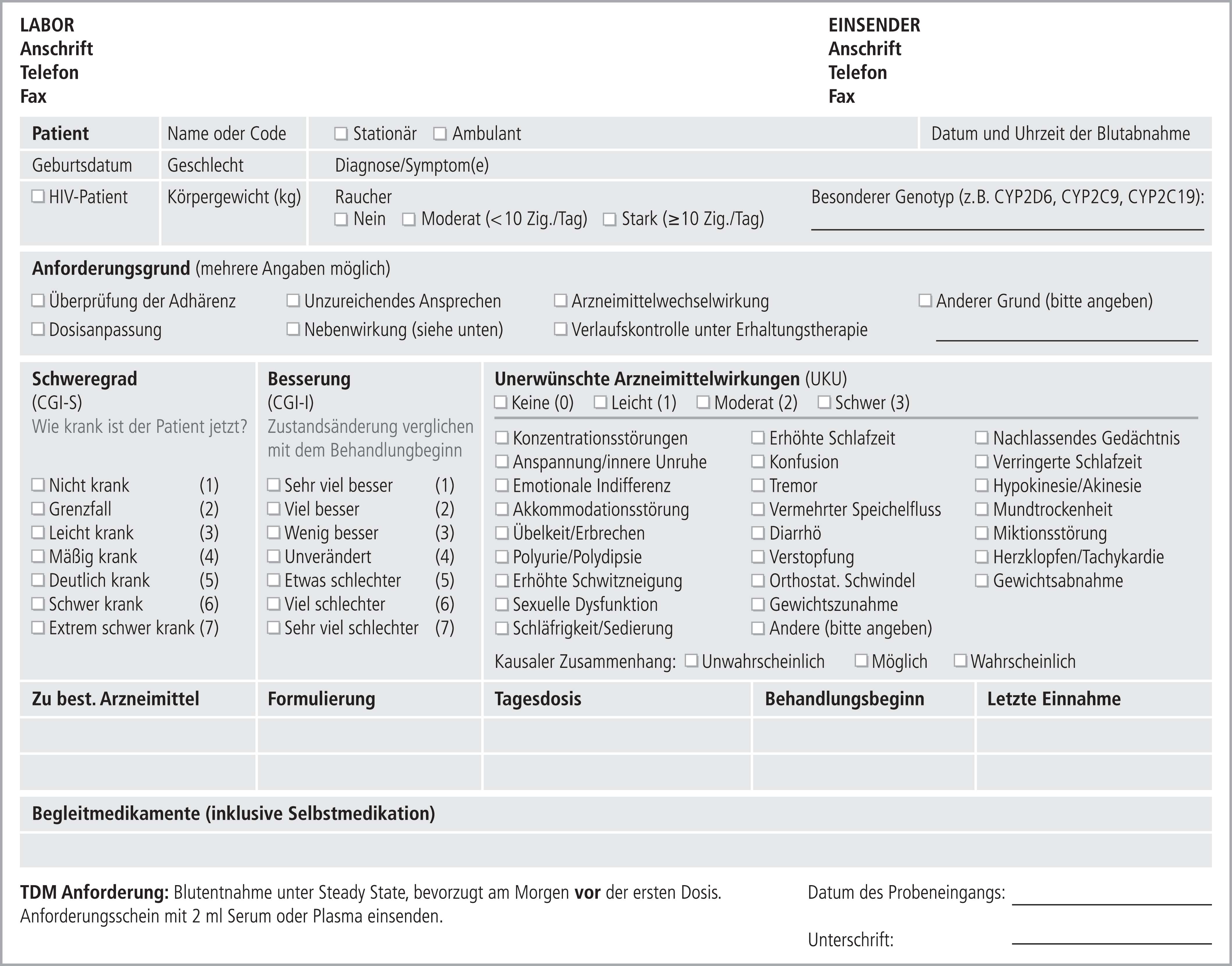
Abb. 2. Formular zur Anforderung einer Blutspiegelmessung
Wie oben ausgeführt, ist es absolut notwendig, das Anforderungsformular ausreichend und vollständig auszufüllen, wenn vom Labor eine Interpretation der Ergebnisse erwartet wird. Dabei hat die Anforderung per EDV Vorteile. Es ist preiswert und übermittelt die für die Interpretation relevanten Informationen auf komfortable Weise. Die computerisierte Anforderung ist jedoch noch nicht weit verbreitet, aber effektive Programme sind entwickelt und werden bald zur Verfügung stehen (z.B. www.konbest.de).
Blutentnahme
Üblicherweise werden für TDM die Arzneistoffkonzentrationen in Plasma- oder Serum-Proben gemessen. Die Analyse von Vollblut, die lange Zeit für Immunsuppressiva mittels Immunoassays verwendet wurde [693], kommt für TDM in der Psychiatrie nicht infrage. Es gibt keinen Konsens, ob Plasma oder Serum bevorzugt werden sollte. Die wenigen verfügbaren Vergleichsuntersuchungen sprechen dafür, dass aus Serum oder Plasma gewonnene Werte gleichgesetzt werden können [308]. Die meisten Psychopharmaka werden intensiv an Blutzellen und Blutproteine gebunden. Für Imipramin wurde gezeigt, dass der Arzneistoff rasch und fast vollständig während einer einzigen Passage durch das Gehirn aus dem Blut ins Hirngewebe extrahiert wird [555]. Die Extraktion wird dabei nicht durch die Anwesenheit von Albumin, Lipoproteine oder Erythrozyten beeinflusst. Dazu passend fand man für Nortriptylin keine signifikante Beziehung zwischen freier Wirkstofffraktion und klinischem Ansprechen [506]. Einen Zusammenhang zwischen klinischem Ansprechen und Blutspiegel ist bisher nur für die Gesamtfraktion der Arzneistoffe gezeigt (gebunden plus frei). Daher beziehen sich die in diesen Leitlinien angegebenen Konzentrationen neuropsychiatrischer Arzneistoffe in Übereinstimmung mit der Literatur jeweils auf die Gesamtfraktion. Die Analyse von Urin, Liquor, Tränenflüssigkeit, Haare oder Muttermilch sind bislang für TDM von Psychopharmaka nicht etabliert. Es gibt bis jetzt keine validen Daten zu therapeutischen Konzentrationen. Die Speichelgewinnung ist eine nicht-invasive Methode [20, 25, 356], was insbesondere bei Kindern vorteilhaft ist. Allerdings entspricht die Wirkstoffkonzentration im Speichel dem nicht proteingebundenen Anteil des Arzneistoffs im Blut. Da für die meisten Psychopharmaka der freie Anteil nur 10% oder weniger der Gesamtkonzentration ausmacht, kann es Nachweisprobleme geben, wenn Speichel statt Plasma oder Serum verwendet wird. In jedem Fall müssen weitere Daten über die Verwendung von Speichel erarbeitet werden.
Bis auf wenige Ausnahmen werden für TDM Talspiegelmessungen unter Steady-State-Bedingungen durchgeführt. Blut sollte daher nach frühestens vier Eliminations-Halbwertszeiten nach Beginn bzw. Änderung einer Dosierung und während der terminalen Beta-Eliminations-Phase abgenommen werden. Für die meisten Psychopharmaka variieren die Halbwertszeiten zwischen 12 und 36 Stunden (Tab. 5). Ausnahmen sind Quetiapin, Trazodon oder Venlafaxin, deren Halbwertszeiten nur etwa 6 Stunden betragen, sowie Fluoxetin und Aripiprazol, die lange Halbwertszeiten haben (Tab. 5). In der klinischen Praxis ist der günstigste Zeitpunkt der Blutentnahme für die meisten Psychopharmaka eine Woche nach Erreichen einer stabilen täglichen Dosierung und unmittelbar vor Einnahme der morgendlichen Dosis. Letztere erfolgt in der Regel im Abstand von 12 bis 16 Stunden nach abendlicher Einnahme oder von 24 Stunden, wenn das Medikament einmal täglich eingenommen wird. Wenn aus logistischen Gründen die Blutentnahme nur am späten Vormittag erfolgen kann, sollte der Patient die morgendliche Medikamenteneinnahme vor der Blutentnahme ausfallen lassen bzw. verschieben. Bei ambulanten Patienten ist es für die Interpretation wichtig, den genauen Zeitpunkt der letzten Dosis anzugeben. Talspiegel können dann durch eine pharmakokinetische Modellrechnung berechnet werden.
Bei Patienten mit einem antipsychotischen Depotpräparat sollte das Blut unmittelbar vor der nächsten Injektion abgenommen werden. Formulierungen von Antipsychotika wie Haloperidoldecanoat oder Risperidon in Mikrosphären sind durch eine langsame Wirkstofffreisetzung nach intramuskulärer Verabreichung charakterisiert. Maximale Plasmakonzentrationen von Depot-Antipsychotika der ersten Generation werden 1 bis 14 Tage nach der Injektion erreicht, und die scheinbare Halbwertszeit liegt bei 2 bis 3 Wochen [647]. Ähnliche Eigenschaften weist das neu eingeführte Paliperidon-Palmitat auf [131]. Für die Freisetzung von Risperidon aus Mikrosphären beträgt die mittlere Zeit bis zur maximalen Konzentration 4 Wochen bei einer Plasmahalbwertszeit von 4 bis 6 Tagen [647]. Für andere Arzneistoffe in retardierten Formulierungen wie Paliperidon [70] oder Quetiapin [212] muss die Zeit der Einnahme der Medikamente für die korrekte Interpretation beachtet werden (siehe Tab. 5). Bei diesen Formulierungen ist der Zeitpunkt der maximalen Plasmakonzentration verzögert, aber die Eliminationshalbwertszeit bleibt im Wesentlichen unverändert. Das lang wirkende Olanzapinpamoat ist eine Depotform, bei der das Salz den Wirkstoff langsam aus dem Muskelgewebe ins Blut freisetzt [399]. Allerdings löst sich der Wirkstoff schnell, wenn das Depot mit Blut in Kontakt kommt. Dann kommt es zu hohen Plasmakonzentrationen und konzentrationsabhängig zu starker Sedierung bis hin zum Delir, dem sogenannten Postinjektionssyndrom [399, 647]. Angesichts dieses speziellen Problems kann es sinnvoll sein, die Plasmakonzentration von Olanzapin kurz, das heißt etwa zwei Stunden nach der Injektion zu messen, um zu prüfen, ob die Blutspiegel rasch ansteigen. Dieser Ansatz erfordert allerdings eine Quantifizierung und Meldung des Olanzapin-Spiegels innerhalb von etwa zwei Stunden.
TDM kann jederzeit nach Medikamenten-Einnahme durchgeführt werden, wenn unerwünschte Arzneimittelwirkungen auftreten. In diesem Falle ist es nicht nötig, Talspiegel zu messen, aber das Dosierungsschema sollte für die Interpretation des Befundes mitgeteilt werden.
Lagerung und Versand
Wenn die Blutproben aufbewahrt oder eingefroren versandt werden sollen, ist es erforderlich, das Serum bzw. Plasma vor dem Einfrieren herzustellen, da es wegen der Hämolyse nicht möglich ist, Serum oder Plasma aus gefrorenem Blut zu gewinnen. Bis auf wenige Ausnahmen können Serum- bzw. Plasmaproben im Dunkeln (bei 4°C) für mindestens 24 Stunden ohne Substanzverlust gelagert werden.
Die meisten Proben können in nicht eingefrorenem Zustand versandt werden [305]. Ausnahmen sind Licht- und/oder Sauerstoff-empfindliche Substanzen. Dies betrifft Bupropion und Methylphenidat. Die Proben müssen sofort nach der Blutentnahme abzentrifugiert oder extrahiert und stabilisiert werden (siehe Tab. 5). Proben für Olanzapin-Bestimmungen müssen tiefgefroren (–20°C) gelagert werden, wenn sie nicht innerhalb von 72 Stunden analysiert werden [305]. Das Labor sollte auf seiner Website und auf dem Anforderungsschein angeben, wie Proben zu sammeln (Blutvolumen, Kennzeichnung der Proben), zu lagern und zu versenden sind.
Laboruntersuchung
Voraussetzung für die erfolgreiche Durchführung von TDM sind selektive und empfindliche Methoden für die quantitative Bestimmung der Arzneistoffe und ihrer Metaboliten (Analyte). Die Methoden müssen validiert sein, das heißt, es muss nachgewiesen werden, dass die verwendete Methode für den vorgesehenen Verwendungszweck zuverlässige und reproduzierbare Ergebnisse erzielt [114, 219]. Als Leistungsmerkmale der Methode sind Angaben zu (1) Genauigkeit, (2) Präzision, (3) Selektivität, (4) Empfindlichkeit, (5) Reproduzierbarkeit und (6) Stabilität erforderlich.
Zum TDM von Psychopharmaka werden üblicherweise chromatographische Techniken wie Gaschromatographie (GC) und Hochleistungs-Flüssigkeitschromatographie (HPLC) in Kombination mit geeigneten Nachweisverfahren bevorzugt [186]. Sie sind ausreichend präzise, genau und robust und für die Analyse der meisten Psychopharmaka geeignet. Nachteilig sind die notwendige Probenvorbereitung vor der chromatographischen Trennung und ein begrenzter Probendurchsatz. Der Durchsatz kann durch automatisierte Probenvorbereitung vor der GC oder HPLC erhöht werden. Einige Labors haben HPLC mit Säulenschaltung eingeführt, was die direkte Injektion von Plasma oder Serum in das HPLC-System erlaubt. Solche Verfahren sind für eine Reihe von Antidepressiva [269, 292–294, 297, 298, 702, 710] und Antipsychotika [368, 369, 571, 572, 711] verfügbar. Eine chromatographische Hochdurchsatz-Methode ist Flüssigkeits-Chromatographie (LC) mit Massenspektrometrie (MS). Mit LC gekoppelt mit einem Tandem-MS (LC-MS/MS) kann praktisch jedes Psychopharmakon einschließlich seiner Metaboliten gemessen werden [577]. Die MS-Methoden sind sehr empfindlich und selektiv und können nach kurzer Probenvorbereitung eingesetzt werden. In einem Lauf können zahlreiche Substanzen identifiziert und quantifiziert werden. Ein hervorragendes Beispiel ist die LC-MS/MS-Methode von Kirchherr und Kühn-Felten [366]. Diese Methode wurde für über 50 psychoaktive Arzneistoffe validiert. Nachteilig für LC-MS/MS-Verfahren sind die hohen Kosten. Darüber hinaus kann die Quantifizierung aufgrund von Ionen-Suppression problematisch sein; hinzu kommt, dass als interne Standards vorzugsweise deuterierte Analoga verwendet werden sollten, die jedoch nur eingeschränkt verfügbar und teuer sind [584].
Bei Verdacht auf Vergiftungen sollte eine Methode mit Analysezeit von 1 bis 2 Stunden verfügbar sein [215]. Dafür eignen sich am besten automatisierte Methoden.
Das Labor sollte nicht nur die Muttersubstanz, sondern auch aktive Metaboliten analysieren, beispielsweise bei Bupropion Hydroxybupropion, bei Clomipramin Norclomipramin, bei Fluoxetin Norfluoxetin, bei Naltrexon Naltrexol, bei Risperidon 9-Hydroxyrisperidon oder bei Venlafaxin O-Desmethylvenlafaxin (Tab. 5). Auch die Bestimmung pharmakologisch nicht aktiver Metaboliten (z.B. Norclozapin, Norsertralin, Normirtazapin, Norcitalopram) ist sinnvoll. Damit kann die Compliance des Patienten beurteilt werden [546] oder es werden Hinweise auf eine individuell auffällige Verstoffwechslung oder auf Wechselwirkungen zwischen Arzneistoffen geliefert (Tab. 2). Die „normalen“ Verhältnisse der Konzentrationen von Metabolit zu Muttersubstanz, die theoretisch bei 68% der Patienten zu erwarten sind, sind in Tabelle 3 aufgeführt. Wenn das Verhältnis außerhalb des „normalen“ Bereichs liegt, sollte dies als Hinweis auf eine mögliche Arzneimittelinteraktion, eine genetisch bedingt veränderte Stoffwechsellage, eine krankheitsbedingt veränderte Leber- oder Nierenfunktion, Non-Compliance oder eine Medikamenteneinnahme kurz vor der Blutentnahme (kein Talspiegel) gewertet werden.
Die Bestimmung der Enantiomere von chiralen Verbindungen erfordert entweder eine stereoselektive Derivatisierung der Arzneistoffe vor ihrer Quantifizierung, oder die Trennung mittels chiraler Chromatographie über spezielle GC- oder HPLC-Säulen. LC-MS ist meist die bevorzugte Methode. Wenn beispielsweise der Nachweis der Enantiomere von Methadon mit Fluoreszenz- oder Ultraviolettspektroskopie geführt wird, kann das Ergebnis durch den Beigebrauch von Drogen gestört werden. Diese Probleme können durch die Verwendung eines Massendetektors umgangen werden, vorzugsweise eines Tandem-Massenspektrometers.
Innerhalb des therapeutischen Referenzbereichs sollten die Variationskoeffizienten der Präzision nicht mehr als 15% betragen, und auch die Richtigkeit sollte nicht mehr als 15% vom Nominalwert abweichen [114, 219].
Um Qualität und Zuverlässigkeit der TDM-Methoden sicherzustellen, sind interne und externe Qualitätskontrollen obligat. Die Proben müssen geeignete interne Standards enthalten und jede Probenserie muss interne Kontrollproben beinhalten. Wenn Standards nicht kommerziell erhältlich sind, sollten sie nicht von den Personen eingewogen und hergestellt werden, die auch für die Durchführung der Tests zuständig sind. Die Weitergabe der TDM-Ergebnisse setzt voraus, dass die Ergebnisse der Qualitätskontrollen innerhalb des erwarteten Bereichs liegen. Wenn Qualitätskontrollen außerhalb des erwarteten Bereichs liegen, muss der Grund für den Ausreißer geklärt und dokumentiert werden.
Das Labor hat sich an einem externen Ringversuch zu beteiligen, obwohl dies nicht in allen Ländern gesetzlich vorgeschrieben ist. Cardiff Bioanalytical Services Ltd. startete 1972 das erste Ringversuchsprogramm für neuropsychiatrische Arzneistoffe [720] und hat 450 Teilnehmer aus 36 Ländern versorgt (www.heathcontrol.com). Im Jahre 2012 wurde der Service von LGC Standards GmbH, Wesel (www.lgcstandards.com) übernommen. Instand e.V. (www.instanddev.de/ringversuche/) ist ein deutscher Anbieter von Ringversuchen, der auf Initiative der TDM-Gruppe der AGNP das Qualitätskontrollprogramm für Psychopharmaka erweiterte. Darüber hinaus werden Ringversuche und externe Referenzmaterialien auch von der Deutschen Gesellschaft für Toxikologie und forensische Chemie angeboten (www.pts-gtfch.de/).
Mitteilung der Ergebnisse
Die Konzentrationen der Arzneistoffe sowie ihrer Metaboliten sollten zusammen mit dem jeweiligen Referenzbereich in Massen- oder Mol-Einheiten berichtet werden (Tab. 5). Wir empfehlen, Masse-Einheiten zu verwenden, um die gemessene Konzentration auf die ebenfalls in Masse-Einheiten angegebene Dosierung beziehen zu können. Die Laboratorien unterscheiden sich in der Darstellung ihrer Ergebnisse zum Teil erheblich. Daher sollte der behandelnde Arzt genau auf die in der Ergebnismitteilung angegebenen Einheiten achten (ng/ml, mg/l, mmol/l oder nmol/l). Dies gilt insbesondere, wenn Befunde aus verschiedenen Laboratorien verglichen oder wenn in der Literatur publizierte Daten herangezogen werden. Um molare Einheiten in Masse-Einheiten umzuwandeln und umgekehrt, sind in Tabelle 5 die Umrechnungsfaktoren angegeben.
Wenn Arzneistoffkonzentrationen unterhalb der Bestimmungsgrenze (BG) gemessen werden, sollte die BG angegeben werden; sie ist durch die niedrigste Konzentration der Standardkurve charakterisiert, die mit mindestens 20% Präzision gemessen werden kann.
Die Ergebnisse sollten für die Entscheidungsfindung innerhalb eines klinisch bedeutsamen Zeitfensters mitgeteilt werden; 24 Stunden sind wünschenswert, eine Bearbeitungszeit von 48 Stunden ist in den meisten Fällen ausreichend. Bei Verdacht auf Vergiftungen muss das Ergebnis allerdings im Einzelfall innerhalb weniger Stunden mitgeteilt werden können [215]. Wenn Arzneistoffkonzentrationen oberhalb der „Warnschwelle des Labors“ gemessen wurden, ist es wichtig, den behandelnden Arzt unmittelbar telefonisch zu informieren, um dem Risiko einer toxischen Reaktion oder Unverträglichkeit rasch entgegenzusteuern. Die Warnschwellen wurden in den vorliegenden Konsens-Leitlinien erstmalig definiert (Tab. 5).
Interpretation der Ergebnisse
Wir empfehlen, jedes Messergebnis zu kommentieren. Um den klinischen Nutzen von TDM auszuschöpfen, ist eine Interpretation durch einen Experten ebenso wichtig wie die angemessene Umsetzung der Informationen in der Klinik. Die dem Ergebnisbericht beigefügten Kommentare und Empfehlungen müssen auf der besten verfügbaren Evidenz beruhen. Um Dosiskorrekturen zu berechnen oder Wechselwirkungen mit anderen Arzneimitteln zu analysieren, ist Expertenwissen notwendig. Für den Kliniker ist es daher vorteilhaft, ein Labor zu wählen, das über die entsprechende Expertise verfügt. Es kann auch ein klinischer Pharmakologe oder ein dafür ausgebildeter Spezialist der Klinik die Aufgabe der Befundinterpretation übernehmen. Der Zugriff auf fachliche Beratung ist auch oft erforderlich, wenn aufgrund des TDM-Ergebnisses eine Genotypisierung empfohlen wird [335].
Die Diagnose und vor allem die Dosis sind wichtig für die Interpretation. Nur so ist es möglich zu prüfen, ob ein Ergebnis plausibel ist oder nicht. Darüber hinaus muss darauf geachtet werden, ob die Blutprobe leitlinienkonform gesammelt wurde. Dies ist vor allem bei ambulanten Patienten bedeutsam. Wenn das Medikament wenige Stunden vor der Blutentnahme eingenommen wurde, kann die Wirkstoffkonzentration um ein Mehrfaches höher sein als der Talspiegel.
Im Zuge der Interpretation der Ergebnisse sollte nicht nur geprüft werden, ob die Plasmakonzentration des Arzneistoffs innerhalb oder außerhalb des „therapeutischen Referenzbereichs“ (Tab. 5) liegt. Es muss auch beachtet werden, ob der Blutspiegel bezogen auf die Dosis plausibel ist (Tab. 4). Die Plasmakonzentration kann außerhalb des therapeutischen Referenzbereichs liegen, weil eine zu niedrige oder zu hohe Dosis eingenommen wurde [285]. Ebenso sollte auch berücksichtigt werden, ob die tägliche Medikamentendosis als Einzel- oder als Mehrfach-Dosis gegeben wurde. Darüber hinaus ist darauf zu achten, wie gut der „therapeutische Referenzbereich“ evaluiert ist (Tab. 5).
Für eine umfassende Interpretation ist es notwendig, die am Metabolismus beteiligten Enzyme sowie Substrat-, Inhibitor- und Induktor-Eigenschaften aller Medikamente des Patienten zu kennen. In den vorliegenden aktualisierten Leitlinien werden dazu unterstützende Informationen in den Tabellen 1 und 3 gegeben.
Immer wenn eine Wirkstoffkonzentration außerhalb der Referenzbereiche gemessen wird, sollte eine Überprüfung stattfinden. Seitens des Patienten ist auf pharmakokinetisch wichtige Genpolymorphismen, veränderte Funktionen der Ausscheidungsorgane Leber und Niere, alters- und/oder krankheitsbedingte Veränderungen und die Compliance (insbesondere bei ambulanten Patienten) zu achten. Aber auch mögliche Störungen der Analytik durch andere Medikamente, die der Patient möglicherweise eingenommen hat, ohne dass sie auf dem Anforderungsschein erwähnt wurden, sind zu bedenken.
Plasmakonzentrationen müssen im Kontext mit der klinischen Situation interpretiert werden. Am häufigsten sind Dosierungsempfehlungen. Informationen, die für den Arzt hilfreich sein können, sind Hinweise auf mögliche genetische Varianten oder pharmakokinetische Interaktionen bei Polypharmazie. Bei Patienten, die einer besonderen Gruppe angehören, beispielsweise Alterspatienten oder Patienten mit Leber- oder Niereninsuffizienz, sollten Hinweise auf pharmakokinetische Besonderheiten gegeben werden. Für die Behandlung von Schmerzen können beispielsweise relativ niedrige Blutspiegel trizyklischer Antidepressiva ausreichend sein. Die Spiegel sollten innerhalb des „dosisabhängigen Referenzbereichs“ (Tab. 4) liegen, können aber außerhalb des „therapeutischen Referenzbereiche“ der Tabelle 5 sein, die für die Indikation Depression erstellt wurde.
Ein Labor kann empfehlen, dass eine weitere Probe abgenommen werden sollte, wenn bei ungewöhnlich niedrigen oder hohen Blutspiegeln eine wiederholte Messung klären kann, ob der Patient das Medikament unregelmäßig einnimmt oder ob eine abnorme Verstoffwechselung vorliegt.
Da die Interpretation eines TDM-Ergebnisses aus komplexen quantitativen Beziehungen erfolgt, ist eine solide Ausbildung in klinischer Psychopharmakologie und Pharmakokinetik für die qualifizierte Befundkommentierung unerlässlich. Regelmäßige Fallkonferenzen mit gemeinsamer Interpretation realer Fälle sind für die Weiterbildung hilfreich. Es wird auch empfohlen, dass angehende Psychiater die Interpretation der Ergebnisse unter Anleitung eines Experten erlernen.
Therapieentscheidung
Der behandelnde Arzt entscheidet, ob eine bestehende Behandlungsstrategie geändert werden soll oder nicht. Der kommentierte Laborbefund ist eine Hilfe für die Dosiseinstellung eines individuellen Patienten. Die Kommentare des Labors erfolgen nach der Datenlage, die dem Labor zugänglich ist [310]. Das Labor hat allerdings begrenzte bis keine Kenntnisse über die klinische Situation. Auf der anderen Seite haben die meisten behandelnden Ärzte begrenzte pharmakokinetische Kenntnisse. Deshalb ist optimales TDM eine interdisziplinäre Aufgabe, die eine enge Kommunikation zwischen Labor und Klinik erfordert.
Wenn die Plasmakonzentration des Arzneistoffs im therapeutischen Referenzbereich liegt, sollte die Dosis oder Medikation nur dann geändert werden, wenn klinische Gründe vorliegen, zum Beispiel eine unerwünschte Arzneimittelwirkung oder fehlendes Ansprechen. Wenn das Labor eine Änderung der Dosis oder Medikation empfiehlt und der Arzt dem nicht folgt, dann muss dies in der Krankenakte dokumentiert werden, damit die Entscheidung beurteilt werden kann, falls der Patient zu Schaden kommt. Für die gutachterliche Bewertung von Psychopharmakaspiegeln im gerichtsabhängigen Schadensfall wurden von der TDM-Gruppe der AGNP Empfehlungen veröffentlicht [741].
Bei Patienten mit ungewöhnlich rascher Elimination kann es sinnvoll sein, eine höhere als die maximal zugelassene Dosis zu verschreiben, da solche Patienten auch bei maximal zugelassener Dosis eine Wirkstoffkonzentration unter dem Referenzbereich haben können. Allerdings sollte die Medikation geändert werden, wenn der Patient für eine ausreichend lange Behandlungszeit, das heißt für mindestens zwei Wochen, ausreichend hohe Wirkstoffkonzentrationen aufgebaut hat und sich die Symptomatik darunter nicht um mindestens 20% verbesserte.
Wenn unter der empfohlenen Dosierung unerwünschte Arzneimittelwirkungen mit einer klinischen Verbesserung assoziiert sind, kann die Messung der Blutspiegel klären, ob die unerwünschten Wirkungen mit hohen Wirkstoffspiegeln im Blut verbunden sind. Wenn Letzteres zutrifft, ist es sinnvoll, die Dosis zu reduzieren. Und es muss nicht befürchtet werden, dass die klinische Wirksamkeit verloren geht.
Für die Behandlung mit Antidepressiva oder Antipsychotika ist umfassend belegt, dass eine ausbleibende klinische Verbesserung innerhalb von zwei Behandlungswochen einen hohen Vorhersagewert für das spätere Ansprechen und die Remission hat [119, 138, 392, 620, 621, 638]. Besonders das Ausbleiben einer frühen Besserung scheint ein zuverlässiger Prädiktor für späteres Therapieversagen zu sein [358]. Für die Dosisfindung von Antidepressiva und Antipsychotika empfehlen wir daher zusätzlich zur Messung der Konzentrationen des Arzneistoffs die Messung der klinischen Symptome zu Beginn der Behandlung und am Ende der zweiten Behandlungswoche [138].
Abbildung 3 fasst die oben genannten Empfehlungen in einem Flussdiagramm zusammen. Wenn weitere Blutspiegelmessungen empfohlen werden, etwa nach Änderung der Dosis oder nach Verschreibung einer Komedikation, die bekanntermaßen zu einer pharmakokinetischen Interaktion führt, sollte abgewartet werden, bis wieder Steady-State-Bedingungen erreicht sind. Dafür ist die terminale Halbwertszeit des Arzneistoffs zu beachten (Tab. 5).
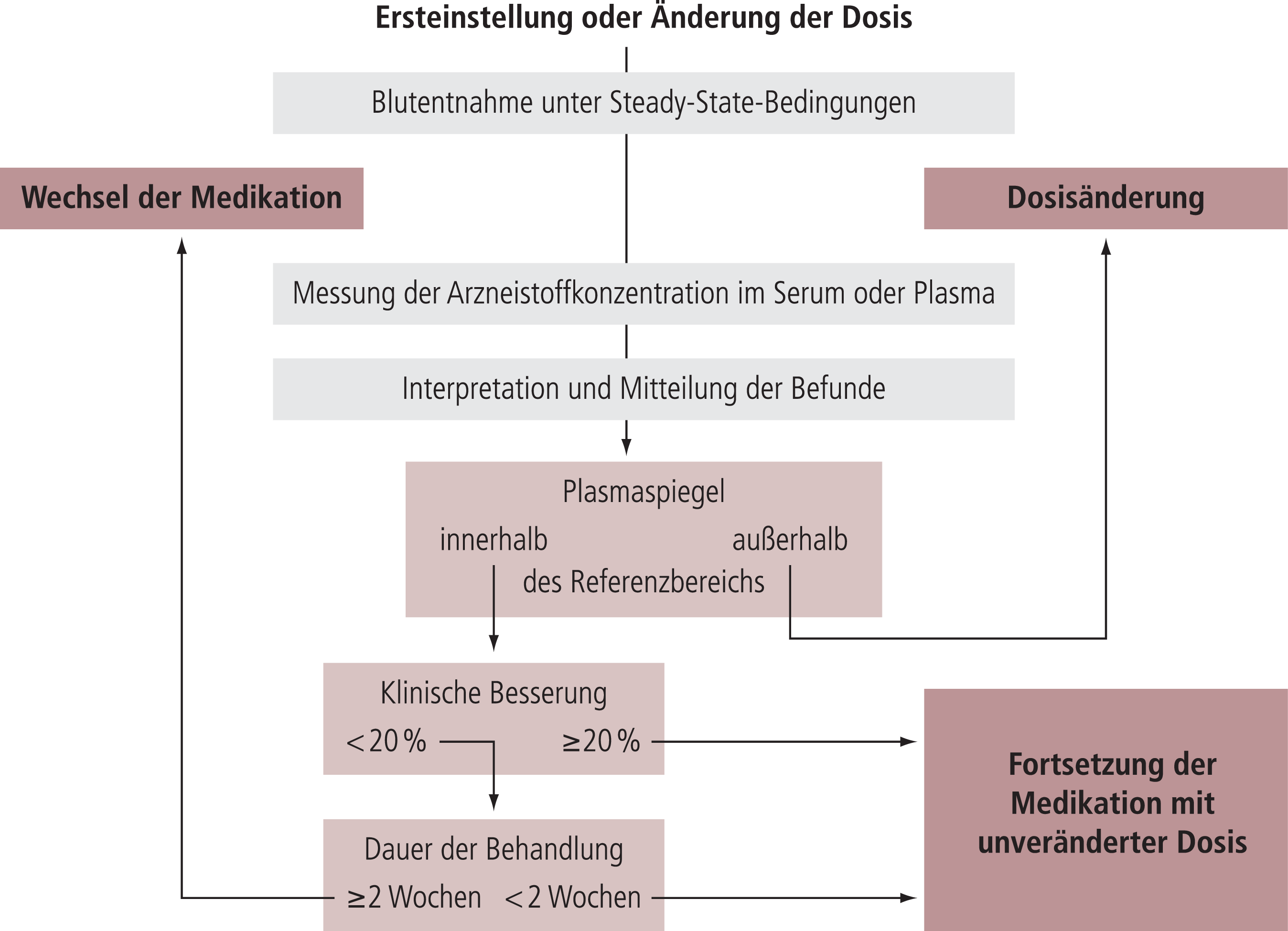
Abb. 3. TDM-geleitete Dosiseinstellung von Antidepressiva oder Antipsychotika [nach 311]. Für die klinische Entscheidungsfindung sind im Wesentlichen klinische Besserung, Dauer der Behandlung und Steady-State-Konzentration des Wirkstoffs zu beachten. Steady State wird nach vier Halbwertszeiten des Arzneistoffs bzw. des aktiven Metaboliten erreicht (siehe Tab. 5).
Pharmakogenetische Tests als Ergänzung zum TDM
Konzentrationen außerhalb des Referenzbereichs können auf einen Genpolymorphismus hinweisen, durch den die Metabolisierung verlangsamt oder beschleunigt ist. Wenn ein solcher Verdacht besteht, sollte das Labor darauf hinweisen, dass ein pharmakogenetischer Test durchgeführt werden sollte [14, 144, 158, 193, 335, 362, 365, 377, 623, 675]. Eine Genotypisierung ist nicht in jedem TDM-Labor verfügbar. Wir empfehlen, ein spezialisiertes Labor zu beauftragen, welches die genetischen Befunde auch interpretieren kann.
Situationen und Fälle, in denen pharmakogenetische Tests sinnvoll mit TDM kombiniert werden können, sind von Jaquenoud Sirot und Mitarbeitern beschrieben [335]. Einige der wichtigsten Indikationen für die Kombination der Genotypisierung mit TDM sind folgende:
- Der Patient wird mit einem Arzneistoff behandelt, von dem eine große interindividuelle Variabilität des Stoffwechsels bekannt ist.
- Der Arzneistoff hat eine enge therapeutische Breite. Im Fall eines genetisch beeinträchtigten Stoffwechsels kann das Risiko einer Intoxikation aufgrund einer verlangsamten Verstoffwechselung des Arzneistoff oder das Risiko von Therapieversagen aufgrund eines beschleunigten Stoffwechsels bestehen.
- Der Patient hat eine abnorme Plasmakonzentration des Wirkstoffs oder seiner Metabolite, für die genetische Faktoren verantwortlich sein können.
- Der Patient leidet an einer chronischen Krankheit, die eine lebenslange Behandlung erfordert.
Bei einem Patienten, der als PM- oder UM-Genotyp identifiziert wurde, sollte nicht, wie von einigen Autoren vorgeschlagen, automatisch das Medikament gewechselt werden, da die Dosis oftmals durch TDM angepasst werden kann.
Schlussfolgerungen und Perspektiven
TDM ist, wenn es adäquat eingesetzt wird, ein wertvolles Instrument für die Optimierung der Pharmakotherapie. In den vergangenen Jahrzehnten gab es umfassende Erkenntnisfortschritte bezüglich Pharmakokinetik und Pharmakodynamik von Psychopharmaka im menschlichen Körper. Die vorliegenden aktualisierten AGNP-Leitlinien beschreiben auf der Basis des aktuellen Wissens die beste Praxis von TDM in der Psychiatrie, um TDM als wirksames Instrument für die Therapieoptimierung einzusetzen.
Obwohl umfangreiche Daten aus Blutspiegeluntersuchungen von Psychopharmaka vorliegen und obwohl unser Wissen über quantitative Beziehungen zwischen Arzneistoffspiegeln im Blut und Ansprechen auf die Therapie gewachsen ist, gibt es weiteren Forschungsbedarf. Für viele Psychopharmaka fehlen kontrollierte und randomisierte Studien zu Konzentrations-Wirkungs-Beziehungen. Sie sind notwendig, um die Qualität der Daten über therapeutische Referenzbereiche zu verbessern. Wir empfehlen die Durchführung von Blutspiegeluntersuchungen während klinischer Studien der Phase III und IV. Viele Produktinformationen müssen mit TDM-Daten ergänzt werden, um die Wirksamkeit und Verträglichkeit der verfügbaren Psychopharmaka TDM-geleitet zu verbessern. Analysen der deutschen [671] und französischen [568] Produktinformationen (SPC, summary of product characteristics) ergaben für Psychopharmaka, dass TDM-relevante Informationen häufig fehlen, obwohl klinisch-wissenschaftliche Evidenz vorliegt. Unbedingter Forschungsbedarf besteht für die Darstellung der Kosten-Effektivität von TDM. Voraussetzung ist bei Studien dazu, dass TDM korrekt eingesetzt wird. Polypharmazie ist in der Psychiatrie sehr verbreitet. Die Untersuchung der Wirksamkeit von Arzneistoff-Kombinationen ist bislang ein zu wenig beachtetes Feld von TDM bzw. der klinischen Pharmakologie.
Die Anwendung der neuen TDM-Leitlinien wird bei vielen psychiatrischen Patienten dazu beitragen, die Pharmakotherapie im klinischen Alltag zu verbessern, insbesondere bei Auftreten pharmakokinetischer Probleme. Dabei sollte immer beachtet werden, dass TDM eine interdisziplinäre Aufgabe ist, die häufig eine respektvolle Diskussion von zunächst widersprüchlich erscheinenden Daten zum Wohle des Patienten erfordert. Die Leitlinien liefern Sachinformationen für solche Diskussionen.
AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2011
Therapeutic drug monitoring (TDM), i.e., the quantification of serum or plasma concentrations of medications for dose optimization, has proven a valuable tool for the patient-matched psychopharmacotherapy. Uncertain drug adherence, suboptimal tolerability, non-response at therapeutic doses, or pharmacokinetic drug-drug interactions are typical situations when measurement of medication concentrations is helpful. Patient populations that may predominantly benefit from TDM in psychiatry are children, pregnant women, elderly patients, individuals with intelligence disabilities, forensic patients, patients with genetically determined pharmacokinetic abnormalities or individuals with pharmacokinetically relevant comorbidities. However, the potential benefits of TDM for optimization of pharmacotherapy can only be obtained if the method is adequately integrated into the clinical treatment process. To promote an appropriate use of TDM, the TDM expert group of the Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP) issued guidelines for TDM in psychiatry in 2004. Since then, knowledge has advanced significantly, and new psychopharmacologic agents have been introduced that are also candidates for TDM. Therefore, the TDM consensus guidelines were updated and extended from 65 psychiatric to 128 neuropsychiatric drugs. Four levels of recommendation for using TDM were defined ranging from “strongly recommended” to “potentially useful”. Evidence-based “therapeutic reference ranges” and “dose related reference ranges” were elaborated after an extensive literature search and a structured internal review process. A “laboratory alert level” was newly introduced, i.e., a plasma level at or above which the laboratory should immediately inform the treating physician. Supportive information such as cytochrome P450 substrate- and inhibitor properties of medications, normal ranges of ratios of concentrations of drug metabolite to parent drug and recommendations for the interpretative services are given. Recommendations when to combine TDM with pharmacogenetic tests are also provided. Following the guidelines will help to improve the outcomes of psychopharmacotherapy of many patients especially in case of pharmacokinetic problems. Thereby, one should never forget that TDM is an interdisciplinary task that sometimes requires the respectful discussion of apparently discrepant data so that, ultimately, the patient can profit from such a joint effort.
Key words: Consensus guidelines, drug analysis, pharmacokinetics, psychotropic drugs, reference range, therapeutic drug monitoring, therapeutic ranges, therapeutic window
Interessenkonflikte
Christoph Hiemke hat ohne persönliche Honorierung Vorträge auf Veranstaltungen gehalten, die von den Firmen AstraZeneca, Bristol-Meyers Squibb, Pfizer, Lilly oder Servier unterstützt wurden, und die Firma Servier beraten. Er ist Geschäftsführer der psiac GmbH, die ein Internet-basiertes Programm für Arzneimittelwechselwirkungen entwickelt hat und über den Springer-Verlag vertrieben wird.
Pierre Baumann wird als Sprecher und Berater von fast allen pharmazeutischen Firmen unterstützt, die Psychopharmaka in der Schweiz zugelassen haben.
Niels Bergemann hat Honorare für Vortrags- oder Beratungstätigkeit von den Firmen AstraZeneca, Bristol-Myers Squibb, Lilly, Novartis, Otsuka, Pfizer, Sanofi-Aventis, Servier und Wyeth erhalten. Er führte Studien für die Firmen AstraZeneca, Bristol-Myers Squibb, Desitin, Janssen-Cilag, Lilly, Otsuka, Pfizer und Servier durch. Seine Forschung wurden von den Firmen AstraZeneca, GlaxoSmithKline, Lilly und Novo Nordisk unterstützt.
Andreas Conca ist Berater der Firmen Lilly, BMS und Pfizer. Er hat Vortragshonorare von den Firmen Lilly, BMS, AstraZeneca, Lundbeck, Italfarma und Janssen erhalten.
Otto Dietmaier hat Vortrags- oder Beraterhonorare von den Firmen Bristol-Myers Squibb, Janssen, Eli Lilly und Lundbeck erhalten.
Ursula Havemann-Reinecke wurde durch die Firmen AstraZeneca, Bristol-Myers Squibb, Cephalon, Essex, Janssen Cilag, Lundbeck, Pfizer, Schering-Plough und Wyeth unterstützt.
Ekkehard Haen wurde als Berater und für Vorträge durch die Firmen Janssen-Cilag, Lilly, Pfizer, GlaxoSmithKline, AstraZeneca, Bristol-Myers Squibb, Otsuka, Bayer Vital, Servier und Südmedica GmbH unterstützt.
Karin Egberts wurde für Vorträge und Kongressreisen von den Firmen Wyeth und Medice unterstützt. Sie hat sich an klinischen Studien der Firmen AstraZeneca, Janssen-Cilag, Lilly und Shire beteiligt.
Gerhard Gründer wurde durch die Firmen AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson und Otsuka für seine Beratertätigkeit unterstützt. Er hat Vortragshonorare von den Firmen AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen Cilag, Otsuka, Pfizer, Servier und Wyeth erhalten. Seine Forschung wurde durch die Firmen Alkermes, Bristol-Myers Squibb, Eli Lilly und Johnson & Johnson unterstützt. Er ist Mitgründer der Pharma-Image Molecular Imaging Technologies GmbH.
Eveline Jaquenoud Sirot ist Geschäftsführerin von mediQ, die ein Internet-basiertes Programm für Arzneimittelwechselwirkungen in der Psychiatrie entwickelt und vertreibt.
Gerd Laux wurde durch die Firmen AstraZeneca, Bayer, Eli Lilly, Lundbeck, Merz, Pfizer, Servier und Wyeth unterstützt.
Bruno Pfuhlmann wurde für Vorträge oder Beratungstätigkeiten von den Firmen AstraZeneca, Janssen und Pfizer honoriert.
Manfred Gerlach wurde für Vorträge, Beratertätigkeit und Forschungsprojekte von den Firmen Boehringer Ingelheim Pharma GmbH & Co. KG, Desitin Arzneimittel GmbH, Janssen Cilag GmbH, Lundbeck GmbH und Merz Pharmaceuticals GmbH honoriert.
Thomas Messer wurde für Tätigkeiten als Vortragender und Berater und für Forschungsprojekte durch die Firmen Eli Lilly, Bristol-Myers Squibb, Janssen, Servier, Pfizer, Lundbeck und Bayer Vital GmbH/Bayer HealthCare Deutschland unterstützt.
Matthias J. Müller wurde durch die Firmen Janssen, Servier, Pfizer und AstraZeneca für Vortrags- und Beratertätigkeiten unterstützt.
Sven Ulrich ist Angestellter der Firma Aristo Pharma GmbH, Berlin, Germany.
Gerald Zernig wurde für Vortrags- und Beratertätigkeiten und für Forschungsaktivitäten durch die Firmen AlcaSynn, AstraZeneca, Bio-Rad, Bristol-Myers Squibb, Eli Lilly, Lundbeck, Mundipharma, Novartis, Pfizer und Wyeth unterstützt.
Niels Bergemann, Mirijam Fric, Christine Greiner, Hartmut Kirchherr, Ulrich C. Lutz, Bernhard Rambeck, Bernd Schoppek, Julia C Stingl, Manfred Uhr, Roland Waschgler und alle oben genannten Autoren erklären, dass keine Interessenkonflikte zu dieser Publikation bestehen.
Danksagung
Die Autoren danken Sonja Brünen, Elnaz Ostad Haji, Christiane Knoth und Viktoria Stieffenhofer für die Hilfe bei der aufwendigen Erstellung der Tabelle 2. Sie bedanken sich bei Ralf Köber für die Mitwirkung bei der Evaluierung der therapeutischen und dosisbezogenen Referenzbereiche von Antidementiva. Ein besonderer Dank gilt Michaela Jahnke, Christiane Kobelt und Nina Wenzel für die außerordentliche Hilfe bei der Erstellung der 749 Referenzen umfassenden Literaturliste für diese Arbeit. Danke auch an Gudrun Hefner und Kathrin Laib für die sorgfältige Durchsicht der Übersetzung.
Literatur
Das ausführliche Literaturverzeichnis finden Sie unter "Archiv", "Literatur", Heft 03/2012
1. Abbott NJ, Patabendige AA, Dolman DE, et al. Structure and function of the blood–brain barrier. Neurobiol Dis 2010;37:13–25.
2. Abernethy DR, Greenblatt DJ, Shader RI. Imipramine and desipramine disposition in the elderly. J Pharmacol Exp Ther 1985;232:183–188.
3. Adam K, Oswald I. Effects of lormetazepam and of flurazepam on sleep. Br J Clin Pharmacol 1984;17:531–538.
4. Adamiak U, Kaldonska M, Klodowska-Duda G, et al. Pharmacokinetic-pharmacodynamic modeling of levodopa in patients with advanced Parkinson disease. Clin Neuropharmacol 2010;33:135–141.
5. Addington D. Best practices: improving quality of care for patients with first–episode psychosis. Psychiatr Serv 2009;60:1164–1166.
6. Adli M, Baethge C, Heinz A, et al. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? Eur Arch Psychiatry Clin Neurosci 2005;55:387–400.
7. Aichhorn W, Marksteiner J, Walch T, et al. Influence of age, gender, body weight and valproate comedication on quetiapine plasma concentrations. Int Clin Psychopharmacol 2006;21:81–85.
8. Aichhorn W, Weiss U, Marksteiner J, et al. Influence of age and gender on risperidone plasma concentrations. J Psychopharmacol 2005;19:395–401.
9. Aichhorn W, Whitworth AB, Weiss ME, et al. Second-generation antipsychotics: Is there evidence for sex differences in pharmacokinetic and adverse effect profiles? Drug Saf 2006;29:587–598.
10. Åkerblad AC, Bengtsson F, Ekselius L, et al. Effects of an educational compliance enhancement programme and therapeutic drug monitoring on treatment adherence in depressed patients managed by general practitioners. Int Clin Psychopharmacol 2003;18:347–354.
11. Åkerblad AC, Bengtsson F, Holgersson M, et al. Identification of primary care patients at risk of nonadherence to antidepressant treatment. Patient Prefer Adherence 2008;2:379–386.
12. Aklillu E, Kalow W, Endrenyi L, et al. CYP2D6 and DRD2 genes differentially impact pharmacodynamic sensitivity and time course of prolactin response to perphenazine. Pharmacogenet Genomics 2007;17:989–993.
13. Akutsu T, Kobayashi K, Sakurada K, et al. Identification of human cytochrome p450 isozymes involved in diphenhydramine N-demethylation. Drug Metab Dispos 2007;35:72–78.
14. Albers LJ, Ozdemir V, Marder SR, et al. Low–dose fluvoxamine as an adjunct to reduce olanzapine therapeutic dose requirements: a prospective dose–adjusted drug interaction strategy. J Clin Psychopharmacol 2005;25:170–174.
15. Alderman J, Wolkow R, Fogel IM. Drug concentration monitoring with tolerability and efficacy assessments during open–label, long–term sertraline treatment of children and adolescents. J Child Adolesc Psychopharmacol 2006;16:117–129.
16. Allen MH, Hirschfeld RM, Wozniak PJ, et al. Linear relationship of valproate serum concentration to response and optimal serum levels for acute mania. Am J Psychiatry 2006;163:272–275.
17. Allqvist A, Miura J, Bertilsson L, et al. Inhibition of CYP3A4 and CYP3A5 catalyzed metabolism of alprazolam and quinine by ketoconazole as racemate and four different enantiomers. Eur J Clin Pharmacol 2007;63:173–179.
18. Altamura AC, Moro AR, Percudani M. Clinical pharmacokinetics of fluoxetine. Clin Pharmacokinet 1994;26:201–214.
19. Althaus M, Retzow A, Castell JV, et al. In vitro identification of the cytochrome P450 isoform responsible for the metabolism of alpha–dihydroergocryptine. Xenobiotica 2000;30:1033–1045.
20. Aman MG, Vinks AA, Remmerie B, et al. Plasma pharmacokinetic characteristics of risperidone and their relationship to saliva concentrations in children with psychiatric or neurodevelopment disorders. Clin Therap 2007;29:1476–1486.
21. Anderson D, Reed S, Lintemoot J, et al. A first look at duloxetine (Cymbalta®) in a post-mortem laboratory. J Analyt Toxicology 2006;30:576–579.
22. Anderson GD. Pregnancy–induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet 2005;44:989–1008.
23. Anderson IM, Ferrier IN, Baldwin RC, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol 2008;22:343–396.
24. Aoki FY, Sitar DS. Clinical pharmacokinetics of amantadine hydrochloride. Clin Pharmacokinet 1988;14:35–51.
25. Aps JK, Martens LC. Review: The physiology of saliva and transfer of drugs into saliva. Forensic Sci Int 2005;150:119–131.
26. Arakawa R, Ito H, Takano A, et al. Dose–finding study of paliperidone ER based on striatal and extrastriatal dopamine D2 receptor occupancy in patients with schizophrenia. Psychopharmacology (Berl) 2008;197:229–235.
27. Aravagiri M, Marder SR, Yuwiler A, et al. Distribution of fluphenazine and its metabolites in brain regions and other tissues of the rat. Neuropsychopharmacology 1995;13:235–247.
28. Aravagiri M, Teper Y, Marder SR. Pharmacokinetics and tissue distribution of olanzapine in rats. Biopharm Drug Dispos 1999;20:369–377.
29. Aravagiri M, Yuwiler A, Marder SR. Distribution after repeated oral administration of different dose levels of risperidone and 9-hydroxy-risperidone in the brain and other tissues of rat. Psychopharmacology 1998;139:356–363.
30. Åsberg M, Cronholm B, Sjöqvist F, et al. Correlation of subjective side effects with plasma concentrations of nortriptyline. Br Med J 1970;5726:18–21.
31. Åsberg M, Crönholm B, Sjöqvist F, et al. Relationship between plasma level and therapeutic effect of nortriptyline. Br Med J 1971;3:331–334.
32. Bachmann CJ, Haberhausen M, Heinzel-Gutenbrunner M, et al. Large intraindividual variability of olanzapine serum concentrations in adolescent patients. Ther Drug Monit 2008;30:108–112.
33. Bachmann CJ, Rieger-Gies A, Heinzel–Gutenbrunner M, et al. Large variability of aripiprazole and dehydroaripiprazole serum concentrations in adolescent patients with schizophrenia. Ther Drug Monit 2008;30:462–466.
34. Bachus R, Bickel U, Thomsen T, et al. The O-demethylation of the antidementia drug galanthamine is catalysed by cytochrome P450 2D6. Pharmacogenetics 1999;9:661–668.
35. Backman JT, Olkkola KT, Ojala M, et al. Concentrations and effects of oral midazolam are greatly reduced in patients treated with carbamazepine or phenytoin. Epilepsia 1996;37:253–257.
36. Bagli M, Höflich G, Rao ML, et al. Bioequivalence and absolute bioavailability of oblong and coated levomepromazine tablets in CYP2D6 phenotyped subjects. Int J Clin Pharmacol Ther 1995;33:646–652.
37. Baker GB, Urichuk LJ, McKenna KF, et al. Metabolism of monoamine oxidase inhibitors. Cell Mol Neurobiol 1999;19:411–426.
38. Bakken GV, Rudberg I, Christensen H, et al. Metabolism of quetiapine by CYP3A4 and CYP3A5 in presence or absence of cytochrome B5. Drug Metab Dispos 2009;37:254–258.
39. Balant LP, Balant–Gorgia AE, Eisele R, et al. Clinical and pharmacokinetic evaluation of zuclopenthixol acetate in Viscoleo. Pharmacopsychiatry 1989;22:250–254.
40. Balant-Gorgia AE, Eisele R, Aeschlimann JM, et al. Plasma flupentixol concentrations and clinical response in acute schizophrenia. Ther Drug Monit 1985;7:411–414.
41. Barbhaiya RH, Shukla UA, Pfeffer M, et al. Disposition kinetics of buspirone in patients with renal or hepatic impairment after administration of single and multiple doses. Eur J Clin Pharmacol 1994;46:41–47.
42. Bareggi SR, Bianchi L, Cavallaro R, et al. Citalopram concentrations and response in obsessive-compulsive disorder – Preliminary results. CNS Drugs 2004;18:329–335.
43. Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev 2008;40:553–624.
44. Baruzzi A, Bordo B, Bossi L, et al. Plasma levels of di–no–propylacetate and clonazepam in epileptic patients. Int J Clin Pharmacol Biopharm 1977;15:403–408.
45. Bates DW, Gawande AA. Improving safety with information technology. N Engl J Med 2003;348:2526–2534.
46. Bauer M, Whybrow PC, Angst J, et al. World Federation of Societies Biological Psychiatry Task Force on Treatment Guidelines for Unipolar Depressive Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Unipolar Depressive Disorders, Part 1:Acute and continuation treatment of major depressive disorder. World J Biol Psychiatry 2002;3:5–43.
47. Bauer S, David Rudd G, Mylius V., et al. Lacosamide intoxication in attempted suicide. Epilepsy Behav 2010;17:549–551 .
48. Baumann P. Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors. Clin Pharmacokinet 1996;31:444–469.
49. Baumann P, Barbe R, Vabre-Bogdalova A, et al. Epileptiform seizure after sertraline treatment in an adolescent experiencing obsessive-compulsive disorder and presenting a rare pharmacogenetic status. J Clin Psychopharmacol 2006;26:679–681.
50. Baumann P, Hiemke C, Ulrich S, et al. Le dosage plasmatique des médicaments psychotropes à des fins thérapeutiques: recommandations du groupe d’experts AGNP–TDM. Rev Med Suisse 2006;2:1413–1418.
51. Baumann P, Hiemke C, Ulrich S, et al. The AGNP-TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry 2004;37:243–265.
52. Baumann P, Ulrich S, Eckermann G, et al. The AGNP–TDM Expert Group Consensus Guidelines: focus on therapeutic monitoring of antidepressants. Dialogues Clin Neurosci 2005;7:231–247.
53. Baumann P, Zullino DF, Eap CB. Enantiomers‘ potential in psychopharmacology – a critical analysis with special emphasis on the antidepressant escitalopram. Eur Neuropsychopharmacol 2002;12:433–444.
54. Bazire S. Psychotropic Drug Directory 2010. Aberdeen: Healthcomm UK Ltd, 2011.
55. Beasley CM Jr, Stauffer VL, Liu–Seifert H, et al. All-cause treatment discontinuation in schizophrenia during treatment with olanzapine relative to other antipsychotics: an integrated analysis. J Clin Psychopharmacol 2007;27:252–258.
56. Bech P, Gex-Fabry M, Aubry JM, et al. Olanzapine plasma level in relation to antimanic effect in the acute therapy of manic states. Nord J Psychiatry 2006;60:181–182.
57. Becquemont L, Mouajjah S, Escaffre O, et al. Cytochrome P-450 3A4 and 2C8 are involved in zopiclone metabolism. Drug Metab Dispos 1999;27:1068–1073.
58. Beedham C, Miceli JJ, Obach RS. Ziprasidone metabolism, aldehyde oxidase, and clinical implications. J Clin Psychopharmacol 2003;23:229–232.
59. Benedetti MS, Whomsley R, Poggesi I, et al. Drug metabolism and pharmacokinetics. Drug Metab Rev 2009;41:344–390.
60. Benetton SA, Fang C, Yang YO, et al. P450 phenotyping of the metabolism of selegiline to desmethylselegiline and methamphetamine. Drug Metab Pharmacokinet 2007;22:78–87.
61. Bengtsson F. Therapeutic drug monitoring of psychotropic drugs. TDM „nouveau“. Ther Drug Monit 2004;26:145–151.
62. Bennett JP Jr, Piercey MF. Pramipexole: a new dopamine agonist for the treatment of Parkinson’s disease. J Neurol Sci 1999;163:25–31.
63. Bergemann N, Frick A, Parzer P, et al. Olanzapine plasma concentration, average daily dose, and interaction with co-medication in schizophrenic patients. Pharmacopsychiatry 2004;37:63–68.
64. Bergemann N, Kopitz J, Kress KR, et al. Plasma amisulpride levels in schizophrenia or schizoaffective disorder. Eur Neuropsychopharmacology 2004;14:245–250.
65. Bergemann N, Rommel F, Conca A. Therapeutisches Drug-Monitoring von Psychopharmaka in der Schwangerschaft. J Neurol Neurochir Psychiatr 2009;10:38–40.
66. Bergmann TK, Bathum L, Brøsen K. Duplication of CYP2D6 predicts high clearance of desipramine but high clearance does not predict duplication of CYP2D6. Eur J Clin Pharmacol 2001;57:123–127.
67. Bergstrom RF, Cerimele BJ. Olanzapine in subjects with and without renal failure (data on file). Lilly Laboratory for Clinical Research. Eli Lilly and Co. 1996.
68. Berry D, Millington C. Analysis of pregabalin at therapeutic concentrations in human plasma/serum by reversed–phase HPLC. Ther Drug Monit 2005;27:451–456.
69. Bertelsen KM, Venkatakrishnan K, von Moltke LL, et al. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Metab Dispos 2003;31: 289–293.
70. Berwaerts J, Cleton A, Rossenu S, et al. A comparison of serum prolactin concentrations after administration of paliperidone extended–release and risperidone tablets in patients with schizophrenia. J Psychopharmacol 2010;24:1011–1018.
71. Bigliani V, Mulligan RS, Acton PD, et al. Striatal and temporal cortical D2/D3 receptor occupancy by olanzapine and sertindole in vivo: a [123I]epidepride single photon emission tomography (SPET) study. Psychopharmacology (Berl) 2000;150:132–140.
72. Birkenhäger TK, van den Broek WW, Moleman P, et al. Imipramine dose in relation to therapeutic plasma level: are clinical trials using imipramine as a positive control flawed? Psychopharmacology (Berl) 2005;181:595–599.
73. Bjerkenstedt L, Flyckt L, Overø KF, et al. Relationship between clinical effects, serum drug concentration and serotonin uptake inhibition in depressed patients treated with citalopram. A double-blind comparison of three dose levels. Eur J Clin Pharmacol 1985;28:553–557.
74. Bjørndal N, Bjerre M, Gerlach J, et al. High dosage haloperidol therapy in chronic schizophrenic patients: a double–blind study of clinical response, side effects, serum haloperidol, and serum prolactin. Psychopharmacology (Berl) 1980;67:17–23.
75. Bockbrader HN. Clinical pharmacokinetics of gabapentin. Drugs Today 1995;31:613–619.
76. Bockbrader HN, Burger P, Knapp L, et al. Population pharmacokinetics of pregabalin in healthy subjects and patients with chronic pain or partial seizures. Epilepsia 2011;52:248–257.
77. Bockbrader HN, Wesche D, Miller R, et al. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin Pharmacokinet 2010;49:661–669.
78. Bogaards JJ, Hissink EM, Briggs M, et al. Prediction of interindividual variation in drug plasma levels in vivo from individual enzyme kinetic data and physiologically based pharmacokinetic modeling. Eur J Pharm Sci 2000;12:117–124.
79. Bomsien S, Aderjan R, Mattern R, et al. Effect of psychotropic medication on the in vitro metabolism of buprenorphine in human cDNA-expressed cytochrome P450 enzymes. Eur J Clin Pharmacol 2006;62:639–643.
80. Bond A, Seijas D, Dawling S, et al. Systemic absorption and abuse liability of snorted flunitrazepam. Addiction 1994;89:821–830.
81. Bondolfi G, Morel F, Crettol, et al. Increased clozapine plasma concentrations and side effects induced by smoking cessation in 2 CYP1A2 genotyped patients. Ther Drug Monit 2005;27:539–543.
82. Bont L, Bosker HA, Brus F, et al. Torsade de pointes after pipamperone intoxication. Pharm World Sci 1998;20:137.
83. Borgstrom L, Larsson H, Molander L. Pharmacokinetics of parenteral and oral melperone in man. Eur J Clin Pharmacol 1982;23:173–176.
84. Borys DJ, Setzer SC, Ling LJ, et al. Acute fluoxetine overdose: a report of 234 cases. Am J Emerg Med 1992;10:115–120.
85. Bosse GM, Spiller HA, Collins AM. A fatal case of venlafaxine overdose. J Med Toxicol 2008;4:18–20.
86. Brachtendorf L, Jetter A, Beckurts KT, et al. Cytochrome P450 enzymes contributing to demethylation of maprotiline in man. Pharmacol Toxicol 2002;90:144–149.
87. Brahmi N, Kouraichi N, Abderrazek H, et al. Clinical experience with carbamazepine overdose: relationship between serum concentration and neurological severity. J Clin Psychopharmacol 2008;28:241–243.
88. Brandt C, Baumann P, Eckermann G, et al. Therapeutic drug monitoring in Epileptologie und Psychiatrie [Therapeutic drug monitoring in epileptology and psychiatry]. Nervenarzt 2008;79:167–174.
89. Bressan RA, Erlandsson K, Jones HM, et al. Is regionally selective D2/D3 dopamine occupancy sufficient for atypical antipsychotic effect? An in vivo quantitative [123I] epidepride SPET study of amisulpride–treated patients. Am J Psychiatry 2003;160:1413–1420.
90. Breyer-Pfaff U. The metabolic fate of amitriptyline, nortriptyline and amitriptyline oxide in man. Drug Metab Rev 2004;36:723–746.
91. Breyer-Pfaff U, Brinkschulte M, Rein W, et al. Prediction and evaluation criteria in perazine therapy of acute schizophrenics pharmacokinetic data. Pharmacopsychiatry 1983;16:160–165.
92. Breyer-Pfaff U, Nill K. Carbonyl reduction of naltrexone and dolasetron by oxidoreductases isolated from human liver cytosol. J Pharm Pharmacol 2004;56:1601–1606.
93. Brockmöller J, Kirchheiner J, Schmider J, et al. The impact of the CYP2D6 polymorphism on haloperidol pharmacokinetics and on the outcome of haloperidol treatment. Clin Pharmacol Ther 2002;72:438–452.
94. Brøsen K. Drug-metabolizing enzymes and therapeutic drug monitoring in psychiatry. Ther Drug Monit 1996;18:393–396.
95. Brøsen K, Gram LF, Klysner R, et al. Steady-state levels of imipramine and its metabolites: significance of dose-dependent kinetics. Eur J Clin Pharmacol 1986;30:43–49.
96. Brøsen K, Klysner R, Gram LF, et al. Steady-state concentrations of imipramine and its metabolites in relation to the sparteine/debrisoquine polymorphism. Eur J Clin Pharmacol 1986;30:679–684.
97. Brøsen K, Naranjo CA. Review of pharmacokinetic and pharmacodynamic interaction studies with citalopram. Eur Neuropsychopharmacol 2001;11:275–283.
98. Bruijn JA, Moleman P, Mulder PG, et al. A double-blind, fixed blood-level study comparing mirtazapine with imipramine in depressed in-patients. Psychopharmacology (Berl) 1996;127:231–237.
99. Brünen S, Vincent DP, Baumann P, et al. Therapeutic Drug Monitoring (TDM) for drugs used in the treatment of substance related disorders. Literature review using a TDM appropriateness rating scale. Ther Drug Monit 2011;in press.
100. Brunton LL, Lazo JS, Parker KL. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th edn. McGraw-Hill: New York, 2006 .
101. Buchanan RW, Kreyenbuhl J, Kelly DL, et al. Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull 2010;36:71–93.
102. Buckley NA, Dawson AH, Whyte IM, et al. Greater toxicity in overdose of dothiepin than of other tricyclic antidepressants. Lancet 1994;343:159–162 .
103. Burke MJ, Preskorn SH. Therapeutic drug monitoring of antidepressants – cost implications and relevance to clinical practice. Clin Pharmacokinet 1999;37:147–165.
104. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatr Serv 2007;58:844–847.
105. Caccia S, Garattini S. Pharmacokinetic and pharmacodynamic significance of antidepressant drug metabolites. Pharmacol Res 1992;26:317–329.
106. Caccia S, Pasia L, Nobili L. New atypical antipsychotics for schizophrenia: iloperidone. Drug Des Devel Ther 2010;4:33–48.
107. Callaghan JT, Bergstrom RF, Ptak LR, et al. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet 1999;37:177–193.
108. Canal M, Desanti CR, Santoni JP. A new oral formulation of tiapride (drops): pharmacokinetic profile and therapeutic applications. Clin Drug Investig 1998;15:455–460.
109. Canal-Raffin M, Déridet E, Titier K, et al. Simplified ultraviolet liquid chromatographic method for determination of sertindole, dehydrosertindone and norsertindole, in human plasma. J Chromatography B 2005;814:61–67.
110. Canal–Raffin M, Titier K, Déridet E, et al. Myocardium distribution of sertindole and its metabolite dehydrosertindole in guinea-pigs. Biopharm Drug Dispos 2006;27:171–179.
111. Carlsson B, Olsson G, Reis M, et al. Enantioselective analysis of citalopram and metabolites in adolescents. Ther Drug Monit 2001;23:658–664.
112. Castberg I, Skogvoll E, Spigset O. Quetiapine and drug interactions: evidence from a routine therapeutic drug monitoring service. J Clin Psychiatry 2007;68:1540–1545.
113. Castberg I, Spigset O. Prescribing pattern and use of therapeutic drug monitoring of psychotropic medication in a psychiatric high-security unit. Ther Drug Monit 2008;30:597–603.
114. Causon R. Validation of chromatographic methods in biomedical analysis. Viewpoint and discussion. J Chromatogr B Biomed Sci Appl 1997;689:175–180.
115. Cawello W, Braun M, Boekens H. Absorption, disposition, metabolic fate, and elimination of the dopamine agonist rotigotine in man: administration by intravenous infusion or transdermal delivery. Drug Metab Dispos 2009;37:2055–2060.
116. Centerholt C, Ekblom M, Odergren T, et al. Pharmacokinetics and sedative effects in healthy subjects and subjects with impaired liver function after continuous infusion of clomethiazole. Eur J Clin Pharmacol 2003;59:117–122.
117. Chakraborty BS, Midha KK, McKay G, et al. Single dose kinetics of thioridazine and its two psychoactive metabolites in healthy humans: a dose proportionality study. J Pharm Sci 1989;78:796–801.
118. Chan V, Morris RG, Ilett KF, et al. Population pharmacokinetics of lamotrigine. Ther Drug Monit 2001;23:630–635.
119. Chang YC, Lane HY, Yang KH, et al. Optimizing early prediction for antipsychotic response in schizophrenia. J Clin Psychopharmacol 2006;26:554–559.
120. Chawarski MC, Schottenfeld RS, O‘Connor PG, et al. Plasma concentrations of buprenorphine 24 to 72 hours after dosing. Drug Alcohol Depend 1999;55:157–163.
121. Chen H, Grover S, Yu L, et al. Bioactivation of lamotrigine in vivo in rat and in vitro in human liver microsomes, hepatocytes, and epidermal keratinocytes: characterization of thioether conjugates by liquid chromatography/mass spectrometry and high field nuclear magnetic resonance spectroscopy. Chem Res Toxicol 2010;23:159–170.
122. Chen P, Tanasijevic MJ, Schoenenberger RA, et al. A computer-based intervention for improving the appropriateness of antiepileptic drug level monitoring. Am J Clin Pathol 2003;119:432–438.
123. Cheng YF, Paalzow LK, Bondesson U, et al. Pharmacokinetics of haloperidol in psychotic patients. Psychopharmacology (Berl) 1987;91:410–414.
124. Chenu F, Batten LA, Zernig G, et al. Comparison of pharmacokinetic profiles of brand-name and generic formulations of citalopram and venlafaxine: a crossover study. J Clin Psychiatry 2009;70:958–966.
125. Chermá MD, Löfgren UB, Almkvist G, et al. Assessment of the prescription of antidepressant drungs in elderly nursing home patients. J Clin Psychopharmacol 2008;28:424–431.
126. Chermá MD, Reis M, Hägg S, et al. Therapeutic drug monitoring of ziprasidone in a clinical treatment setting. Ther Drug Monit 2008;30:682–688.
127. Chetty M, Gouws E, Miller R, et al. The use of a side effect as a qualitative indicator of plasma chlorpromazine levels. Eur Neuropsychopharmacol 1999;9:77–82 .
128. Choc MG, Hsuan F, Honigfeld G, et al. Single- vs multiple–dose pharmacokinetics of clozapine in psychiatric patients. Pharm Res 1990;7:347–351.
129. Choong E, Dobrinas M, Carrupt PA, et al. The permeability P-glycoprotein: a focus on enantioselectivity and brain distribution. Expert Opin Drug Metab Toxicol 2010;6:953–965.
130. Ciraulo DA, Hitzemann RJ, Somoza E, et al. Pharmacokinetics and pharmacodynamics of multiple sublingual buprenorphine tablets in dose–escalation trials. J Clin Pharmacol 2006;46:179–192.
131. Citrome L. Paliperidone palmitate – review of the efficacy, safety and cost of a new second-generation depot antipsychotic medication. Int J Clin Pract 2010;64:216–239.
132. Citrome L, Stauffer VL, Chen L, et al. Olanzapine plasma concentrations after treatment with 10, 20, and 40 mg/d in patients with schizophrenia. J Clin Psychopharmacol 2009;29:278–283.
133. Collins N, Barnes TR, Shingleton–Smith A, et al. Standards of lithium monitoring in mental health trusts in the UK. BMC Psychiatry 2010;10:80.
134. Conca A, Schmidt E, Pastore M, et al. Therapeutic drug monitoring in Italian psychiatry. Pharmacopsychiatry 2011;this issue .
135. Contin M, Riva R, Martinelli P et al: Effect of meal timing on the kinetic–dynamic profile of levodopa/carbidopa controlled is release in parkinsonian patients. Eur J Clin Pharmacol 1998;54:303–308.
136. Cooper TB. Plasma level monitoring of antipsychotic drugs. Clin Pharmacokinet 1978;3:14–38.
137. Coppen A, Kopera H. Workshop on the clinical pharmacology and efficacy of mianserin. Br J Clin Pharmacol 1978;5:91S–99S.
138. Correll CU, Malhotra AK, Kaushik S, et al. Early prediction of antipsychotic response in schizophrenia. Am J Psychiatry 2003;160:2063–2065.
139. Couchman L, Morgan PE, Spencer EP, et al. Plasma clozapine and norclozapine in patients prescribed different brands of clozapine (Clozaril, Denzapine, and Zaponex). Ther Drug Monit 2010;32:624–627.
140. Couchman L, Morgan PE, Spencer EP, et al. Plasma clozapine, norclozapine, and the clozapine: norclozapine ratio in relation to prescribed dose and other factors: data from a therapeutic drug monitoring service, 1993–2007. Ther Drug Monit 2010;32:438–447.
141. Coulomb F, Ducret F, Laneury JP, et al. Pharmacokinetics of single-dose reboxetine in volunteers with renal insufficiency. J Clin Pharmacol 2000;40:482–487.
142. Cournoyer G, De Montigny C, Ouellette J, et al. A comparative double-blind controlled study of trimipramine and amitriptyline in major depression: lack of correlation with 5-hydroxytryptamine reuptake blockade. J Clin Psychopharmacol 1987;7:385–393.
143. Court MH. Interindividual variability in hepatic drug glucuronidation: studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab Rev 2010;42:202–217.
144. Crettol S, Besson J, Croquette-Krokar M, et al. Association of dopamine and opioid receptor genetic polymorphisms with response to methadone maintenance treatment. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:1722–1727.
145. Crettol S, Déglon JJ, Besson J, et al. ABCB1 and cytochrome P450 genotypes and phenotypes: influence on methadone plasma levels and response to treatment. Clin Pharmacol Ther 2006;80:668–681.
146. Crettol S, Déglon JJ, Besson J, et al. Methadone enantiomer plasma levels, CYP2B6, CYP2C19, and CYP2C9 genotypes, and response to treatment. Clin Pharmacol Ther 2005;78:593–604.
147. Cummings J, Lefèvre G, Small G, et al. Pharmacokinetic rationale for the rivastigmine patch. Neurology 2007;69 Suppl 1:S10–S13 .
148. Dahl SG. Active metabolites of neuroleptic drugs: possible contribution to therapeutic and toxic effects. Ther Drug Monit 1982;4:33–40.
149. Dahl SG, Strandjord RE, Sigfusson S. Pharmacokinetics and relative bioavailability of levomepromazine after repeated administration of tablets and syrup. Eur J Clin Pharmacol 1977;11:305–310.
150. Darby JK, Pasta DJ, Wilson MG, et al. Long-term therapeutic drug monitoring of risperidone and olanzapine identifies altered Steady-state pharmacokinetics: a clinical, two-group, naturalistic study. Clin Drug Investig 2008;28:553–564.
151. Daviss WB, Perel JM, Birmaher B, et al. Steady-state clinical pharmacokinetics of bupropion extended-release in youths. J Am Acad Child Adolesc Psychiatry 2006;45:1503–1509.
152. Daviss WB, Perel JM, Brent DA, et al. Acute antidepressant response and plasma levels of bupropion and metabolites in a pediatric-aged sample: an exploratory study. Ther Drug Monit 2006;28:190–198.
153. Dawling S. Monitoring of tricyclic antidepressant therapy. Clin Biochem 1982;15:56–61.
154. de Lange EC. Potential role of ABC transporters as a detoxification system at the blood-CSF barrier. Adv Drug Deliv Rev 2004;56:1793–1809.
155. de Leon J. Glucuronidation enzymes, genes and psychiatry. Int J Neuropsychopharmacol 2003;6:57–72.
156. de Leon J. Incorporating pharmacogenetics into clinical practice: reality of a new tool in psychiatry. Current issues in clinical implementation. CNS Spectr 2006;11 Suppl 3:8–12.
157. de Leon J. The crucial role of the therapeutic window in understanding the clinical relevance of the poor versus the ultrarapid metabolizer phenotypes in subjects talking drugs metabolized by CYP2D6 or CYP2C19. J Clin Psychopharmacol 2007;27: 241–245.
158. de Leon J, Greenlee B, Barber J, et al. Practical guidelines for the use of new generation antipsychotic drugs (except clozapine) in adult individuals with intellectual disabilities. Res Dev Disabil 2009;30:613–669.
159. de Leon J, Susce MT, Pan RM, et al. A study of genetic (CYP2D6 and ABCB1) and environmental (drug inhibitors and inducers) variables that may influence plasma risperidone levels. Pharmacopsychiatry 2007;40:93–102.
160. de Leon J, Susce MT, Pan RM, et al. The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J Clin Psychiatry 2005;66:15–27.
161. de Leon J, Wynn G, Sandson NB. The pharmacokinetics of paliperidone versus risperidone. Psychosomatics 2010;51:80–88.
162. de Mey C, Althaus M, Ezan E, et al. Erythromycin increases plasma concentrations of alpha-dihydroergocryptine in humans. Clin Pharmacol Ther 2001;70:142–148.
163. de Vries MH, Raghoebar M, Mathlener IS, et al. Single and multiple oral dose fluvoxamine kinetics in young and elderly subjects. Ther Drug Monit 1992;14: 493–498.
164. de Wit M, Best AM, Epstein SK, et al. Lorazepam concentrations, pharmacokinetics and pharmacodynamics in a cohort of mechanically ventilated ICU patients. Int J Clin Pharmacol Ther 2006;44:466–473.
165. Degen J, Wölke E, Seiberling M, et al. Comparative study of the pharmacokinetics of amitriptyline oxide and trimipramine after single administration in healthy male probands and patients with renal failure. Med Klin (Munich) 1993;88:129–133.
166. Degner D, Grohmann R, Kropp S, et al. Severe adverse drug reactions of antidepressants: results of the German multicenter drug surveillance program AMSP. Pharmacopsychiatry. 2004;37 Suppl 1:S39–S45.
167. Del Dotto P, Bonuccelli U. Clinical pharmacokinetics of cabergoline. Clin Pharmacokinet 2003;42:633–645.
168. Deleu D, Northway MG, Hanssens Y. Clinical pharmacokinetic and pharmacodynamic properties of drugs used in the treatment of Parkinson‘s disease. Clin Pharmacokinet 2002;41:261–309.
169. Deligiannidis KM. Therapeutic drug monitoring in pregnant and postpartum women: recommendations for SSRIs, lamotrigine, and lithium. J Clin Psychiatry 2010;71:649–650.
170. Delva NJ, Hawken ER. Preventing lithium intoxication. Guide for physicians. Can Fam Physician 2001;47:1595–1600.
171. Desta Z, Kerbusch T, Soukhova N, et al. Identification and characterization of human cytochrome P450 isoforms interacting with pimozide. J Pharmacol Exp Ther 1998;285:428–437.
172. Deuschle M, Härtter S, Hiemke C, et al. Doxepin and its metabolites in plasma and cerebrospinal fluid in depressed patients. Psychopharmacology (Berl) 1997;131:19–22.
173. DeVane CL, Boulton DW, Miller LF, et al. Pharmacokinetics of trazodone and its major metabolite m-chlorophenylpiperazine in plasma and brain of rats. Int J Neuropsychopharmacol 1999;2:17–23.
174. DeVane CL, Stowe ZN, Donovan JL, et al. Therapeutic drug monitoring of psychoactive drugs during pregnancy in the genomic era: challenges and opportunities. J Psychopharmacol 2006;20 Suppl: 54–59.
175. Diaz FJ, de Leon J, Josiassen RC, et al. Plasma clozapine concentration coefficients of variation in a long-term study. Schizophr Res 2005;72:131–135.
176. Diaz FJ, Santoro V, Spina E, et al. Estimating the size of the effects of co-medications on plasma clozapine concentrations using a model that controls for clozapine doses and confounding variables. Pharmacopsychiatry 2008;41:81–91.
177. Dingemanse J, Jorga K, Zürcher G, et al. Multiple-dose clinical pharmacology of the catechol-O-methyl-transferase inhibitor tolcapone in elderly subjects. Eur J Clin Pharmacol 1996;50:47–55.
178. Dockens RC, Salazar DE, Fulmor IE, et al. Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range. J Clin Pharmacol 2006;46:1308–1312.
179. Doose DR, Walker SA, Gisclon LG, et al. Single-dose pharmacokinetics and effect of food on the bioavailability of topiramate, a novel antiepileptic drug. J Clin Pharmacol 1996;36:884–891.
180. Doran A, Obach RS, Smith BJ, et al. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos 2005;33:165–174.
181. dos Santos FM, Gonçalves JC, Caminha R, et al. Pharmacokinetic/pharmacodynamic modeling of psychomotor impairment induced by oral clonazepam in healthy volunteers. Ther Drug Monit 2009;31:566–574.
182. Dunbar JL, Turncliff RZ, Hayes SC, et al. Population pharmacokinetics of extended–release injectable naltrexone (XR-NTX) in patients with alcohol dependence. J Stud Alcohol Drugs 2007;68:862–870.
183. Dutheil F, Jacob A, Dauchy S, et al. ABC transporters and cytochromes P450 in the human central nervous system: influence on brain pharmacokinetics and contribution to neurodegenerative disorders. Expert Opin Drug Metab Toxicol 2010;6: 1161–1174.
184. Dvorchik BH, Vesell ES. Pharmacokinetic interpretation of data gathered during therapeutic drug monitoring. Clin Chem 1976;22:868–878.
185. Eagles JM, McCann I, MacLeod TN, et al. Lithium monitoring before and after the distribution of clinical practice guidelines. Acta Psychiatr Scand 2000;101:349–353.
186. Eap CB, Baumann P. Analytical methods for the quantitative determination of selective serotonin reuptake inhibitors for therapeutic drug monitoring purposes in patients. J Chromatogr B Biomed Appl 1996;686:51–63.
187. Eap CB, Bender S, Gastpar M, et al. Steady state plasma levels of the enantiomers of trimipramine and of its metabolites in CYP2D6-, CYP2C19- and CYP3A4/5-phenotyped patients. Ther Drug Monit 2000;22:209–214.
188. Eap CB, Bertschy G, Baumann P, et al. High interindividual variability of methadone enantiomer blood levels to dose ratios. Arch Gen Psychiatry 1998;55:89–90.
189. Eap CB, Buclin T, Baumann P. Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin Pharmacokinet 2002;41:1153–1193.
190. Eap CB, Finkbeiner T, Gastpar M, et al. Replacement of R–methadone by a double dose of (R,S)-methadone in addicts: interindividual variability of the R/S ratios and evidence of adaptve changes in methadone pharmacokinetics. Eur J Clin Pharmacol 1996;50:385–389.
191. Eap CB, Koeb L, Baumann P. Determination of trimipramine and its demethylated and hydroxylated metabolites in plasma by gas chromatography-mass spectrometry. J Chromatogr 1994;652:97–103.
192. Eap CB, Lima CA, Macciardi F, et al. Steady state concentrations of the enantiomers of mianserin and desmethylmianserin in poor and in homozygous and heterozygous extensive metabolizers of debrisoquine. Ther Drug Monit 1998;20:7–13.
193. Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annu Rev Med 2006;57:119–137 .
194. Egberts K, Mehler-Wex C, Gerlach M. 2011 Therapeutic drug monitoring in child and adolescent psychiatry. Pharmacopsychiatry 2011;this issue.
195. Eggert Hansen C, Rosted Christensen T, Elley J, et al. Clinical pharmacokinetic studies of perphenazine. Br J Clin Pharmacol 1976;3:915–923.
196. Ellinwood EH JR, Heatherly DG, Nikaido AM, et al. Comparative pharmacokinetics and pharmacodynamics of lorazepam, alprazolam and diazepam. Psychopharmacology 1985;86:392–399.
197. Ereshefsky L, Jann MW, Saklad SR, et al. Effects of smoking on fluphenazine clearance in psychiatric inpatients. Biol Psychiatry 1985;20:329–332.
198. Evans LE, Bett JH, Cox JR, et al. The bioavailability of oral and parenteral chlorimipramine (Anafranil). Prog Neuropsychopharmacol 1980;4:293–302.
199. Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science 1999;286:487–491.
200. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther 2004;76:178–184.
201. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why, how, and when? Basic Clin Pharmacol Toxicol 2005;97:125–134.
202. Faessel HM, Gibbs MA, Clarc DJ, et al. Multiple–does pharmacokinetics of the selective nicotinic receptor partial agonist, varenicline, in healthy smokers. J Clin Pharmacol 2006;46:1439–1448.
203. Faiman MD, Jensen JC, Lacoursiere RB. Elimination kinetics of disulfiram in alcoholics after single and repeated doses. Clin Pharmacol Ther 1984;36:520–526 .
204. Falkai P, Wobrock T, Lieberman J, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, Part 1:acute treatment of schizophrenia. World J Biol Psychiatry 2005;6:132–191.
205. Falkai P, Wobrock T, Lieberman J, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2:long-term treatment of schizophrenia. World J Biol Psychiatry 2006;7:5–40.
206. Fanton L, Bévalot F, Grait H, et al. Fatal intoxication with milnacipran. J Forensic Leg Med 2008;15:388–390.
207. Farde L, Nordström AL, Wiesel FA, et al. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Arch Gen Psychiatry 1992;49:538–544.
208. Feng Y, Pollock BG, Coley K, et al. Population pharmacokinetic analysis for risperidone using highly sparse sampling measurements from the CATIE study. J Clin Pharmacol 2008;66:629–639.
209. Feng Y, Pollock BG, Ferrell RE, et al. Paroxetine: population pharmacokinetic analysis in late–life depression using sparse concentration sampling. Br J Clin Pharmacol 2006;61:558–569 .
210. Fenner KS, Troutman MD, Kempshall S, et al. Drug-drug interactions mediated through P-glycoprotein: clinical relevance and in vitro-in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther 2009;85:173–181.
211. Ferrari A, Bertolotti M, Dell’Utri A, et al. Serum time course of naltrexone and 6β-naltrexol levels during long term treatment in drug addicts. Drug Alcohol Depend 1998;52:211–220.
212. Figueroa C, Brecher M, Hamer–Maansson JE, et al. Pharmacokinetic profiles of extended release quetiapine fumarate compared with quetiapine immediate release. Prog Neuropsychopharmacol Biol Psychiatry 2009;33:199–204.
213. Findling RL, Reed MD, Myers C, et al. Paroxetine pharmacokinetics in depressed children and adolescents. J Am Acad Child Adolesc Psychiatry 1999;38:952–959.
214. Fitzgerald PB, Kapur S, Remington G, et al. Predicting haloperidol occupancy of central dopamine D2 receptors from plasma levels. Psychopharmacology (Berl) 2000;149: 1–5.
215. Flanagan RJ. Developing an analytical toxicology service: principles and guidance. Toxicol Rev 2004;23:251–263.
216. Fleming J, Chetty M. Therapeutic monitoring of valproate in psychiatry: how far have we progressed? Clin Neuropharmacol 2006;29:350–360 .
217. Fogelman SM, Schmider J, Venkatakrishnan K, et al. O- and N-demethylation of venlafaxine in vitro by human liver microsomes and by microsomes from cDNA-transfected cells: effect of metabolic inhibitors and SSRI antidepressants. Neuropsychopharmacology 1999;20:480–490.
218. Fontaine R, Mercier P, Beaudry P, et al. Bromazepam and lorazepam in generalized anxiety: a placebo–controlled study with measurement of drug plasma concentrations. Acta Psychiatr Scand 1986;74:451–458.
219. Food and Drug Administration. Guidance for industry: bioanalytical method validation 2001. www.fda.gov/cvm .
220. Foti RS, Rock DA, Wienkers LC, et al. Selection of alternative CYP3A4 probe substrates for clinical drug interaction studies using in vitro data and in vivo simulation. Drug Metab Dispos 2010;38:981–987.
221. Fountoulakis KN. An update of evidence-based treatment of bipolar depression: where do we stand? Curr Opin Psychiatry 2010;23:19–24 .
222. Fric M, Pfuhlmann B, Laux G, et al. The influence of smoking on the serum level of duloxetine. Pharmacopsychiatry 2008;41:151–155.
223. Frieboes RM, Sonntag A, Yassouridis A, et al. Clinical outcome after trimipramine in patients with delusional depression – a pilot study. Pharmacopsychiatry 2003;36:12–17.
224. Friedman H, Greenblatt DJ, Peters GR, et al. Pharmacokinetics and pharmacodynamics of oral diazepam: effect of dose, plasma concentration, and time. Clin Pharmacol Ther 1992;52:139–150.
225. Fritze J, Laux G, Sofic E, et al. Plasma moclobemide and metabolites: lack of correlation with clinical response and biogenic amines. Psychopharmacology (Berl) 1989;99:252–256.
226. Fröscher W, Schier KR, Hoffmann M, et al. Topiramate: a prospective study on the relationship between concentration, dosage and adverse events in epileptic patients on combination therapy. Epileptic Disord 2005;7:237–248.
227. Fudio S, Borobia AM, Piñana E, et al. Evaluation of the influence of sex and CYP2C19 and CYP2D6 polymorphisms in the disposition of citalopram. Eur J Pharmacol 2010;626:200–204.
228. Fukasawa T, Suzuki A, Otani K. Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J Clin Pharm Ther 2007;32:333–341.
229. Furlanut M, Montanari G, Benetello P, et al. Steady-state serum concentrations of imipramine, its main metabolites and clinical response in primary enuresis. Pharmacol Res 1989;21:561–566 .
230. Furukori H, Kondo T, Yasui N, et al. Effects of itraconazole on the Steady-state plasma concentrations of bromperidol and reduced bromperidol in schizophrenic patients. Psychopharmacology (Berl) 1999;145:189–192.
231. Gaertner HJ, Golfinopoulos G, Breyer–Pfaff U. Response to Maprotiline treatment in depressive patients, relationship to urinary MHPG excretion, and plasma drug level. Pharmacopsychiatria 1982;15:170–174.
232. Gaertner I, Gaertner HJ, Vonthein R, et al. Therapeutic drug monitoring of clozapine in relapse prevention: a five-year prospective study. J Clin Psychopharmacol 2001;21:305–310.
233. Garnock–Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs 2009;11:203–226.
234. Gelenberg AJ. A review of the current guidelines for depression treatment. J Clin Psychiatry 2010;71:e15.
235. Genton P, Guerrini R, Perucca E. Tiagabine in clinical practice. Epilepsia 2001;42 Suppl 3:42–45.
236. Gerlach M, Hünnerkopf R, Rothenhöfer S, et al. Therapeutic drug monitoring of quetiapine in adolescents with psychotic disorders. Pharmacopsychiatry 2007;40:72–76 .
237. Gerstenberg G, Aoshima T, Fukasawa T, et al. Relationship between clinical effects of fluvoxamine and the Steady-state plasma concentrations of fluvoxamine and its major metabolite fluvoxamino acid in Japanese depressed patients. Psychopharmacology (Berl) 2003;167:443–448.
238. Gervasini G, Carrillo JA, Benitez J. Potential role of cerebral cytochrome P450 in clinical pharmacokinetics: modulation by endogenous compounds. Clin Pharmacokinet 2004;43:693–706.
239. Gex–Fabry M, Balant-Gorgia AE, Balant LP. Clomipramine concentration as a predictor of delayed response: a naturalistic study. Eur J Clin Pharmacol 1999;54:895–902.
240. Gex-Fabry M, Balant-Gorgia AE, Balant LP. Therapeutic drug monitoring of olanzapine: the combined effect of age, gender, smoking, and comedication. Ther Drug Monit 2003;25:46–53.
241. Gex-Fabry M, Balant–Gorgia AE, Balant LP, et al. Time course of clinical response to venlafaxine: relevance of plasma level and chirality. Eur J Clin Pharmacol 2004;59:883–891.
242. Gex-Fabry M, Gervasoni N, Eap CB, et al. Time course of response to paroxetine: influence of plasma level. Prog Neuropsychopharmacol Biol Psychiatry 2007;31:892–900.
243. Gilles M, Deuschle M, Kellner S, et al. Paroxetine serum concentrations in depressed patients and response to treatment. Pharmacopsychiatry 2005;38:118–121.
244. Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol 2007;151: 737–748.
245. Glassman AH, Perel JM, Shostak M, et al. Clinical implications of imipramine plasma levels for depressive illness. Arch Gen Psychiatry 1977;34:197–204.
246. Glassmann AH, Schildkraut JJ, Orsulak PJ, et al. Tricyclic antidepressants, blood level measurements and clinical outcome: an APA task force report. Am J Psychiat 1985;142:155–162.
247. Glauser TA, Pippenger CE. Controversies in blood-level monitoring: reexamining its role in the treatment of epilepsy. Epilepsia 2000;41 Suppl 8:S6–S15.
248. Gleason MM, Egger HL, Emslie GJ, et al. Psychopharmacological treatment for very young children: contexts and guidelines. J Am Acad Child Adolesc Psychiatry 2007;46:1532–1572.
249. Glotzbach RK, Preskorn SH. Brain concentrations of tricyclic antidepressants: Single–dose kinetics and relationship to plasma concentrations in chronically dosed rats. Psychopharmacology 1982;78:25–27.
250. Goeringer KE, Raymon L, Christian GD, et al. Postmortem forensic toxicology of selective serotonin reuptake inhibitors: a review of pharmacology and report of 168 cases. J Forensic Sci 2000;45:633–648.
251. Gomolin IH, Smith C, Jeitner TM. Once-daily memantine: pharmacokinetic and clinical considerations. J Am Geriatr Soc 2010;58:1812–1813.
252. Gonzalez JP, Brogden RN. Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence. Drugs 1988;35:192–213.
253. Goodnick PJ, Dominguez RA, DeVane CL, et al. Bupropion slow-release response in depression: diagnosis and biochemistry. Biol Psychiatry 1998;44:629–632.
254. Goodwin GM;Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for treating bipolar disorder: revised second edition–recommendations from the British Association for Psychopharmacology. J Psychopharmacol 2009;23:346–388.
255. Gram LF, Guentert TW, Grange S, et al. Moclobemide, a substrate of CYP2C19 and an inhibitor of CYP2C19, CYP2D6, and CYP1A2:a panel study. Clin Pharmacol Ther 1995;57:670–677.
256. Grandjean EM, Aubry JM. Lithium: updated human knowledge using an evidence–based approach. Part II: Clinical pharmacology and therapeutic monitoring. CNS Drugs 2009;23:331–349.
257. Grasmäder K, Verwohlt PL, Kühn KU, et al. Relationship between mirtazapine dose, plasma concentration, response, and side effects in clinical practice. Pharmacopsychiatry 2005;38:113–117.
258. Grasmäder K, Verwohlt PL, Rietschel M, et al. Impact of polymorphisms of cytochrome-P450 isoenzymes 2C9, 2C19 and 2D6 on plasma concentrations and clinical effects of antidepressants in a naturalistic clinical setting. Eur J Clin Pharmacol 2004;60:329–336.
259. Greenblatt DJ, Blaskovich PD, Nuwayser ES, et al. Clonazepam pharmacokinetics: comparison of subcutaneous microsphere injection with multiple-dose oral administration. J Clin Pharmacol 2005;45: 1288–1293.
260. Greenblatt DJ, Divoll M, Harmatz JS, et al. Oxazepam kinetics: effects of age and sex. J Pharmacol Exp Ther 1980;215:86–91.
261. Greenblatt DJ, Ehrenberg BL, Gunderman J, et al. Pharmacokinetic and electroencephalographic study of intravenous diazepam, midazolam, and placebo. Clin Pharmacol Ther 1989;45:356–365.
262. Greenblatt DJ, Friedman H, Burstein ES, et al. Trazodone kinetics: effect of age, gender, and obesity. Clin Pharmacol Ther 1987;42:193–200.
263. Greenblatt DJ, Gan L, Harmatz JS, et al. Pharmocokinetics and pharmacodynamics of single–dose triazolam: electroencephalography compared with the Digit-Symbol Substitution Test. Br J Clin Pharmacol 2005;60:244–248.
264. Greenblatt DJ, Harmatz JS, Friedman H, et al. A large-sample study of diazepam pharmacokinetics. Ther Drug Monit 1989;11:652–657.
265. Greenblatt DJ, Harmatz JS, von Moltke LL, et al. Comparative kinetics and dynamics of zaleplon, zolpidem, and placebo. Clin Pharmacol Ther 1998;64:553–561.
266. Greenblatt DJ, Shader RI, Franke K, et al. Pharmacokinetics and bioavailability of intravenous, intramuscular, and oral lorazepam in humans. J Pharm Sci 1979;68:57–63.
267. Greenblatt DJ, von Moltke LL, Ehrenberg BL, et al. Kinetics and dynamics of lorazepam during and after continuous intravenous infusion. Crit Care Med 2000;28:2750–2757.
268. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Short-term exposure to low-dose ritonavir impairs clearance and enhances adverse effects of trazodone. J Clin Pharmacol 2003;43:414–422.
269. Greiner C, Hiemke C, Bader W, et al. Determination of citalopram and escitalopram together with their active main metabolites desmethyl(es-)citalopram in human serum by column-switching high performance liquid chromatography (HPLC) and spectrophotometric detection. J Chromatogr B Analyt Technol Biomed Life Sci 2007;848:391–394.
270. Grimaldi R, Perucca E, Ruberto G, et al. Pharmacokinetic and pharmacodynamic studies following the intravenous and oral administration of the antiparkinsonian drug biperiden to normal subjects. Eur J Clin Pharmacol 1986;29:735–737.
271. Grohmann R, Engel RR, Rüther E, et al. The AMSP drug safety program: methods and global results. Pharmacopsychiatry 2004;37 Suppl 1:S4–S11.
272. Gründer G, Carlsson A, Wong DF. Mechanism of new antipsychotic medications. Occupancy is not just antagonism. Arch Gen Psychiatry 2003;60:974–977.
273. Gründer G, Fellows C, Janouschek H, et al. Brain and plasma pharmacokinetics of aripiprazole in patients with schizophrenia: an [18F]fallypride PET study. Am J Psychiatry 2008;165:988–995.
274. Gründer G, Hiemke C, Paulzen M, et al. Therpaeutic drug concentrations of antidepressants and antipsychotics: Guidance from PET imaging. Pharmacopsychiatry 2011;this issue.
275. Gründer G, Hippius H, Carlsson A. The ‚atypicality‘ of antipsychotics: a concept re–examined and re–defined. Nat Rev Drug Discov 2009;8:197–202.
276. Grunze H, Vieta E, Goodwin GM. The World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry 2009;10:85–116.
277. Guay DR. Rasagiline (TVP-1012): a new selective monoamine oxidase inhibitor for Parkinson‘s disease. Am J Geriatr Pharmacother 2006;4:330–346.
278. Guberman A, Couture M, Blaschuk K, et al. Add-on trial of clobazam in intractable adult epilepsy with plasma level correlations. Can J Neurol Sci 1990;17:311–316 .
279. Gunes A, Spina E, Dahl ML, et al. ABCB1 polymorphisms influence Steady-state plasma levels of 9-hydroxyrisperidone and risperidone active moiety. Ther Drug Monit 2008;30:628–633.
280. Gupta N. Guidelines for lithium monitoring: are they ideal? Acta Psychiatr Scand 2001;104:76–77 .
281. Gupta RN, Dziurdzy SA. Therapeutic monitoring of sertraline. Clin Chem 1994;40:498–499.
282. Gupta SK, Shah JC, Hwang SS. Pharmacokinetic and pharmacodynamic characterization of OROS and immediate-release amitriptyline. Br J Clin Pharmacol 1999;48:71–78.
283. Guy W, editor. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare;1976.
284. Haberstroh J, Hampel H, Pantel J. Optimal management of Alzheimer‘s disease patients: Clinical guidelines and family advice. Neuropsychiatr Dis Treat 2010;6:243–253.
285. Haen E, Greiner C, Bader W, et al. Wirkstoffkonzentrationsbestimmungen zur Therapieleitung. Ergänzung therapeutischer Referenzbereiche durch dosisbezogene Referenzbereiche. Nervenarzt 2008;79:558–566.
286. Hallett C, Dean BC. Bromazepam: acute benefit-risk assessment in general practice. Curr Med Res Opin 1984;8:683–688.
287. Hammarberg A, Beck O, Eksborg S, et al. Acamprosate determinations in plasma and cerebrospinal fluid after multiple dosing measured by liquid chromatography–mass spectroscopy: a pharmacokinetic study in healthy volunteers. Ther Drug Monit 2010;32:489–496.
288. Hammarberg A, Jayaram–Lindström N, Berck O, et al. The effects of acomprosate on alcohol–cue reactivity and alcohol priming in dependent patients: a randomized controlled trial. Psychopharmacol 2009;205:53–62.
289. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982;143:29–36.
290. Harden CL, Trifiletti R, Kutt H. Felbamate levels in patients with epilepsy. Epilepsia 1996;37:280–283.
291. Härtter S, Dingemanse J, Baier D, et al. The role of cytochrome P450 2D6 in the metabolism of moclobemide. Eur Neuropsychopharmacol 1996;6:225–230.
292. Härtter S, Hermes B, Hiemke C. Automated determination of trimipramine and N-desmethyl–trimipramine in human plasma or serum by HPLC with on–line solid phase extraction. J Liq Chromatogr 1995;18:3495–3505.
293. Härtter S, Hermes B, Szegedi A, et al. Automated determination of paroxetine and its main metabolite by column switching and on–line high–performance liquid chromatography. Ther Drug Monit 1994;16:400–406.
294. Härtter S, Hiemke C. Column switching and high–performance liquid chromatography in the analysis of amitriptyline, nortriptyline and hydroxylated metabolites in human plasma or serum. J Chromatogr 1992;578:273–282.
295. Härtter S, Tybring G, Friedberg T, et al. The N-demethylation of the doxepin isomers is mainly catalyzed by the polymorphic CYP2C19. Pharm Res 2002;19:1034–1037.
296. Härtter S, Wang X, Weigmann H, et al. Differential effects of fluvoxamine and other antidepressants on the biotransformation of melatonin. J Clin Psychopharmacol 2001;21:167–174.
297. Härtter S, Weigmann H, Hiemke C. Automated determination of reboxetine by high-performance liquid chromatography with column-switching and ultraviolet detection. J Chromatogr B Biomed Sci Appl 2000;740:135–140.
298. Härtter S, Wetzel H, Hiemke C. Automated determination of fluvoxamine in plasma by column-switching high-performance liquid chromatography. Clin Chem 1992;38:2082–2086.
299. Hasselstrøm J, Linnet K. Quetiapine serum concentrations in psychiatric patients: the influence of comedication. Ther Drug Monit 2004;26:486–491.
300. Hässler F, Reis O. Pharmacotherapy of disruptive behavior in mentally retarded subjects: A review of the current literature. Dev Disabil Res Rev 2010;16:265–272.
301. Haymond J, Ensom MH. Does valproic acid warrant therapeutic drug monitoring in bipolar affective disorder? Ther Drug Monit 2010;32:19–29.
302. Hazell P, Becker K, Nikkanen EA, et al. Relationship between atomoxetine plasma concentration, treatment response and tolerability in attention–deficit/hyperactivity disorder and comorbid oppositional defiant disorder. Atten Defic Hyperact Disord 2009;1:201–210.
303. Hegerl U, Bottlender R, Gallinat J, et al. The serotonin syndrome scale: first results on validity. Eur Arch Psychiatry Clin Neuroci 1998;248:96–103.
304. Heikkinen H, Saraheimo M, Antila S, et al. Pharmacokinetics of entacapone, a peripherally acting catechol–O–methyltransferase inhibitor, in man. A study using a stable isotope techique. Eur J Clin Pharmacol 2001;56:821–826.
305. Heller S, Hiemke C, Stroba G, et al. Assessment of storage and transport stability of new antidepressant and antipsychotic drugs for a nationwide TDM service. Ther Drug Monit 2004;26:459–461.
306. Hendset M, Hermann M, Lunde H, et al. Impact of the CYP2D6 genotype on Steady-state serum concentrations of aripiprazole and dehydroaripiprazole. Eur J Clin Pharmacol 2007;63:1147–1151.
307. Herman BD, Fleishaker JC, Brown MT. Ketoconazole inhibits the clearance of the enantiomers of the antidepressant reboxetine in humans. Clin Pharmacol Ther 1999;66:374–379.
308. Hermida J, Paz E, Tutor JC. Clozapine and norclozapine concentrations in serum and plasma samples from schizophrenic patients. Ther Drug Monit 2008;30:41–45.
309. Hesse LM, He P, Krishnaswamy S, et al. Pharmacogenetic determinants of interindividual variability in bupropion hydroxylation by cytochrome P450 2B6 in human liver microsomes. Pharmacogenetics 2004;14:225–238.
310. Hiemke C. Clinical utility of drug measurement and pharmacokinetics – therapeutic drug monitoring in psychiatry. Eur J Clin Pharmacol 2008;64:159–166.
311. Hiemke C. Therapeutic drug monitoring in neuropharmacology: does it hold its promises? Eur Arch Psychiatry Clin Neurosci 2008;258 Suppl 1:21–27.
312. Hiemke C, Baumann P, Laux G, et al. Therapeutisches Drug–Monitoring in der Psychiatrie. Konsensus–Leitlinie der AGNP. Psychopharmakotherapie 2005;12:166–182.
313. Hiemke C, Dragicevic A, Gründer G, et al. Therapeutic monitoring of new antipsychotic drugs. Ther Drug Monit 2004;26:156–160.
314. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther 2000;85:11–28.
315. Higuchi H, Yoshida K, Takahashi H, et al. Milnacipran plasma levels and antidepressant response in Japanese major depressive patients. Hum Psychopharmacol 2003;18:255–259.
316. Höjer J, Hulting J, Salmonson H. Fatal cardiotoxicity induced by venlafaxine overdosage. Clin Toxicol (Phila) 2008;46:336–337.
317. Holbrook JM, Parks–Veal P, Mimbs J. Clinical monitoring guidelines for neuroleptic and antidepressant drugs. Central State Hospital, Milledgeville, Georgia. Hosp Pharm 1991;26:783–784, 787–793.
318. Holzer L, Preuss U, Baumgartner L, et al. Quetiapine in adolescents with non-affective psychotic disorders: An open-label trial. Pharmacopsychiatry 2011;44:87–95.
319. Hooper WD, Dickinson RG, Dunstan PR, et al. Oxcarbazepine: preliminary clinical and pharmacokinetic studies on a new anticonvulsant. Clin Exp Neurol 1987;24:105–112.
320. Horadam VW, Sharp JG, Smilack JD, et al. Pharmacokinetics of amantadine hydrochloride in subjects with normal and impaired renal function. Ann Intern Med 1981;94:454–458.
321. Hrdina PD, Lapierre YD. Plasma levels of maprotiline and zimelidine and their relationship to clinical response in depressed patients. Ther Drug Monit 1986;8:400–406.
322. Hsieh YH, Yang YH, Yeh HH, et al. Simultaneous determination of galantamine, rivastigmine and NAP 226–90 in plasma by MEKC and its application in Alzheimer‘s disease. Electrophoresis 2009;30:644–653.
323. Hughes J, Gill AM, Mulhearn H, et al. Steady-state plasma concentrations of midazolam in critically ill infants and children. Ann Pharmacother 1996;30:27–30.
324. Hui WK, Mitchell LB, Kavanagh KM, et al. Melperone: electrophysiologic and antiarrhythmic activity in humans. J Cardiovasc Pharmacol 1990;15:144–149.
325. Ilett KF, Blythe TH, Hackett LP, et al. Plasma concentrations of dothiepin and its metabolites are not correlated with clinical efficacy in major depressive illness. Ther Drug Monit 1993;15:351–357.
326. Isacsson G, Holmgren P, Druid H, et al. The utilization of antidepressants–a key issue in the prevention of suicide: an analysis of 5281 suicides in Sweden during the period 1992–1994. Acta Psychiatr Scand 1997;96:94–100.
327. Isbister GK, Hackett LP, Dawson AH, et al. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003;56:441–450.
328. Ishida M, Otani K, Kaneko S, et al. Effects of various factors on steady state plasma concentrations of trazodone and its active metabolite m–chlorophenylpiperazine. Int Clin Psychopharmacol 1995;10:143–146.
329. Iwersen S, Schmoldt A. One fatal and one nonfatal intoxication with tranylcypromine. Absence of amphetamines as metabolites. J Anal Toxicol 1996;20:301–304.
330. Jaanson P, Marandi T, Kiivet RA, et al. Maintenance therapy with zuclopenthixol decanoate: associations between plasma concentrations, neurological side effects and CYP2D6 genotype. Psychopharmacology (Berl) 2002;162:67–73.
331. Janis GC, Markowitz JS. Influence of ethanol and gender on methylphenidate pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2007;81:346–353.
332. Jann MW, Grimsley SR, Gray EC, et al. Pharmacokinetics and pharmacodynamics of clozapine. Clin Pharmacokinet 1993;24:161–176.
333. Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet 2002;41:719–739 .
334. Jaquenoud Sirot E, Knezevic B, Morena GP, et al. ABCB1 and cytochrome P450 polymorphisms: clinical pharmacogenetics of clozapine. J Clin Psychopharmacol 2009;29:319–326.
335. Jaquenoud Sirot E, van der Velden JW, Rentsch K, et al. Therapeutic drug monitoring and pharmacogenetic rests as tools in pharmacovigilance. Drug Safety 2006;29:735–768.
336. Jefferson JW, Pradko JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Therap 2005;27:1685–1695.
337. Jerling M, Dahl ML, Aberg–Wistedt A, et al. The CYP2D6 genotype predicts the oral clearance of the neuroleptic agents perphenazine and zuclopenthixol. Clin Pharmacol Ther 1996;59:423–428.
338. Ji P, Damle B, Xie J, et al. Pharmacokinetic interaction between efavirenz and carbamazepine after multiple–dose administration in healthy subjects. J Clin Pharmacol 2008;48:948–956.
339. Jimmink A, Caminada K, Hunfeld NG, et al. Clinical toxicology of citalopram after acute intoxication with the sole drug or in combination with other drugs: overview of 26 cases. Ther Drug Monit 2008;30: 365–371.
340. Jin Y, Pollock BG, Frank E, et al. The effect of reporting methods for dosing times on the estimation of pharmacokinetic parameters of escitalopram. J Clin Pharmacol 2009;49:176–184.
341. Jochemsen R, Wesselman JG, Hermans J, et al. Pharmacokinetics of brotizolam in healthy subjects following intravenous and oral administration. Br J Clin Pharmacol 1983;16 Suppl 2:285S–290S.
342. Johannessen SI, Battino D, Berry DJ, et al. Therapeutic drug monitoring of the newer antiepileptic drugs. Ther Drug Monitor 2003;25:347–363.
343. Johannessen SI, Tomson T. Pharmacokinetic variability of newer antiepileptic drugs: when is monitoring needed? Clin Pharmacokinet 2006;45:1061–1075.
344. Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl 1992;369:15–26.
345. Johnston JA, Fiedler–Kelly J, Glover ED, et al. Relationship between drug exposure and the efficacy and safety of bupropion sustained release for smoking cessation. Nicotine Tob Res 2001;3:131–140.
346. Jorga KM, Fotteler B, Heizmann P, et al. Pharmacokinetics and pharmacodynamics after oral and intravenous administration of tolcapone, a novel adjunct to Parkinson‘s disease therapy. Eur J Clin Pharmacol 1998;54:443–447.
347. Jorga KM, Larsen JP, Beiske A, et al. The effect of tolcapone on the pharmacokinetics of benserazide. Eur J Neurol 1999;6:211–219.
348. Jørgensen A. Pharmacokinetic studies in volunteers of intravenous and oral cis(Z)-flupentixol and intramuscular cis (Z)-flupentixol decanoate in Viscoleo. Eur J Clin Pharmacol 1980;18:355–360.
349. Jornil J, Jensen KG, Larsen F, et al. Identification of cytochrome P450 isoforms involved in the metabolism of paroxetine and estimation of their importance for human paroxetine metabolism using a population-based simulator. Drug Metab Dispos 2010;38:376–385.
350. Kandasamy M, Srinivas P, Subramaniam K, et al. Differential outcomes from metabolic ratios in the identification of CYP2D6 phenotypes-focus on venlafaxine and O-desmethylvenlafaxine. Eur J Clin Pharmacol 2010;66:879–887.
351. Kane JM, Leucht S, Carpenter D, et al. The expert consensus guideline series. Optimizing pharmacologic treatment of psychotic disorders. Introduction: methods, commentary, and summary. J Clin Psychiatry 2003;12 Suppl: 5–19.
352. Kaplan SA, Jack ML, Weinfeld RE, et al. Biopharmaceutical and clinical pharmacokinetic profile of bromazepam. J Pharmacokinet Biopharm 1976;4:1–16.
353. Kasper S, Dötsch M, Kick H, et al. Plasma concentrations of fluvoxamine and maprotiline in major depression: implications on therapeutic efficacy and side effects. Eur Neuropsychopharmacol 1993;3:13–21.
354. Katoh Y, Uchida S, Kawai M, et al. Effects of cigarette smoking and cytochrome P450 2D6 genotype on fluvoxamine concentration in plasma of Japanese patients. Biol Pharm Bull 2010;33:285–288.
355. Katon W, Cantrell CR, Sokol MS, et al. Impact of antidepressant drug adherence on comorbid medication use and resource utilization. Arch Intern Med 2005;165:2497–2503.
356. Kaufman E, Lamster IB. The diagnostic applications of saliva – a review. Crit Rev Oral Biol Med 2002;13:197–212.
357. Kaye CM, Nicholls B. Clinical pharmacokinetics of ropinirole. Clin Pharmacokinet 2000;39:243–254.
358. Kemp DE, Ganocy SJ, Brecher M, et al. Clinical value of early partial symptomatic improvement in the prediction of response and remission during short-term treatment trials in 3369 subjects with bipolar I or II depression. J Affect Disord 2011;130:171–179.
359. Kennedy MC. Post mortem drug concentrations. Intern Med J 2010;40:183–187.
360. Kerr BM, Thummel KE, Wurden CJ, et al. Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation. Biochem Pharmacol 1994;47:1969–1979 .
361. Kinirons MT, O‘Mahony MS. Drug metabolism and ageing. Br J Clin Pharmacol 2004;57:540–544.
362. Kirchheiner J. CYP2D6 phenotype prediction from genotype: which system is the best? Clin Pharmacol Ther 2008;83:225–227.
363. Kirchheiner J, Meineke I, Müller G, et al. Contributions of CYP2D6, CYP2C9 and CYP2C19 to the biotransformation of E- and Z-doxepin in healthy volunteers. Pharmacogenetics 2002;12:571–580 .
364. Kirchheiner J, Müller G, Meineke I, et al. Effects of polymorphisms in CYP2D6, CYP2C9, and CYP2C19 on trimipramine pharmacokinetics. J Clin Psychopharmacol 2003;23:459–466.
365. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442–473.
366. Kirchherr H, Kühn-Velten WN. Quantitative determination of forty-eight antidepressants and antipsychotics in human serum by HPLC tandem mass spectrometry: a multi-level, single-sample approach. J Chromatogr B 2006;843:100–113.
367. Kirkton C, McIntyre IM. Therapeutic and toxic concentrations of mirtazapine. J Anal Toxicol 2006;30:687–691.
368. Kirschbaum KM, Müller MJ, Malevani J, et al. Serum levels of aripiprazole and dehydroaripiprazole, clinical response and side effects. World J Biol Psychiatry 2008;9:212–218.
369. Kirschbaum KM, Müller MJ, Zernig G, et al. Therapeutic monitoring of aripiprazole by HPLC with column–switching and spectrophotometric detection. Clin Chem 2005;51:1718–1721.
370. Kirschbaum KM, Uhr M, Holthoewer D, et al. Pharmacokinetics of acute and sub-chronic aripiprazole in P-glycoprotein deficient mice. Neuropharmacology. 2010;59:474–479.
371. Kjolbye M, Thomsen K, Rogne T, et al. Search for a therapeutic range for serum zuclopenthixol concentrations in schizophrenic patients. Ther Drug Monit 1994;16:541–547.
372. Klamerus KJ, Maloney K, Rudolph RL, et al. Introduction of a composite parameter to the pharmacokinetics of venlafaxine and its active O-desmethyl metabolite. J Clin Pharmacol 1992;32:716–724.
373. Klampfl K, Taurines R, Preuss A, et al. Serum concentrations, therapeutic response and side effects in children and adolescents with impulsive-aggressive symptoms during risperidone therapy. Pharmacopsychiatry 2010;43:58–65.
374. Klotz U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab Rev 2009;41:67–76.
375. Koepp MJ, Patsalos PN, Sander JW. Sulthiame in adults with refractory epilepsy and learning disability: an open trial. Epilepsy Res 2002;50:277–282.
376. Kondo T, Otani K, Ishida M, et al. Adverse effects of zotepine and their relationship to serum concentrations of the drug and prolactin. Ther Drug Monit 1994;16:120–124.
377. Koostra–Ros JE, Van Weelden MJ, Hinrichs JW, et al. Therapeutic drug monitoring of antidepressants and cytochrome P450 genotyping in general practice. J Clin Pharmacol 2006;46:1320–1327.
378. Kornhuber J, Quack G, Danysz W, et al. Therapeutic brain concentration of the NMDA receptor antagonist amantadine. Neuropharmacology 1995;34:713–721.
379. Koyama E, Chiba K, Tani M, et al. Identification of human cytochrome P450 isoforms involved in the stereoselective metabolism of mianserin enantiomers. J Pharmacol Exp Ther 1996;278:21–30.
380. Kozer E, Parvez S, Minassian BA, et al. How high can we go with phenytoin? Ther Drug Monit 2002;24:386–389.
381. Krska J, Corner DA. Serum drug level monitoring in affective disorders. J Clin Pharm Ther 1992;17:357–363.
382. Kugelberg FC, Druid H, Carlsson B, et al. Postmortem redistribution of the enantiomers of citalopram and its metabolites: an experimental staudy in rats. J Analyt Toxicol 2004;28:631–637.
383. Kuhlman JJ JR, Levine B, Johnson RE, et al. Relationship of plasma buprenorphine and norbuprenorphine to withdrawal symptoms during dose induction, maintenance and withdrawal from sublingual buprenorphine. Addiction 1998;93:549–559.
384. Kuss HJ, Feistenauer E. Quantitative high–performance liquid chromatographic assay for the determination of maprotiline and oxaprotiline in human plasma. J Chromatogr 1981;204:349–353.
385. Kvist EE, Al–Shurbaji A, Dahl ML, et al. Quantitative pharmacogenetics of nortriptyline: a novel approach. Clin Pharmacokinet 2001;40:869–877.
386. Lappenberg–Pelzer M. Identification and determination of opipramol metabolites in plasma and urine. J Anal Toxicol 1998;22:215–219.
387. Lautala P, Ethell BT, Taskinen J, et al. The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP–glucuronosyltransferases. Drug Metab Dispos 2000;28:1385–1389.
388. Le Bloc‘h Y, Woggon B, Weissenrieder H, et al. Routine therapeutic drug monitoring in patients treated with 10–360 mg/day citalopram. Ther Drug Monit 2003;25: 600–608.
389. Lee CA, Cook JA, Reyner EL, et al. P-glycoprotein related drug interactions: clinical importance and a consideration of disease states. Expert Opin Drug Metab Toxicol 2010;6:603–619.
390. Lee SY, Kim YG, Kim HG, et al. Pharmacokineitc parameters of bromperidol in Korean subjects. Hum Psychopharmacol 2006;21:409–412.
391. Lefèvre G, Büche M, Sedek G, et al. Similar rivastigmine pharmacokinetics and pharmacodynamics in Japanese and white healthy participants following the application of novel rivastigmine patch. J Clin Pharmacol 2009;49:430–443.
392. Leucht S, Busch R, Kissling W, et al. Early prediction of antipsychotic nonresponse among patients with schizophrenia. J Clin Psychiatry 2007;68:352–360.
393. Leucht S, Steimer W, Kreuz S, et al. Doxepin plasma concentrations: is there really a therapeutic range? J Clin Psychopharmacol 2001;21:432–439.
394. LeWitt PA, Jennings D, Kelly EL, et al. Pharmacokinetic-pharmacodynamic crossover comparison of two levodopa extension strategies. Mov Disord 2009;24:1319–1324.
395. Licht RW, Vestergaard P, Kessing LV, et al. Psychopharmacological treatment with lithium and antiepileptic drugs: suggested guidelines from the Danish Psychiatric Association and the Child and Adolescent Psychiatric Association in Denmark. Acta Psychiatr Scand Suppl 2003;419:1–22.
396. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of Antipsychotic Drugs in Patients with Chronic Schizophrenia. N Engl J Med 2005;353:1209–1223.
397. Lind AB, Reis M, Bengtsson F, et al. Steady-state concentrations of mirtazapine, N-desmethylmirtazapine, 8-hydroxymirtazapine and their enantiomers in relation to cytochrome P450 2D6 genotype, age and smoking behaviour. Clin Pharmacokinet 2009;48:63–70 .
398. Lindberger M, Luhr O, Johannessen SI, et al. Serum concentrations and effects of gabapentin and vigabatrin: obersvations from a dose titration study.Ther Drug Monit 2003;25:457–462.
399. Lindenmayer J. Long-acting injectable antipsychotics: focus on Olanzapine pamoate. Neuropsychiatr Dis Treat 2010;6:261–267.
340. Linder MW, Keck PE JR. Standards of laboratory practice: antidepressant drug monitoring. National Academy of Clinical Biochemistry. Clin Chem 1998;44:1073–1084.
401. Lingam R, Scott J. Treatment non-adherence in affective disorders. Acta Psychiatr Scand 2002;105:164–172.
402. Lingjaerde O, Ahlfors UG, Bech P, et al. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross–sectional study of side effects in neuroleptic–treated patients. Acta Psychiatr Scand Suppl 1987;334:1–100.
403. Liu Y, Jiao J, Zhang C, et al. A simplified method to determine five cytochrome p450 probe drugs by HPLC in a single run. Biol Pharm Bull 2009;32:717–720.
404. Liu ZQ, Shu Y, Huang SL, et al. Effects of CYP2C19 genotype and CYP2C9 on fluoxetine N–demethylation in human liver microsomes. Acta Pharmacol Sin 2001;22:85–90 .
405. Lobo ED, Bergstrom RF, Reddy S, et al. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin Pharmacokinet 2008;47:191–202.
406. Locatelli I, Kstelic M, Kores–Plesnicar, et al. A population pharmacokinetic evalutation of the influence of CYP2D6 genotype on risperidone metabolism in patients with acute episode schizophrenia. Eur J Pharm Sci 2010;41:289–298.
407. Lotrich FE, Pollock BG. Aging and clinical pharmacology: implications for antidepressants. J Clin Pharmacol 2005;45:1106–1122.
408. Lucek R & Dixon R. Chlordiazepoxide concentrations in saliva and plasma measured by radioimmunoassay. Res Commun Chem Pathol Pharmacol 1980;27:397–400.
409. Lundberg J, Christophersen JS, Petereson KB, et al. Int J Neuropsychopharmacol 2007;10:777–785 .
410. Lundmark J, Bengtsson F, Nordin C, et al. Therapeutic drug monitoring of selective serotonin reuptake inhibitors influences clinical dosing strategies and reduces drug costs in depressed elderly patients. Acta Psychiatr Scand 2000;101:354–359.
411. Luurila H, Olkkola KT. Pharmacokinetic–pharmacodynamic modelling of zopiclione effects on human central nervous system. Pharmacol Toxicol 1996;78:348–353.
412 Madan A, Parkinson A, Faiman MD. Identification of the human P–450 enzymes responsible for the sulfoxidation and thiono–oxidation of diethyldithiocarbamate methyl ester: role of P–450 enzymes in disulfiram bioactivation. Alcohol Clin Exp Res 1998;22:1212–1219 .
413. Madsen H, Nielsen KK, Brøsen K. Imipramine metabolism in relation to the sparteine and mephenytoin oxidation polymorphisms–a population study. Br J Clin Pharmacol 1995;39:433–439.
414. Maguire KP, Burrows GD, Norman TR, et al. Metabolism and pharmacokinetics of dothiepin. Br J Clin Pharmacol 1981;12:405–409.
415. Maguire KP, Norman TR, Burrows GD, et al. An evaluation of maprotiline intravenous kinetics and comparison of two oral doses. Eur J Clin Pharmacol 1980;18:249–254.
416. Mahmood I, Sahajwalla C. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin Pharmacokinet 1999;36:277–287.
417. Mallikaarjun S, Salazar DE, Bramer SL. Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J Clin Pharmacol 2004;44:179–187.
418. Mamo D, Kapur S, Keshavan M, et al. D2 receptor occupancy of olanzapine pamoate depot using positron emission tomography: an open–label study in patients with schizophrenia. Neuropsychopharmacology 2008;33:298–304.
419. Mamo D, Kapur S, Shammi CM, et al. A PET study of dopamine D2 and serotonin 5–HT2 receptor occupancy in patients with schizophrenia treated with therapeutic doses of ziprasidone. Am J Psychiatry 2004;161:818–825.
420. Mann K, Hiemke C, Lotz J, et al. Appropriateness of plasma level determinations for lithium and valproate in routine care of psychiatric inpatients with affective disorders. J Clin Psychopharmacol 2006;26:671–673.
421. Mann K, Hiemke C, Schmidt LG, et al. Appropriateness of therapeutic drug monitoring for antidepressants in routine sychiatric inpatient care. Ther Drug Monit 2006;28:83–88 .
422. Markowitz J;Patrick K. Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter? J Clin Psychopharmacol 2008;28 Suppl 2:S54–S61.
423. Martines C, Gatti G, Sasso E, et al. The disposition of primidone in elderly patients. Br J Clin Pharmacol 1990;30:607–611.
424. Mason BJ, Goodman AM, Dixon RM, et al. A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology 2002;27:596–606.
425. Mattila MA, Larni HM. Flunitrazepam: a review of its pharmacological properties and therapeutic use. Drugs 1980;20:353–374.
426. Mauri MC, Laini V, Boscati L, et al. Long–term treatment of chronic schizophrenia with risperidone: a study with plasma levels. Eur Psychiatry 2001;16:57–63.
427. Mauri MC, Volonteri LS, Colasanti A, et al. Clinical pharmacokinetics of atypical antipsychotics. A critical review of the relationship between plasma concentrations and clinical response. Clin Pharmacokinet 2007;46:359–388.
428. May TW, Korn–Merker E, Rambeck B. Clinical pharmacokinetics of oxcarbazepine. Clin Pharmacokinet 2003;42:1023–1042.
429. May TW, Korn–Merker E, Rambeck B, et al. Pharmacokinetics of sulthiame in epileptic patients. Ther Drug Monit 1994;16:251–257.
430. May TW, Rambeck B, Jürgens U. Serum concentrations of levetiracetam in epileptic patients: the influence of dose and co–medication. Ther Drug Monit 2003;25:690–699.
431. May TW, Rambeck B, Jürgens U. Serum concentrations of topiramate in patients with epilepsy: influence of dose, age, and comedication. Ther Drug Monit 2002;24:366–374.
432. May TW, Rambeck B, Neb R, et al. Serum concentrations of pregabalin in patients with epilepsy: the influence of dose, age, and comedication. Ther Drug Monit 2007;29:789–794.
433. Mayo BC, Biggs SR, Chasseaud LF, et al. The metabolic fate of Sormodren (bornaprine hydrochloride) in animals and humans. Xenobiotica 1980;10:873–888.
434. McAlpine DE, Biernacka JM, Mrazek DA, et al. Effect of cytochrome P450 enzyme polymorphisms on pharmacokinetics of venlafaxine. Ther Drug Monit 2011;33:14–20.
435. McConville BJ, Arvanitis LA, Thyrum PT, et al. Pharmacokinetics, tolerability, and clinical effectiveness of quetiapine fumarate: an open–label trial in adolescents with psychotic disorders. J Clin Psychiatry 2000;61:252–260.
436. McKenzie ME, Roswell–Harris D. A controlled trial of prothipendyl (tolnate) in mentally subnormal patients. Br J Psychiatry 1966;112:95–100 .
437. Medori R, Mannaert E, Gründer G. Plasma antipsychotic concentration and receptor occupancy, with special focus on risperidone long–acting injectable. Eur Neuropsychopharmacol 2006;16:233–240.
438. Mehler–Wex C, Kölch M, Kirchheiner J, et al. Drug monitoring in child and adolescent psychiatry for improved efficacy and safety of psychopharmacotherapy. Child Adolesc Psychiatry Ment Health 2009;3:14 .
439. Meijer WE, Bouvy ML, Heerdink ER, et al. Spontaneous lapses in dosing during chronic treatment with selective serotonin reuptake inhibitors. Br J Psychiatry 2001;179:519–522.
440. Meineke I, Kress I, Poser W, et al. Therapeutic drug monitoring and its metabolite desmethylmirtazapine by HPLC with fluorescence detection. Ther Drug Monit 2004;26:277–283.
441. Meisenzahl EM, Schmitt G, Gründer G, et al. Striatal D2/D3 receptor occupancy, clinical response and side effects with amisulpride: an iodine–123–iodobenzamide SPET study. Pharmacopsychiatry 2008;41:169–175.
442. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J Psychiatry Neurosci 2007;32:86–102.
443. Meyer JH, Wilson AA, Sagrati S, et al. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry 2004;161:826–835.
444. Meyer RP, Gehlhaus M, Knoth R, et al. Expression and function of cytochrome p450 in brain drug metabolism. Curr Drug Metab 2007;8:297–306.
445. Meyer–Barner M, Meineke I, Schreeb KH, et al. Pharmacokinetics of doxepin and desmethyldoxepin: an evaluation with the population approach. Eur J Clin Pharmacol 2002;58:253–257.
446. Michelson D, Read HA, Ruff DD, et al. CYP2D6 and clinical response to atomoxetine in children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry 2007;46:242–251.
447. Mihara K, Yasui–Furukori N, Kondo T, et al. Relationship between plasma concentrations of trazodone and its active metabolite, m–chlorophenylpiperazine, and its clinical effect in depressed patients. Ther Drug Monit 2002;24:563–566.
448. Mimaki T. Clinical pharmacology and therapeutic drug monitoring of zonisamide. Ther Drug Monit 1998;20:593–597.
449. Miura H. Zonisamide monotherapy with once–daily dosing in children with cryptogenic localization–related epilepsies: clinical effects and pharmacokinetic studies. Seizure 2004;13 Suppl 1:S17–S23.
450. Miura M, Ohkubo T. Identification of human cytochrome P450 enzymes involved in the major metabolic pathway of fluvoxamine. Xenobiotica 2007;37:169–179.
451. Moffat AC, editor. Clarke‘s analysis of drugs and poisons. London: Pharmaceutical Press;2003. p. 1468–1469.
452. Molden E, Lunde H, Lunder N, et al. Pharmacokinetic variability of aripiprazole and the active metabolite dehydroaripiprazone in psychiatric patients. Ther Drug Monit 2006;28:744–749.
453. Montgomery SA, McAuley R, Montgomery DB. Relationsship between mianserin plasma levels and antidepressant effect in a double-blind trial comparing a single night-time and divided daily dose regimens. Br J Clin Pharmacol 1978;5:71S–76S.
454. Moody DE, Chang Y, Huang W, et al. The in vivo response of novel buprenorphine metabolites, M1 and M3, to antiretroviral inducers and inhibitors of buprenorphine metabolism. Basic Clin Pharmacol Toxicol 2009;105:211–215.
455. Morris RG, Black AB, Harris AL, et al. Lamotrigine and therapeutic drug monitoring: retrospective survey following the introduction of a routine service. Br J Clin Pharmacol 1998;46:547–551.
456. Morris RG, Lee MY, Cleanthous X, et al. Long–term follow–up using a higher target range for lamotrigine monitoring. Ther Drug Monit 2004;26:626–632.
457. Mrazek DA. Psychiatric pharmacogenomic testing in clinical practice. Dialogues Clin Neurosci 2010;12:69–76.
458. Müller H, Eusterschulte B, Havemann–Reinecke U, et al. Forensische Aspekte des therapeutischen Drug–Monitorings (TDM) in der Psychiatrie. Psychopharmakotherapie 2009;16:52–56.
459. Müller MJ, Dragicevic A, Fric M, et al. Therapeutic drug monitoring of tricyclic antidepressants: how does it work under clinical conditions? Pharmacopsychiatry 2003;36:98–104.
460. Müller MJ, Härtter S, Köhler D, et al. Serum levels of sulpiride enantiomers after oral treatment with racemic sulpiride in psychiatric patients: a pilot study. Pharmacopsychiatry 2001;34:27–32.
461. Müller MJ, Regenbogen B, Härtter S, et al. Therapeutic drug monitoring for optimizing amisulpride therapy in patients with schizophrenia. J Psychiat Res 2007;41: 673–679.
462. Müller-Isberner R, Freese R, Jöckel D, et al. Forensic psychiatric assessment and treatment in Germany. Legal framework, recent developments, and current practice. Int J Law Psychiatry 2000;23:467–480.
463. Nagy CF, Kumar D, Cullen EI, et al. Steady-state pharmacokinetics and safety of donepezil HCl in subjects with moderately impaired renal function. Br J Clin Pharmacol 2004;58 Suppl 1:18–24 .
464. Naito H, Wachi M, Nishida M. Clinical effects and plasma concentrations of long–term clonazepam monotherapy in previously untreated epileptics. Acta Neurol Scand 1987;76:58–63.
465. Nakamura K, Yokoi T, Inoue K, et al. CYP2D6 is the principal cytochrome P450 responsible for metabolism of the histamine H1 antagonist promethazine in human liver microsomes. Pharmacogenetics 1996;6:449–457 .
466. Nazirizadeh Y, Vogel F, Bader W, et al. Serum concentrations of paliperidone versus risperidone and clinical effects. Eur J Clin Pharmacol 2010;66:797–803.
467. Neels HM, Sierens AC, Naelaerts K., et al. Therapeutic drug monitoring of old and newer anti-epileptic drugs. Clin chem Lab Med 2004;42:1228–1255.
468. Nemoda Z, Angyal N, Tarnok Z, et al. Carboxylesterase 1 gene polymorphism and methylphenidate response in ADHD. Neuropharmacology 2009;57:731–733.
469. Nesvag R, Hendset M, Refsum H, et al. Serum concentrations of risperidone and 9-OH risperidone following intramuscular injection of long–acting risperidone compared with oral risperidone medication. Acta Psychiatr Scand 2006;114:21–26.
470. Nikisch G, Baumann P, Oneda B, et al. Cytochrome P450 and ABCB1 genetics: association with quetiapine and norquetiapine plasma and cerebrospinal fluid concentrations and with clinical response in patients suffering from schizophrenia. A pilot study. J Psychopharmacol 2010 Dec 8. [Epub ahead of print].
471. Nikisch G, Mathé AA, Czernik A, et al. Stereoselective metabolism of citalopram in plasma and cerebrospinal fluid of depressive patients: relationship with 5-HIAA in CSF and clinical response. J Clin Psychopharmacol 2004;24:283–290 .
472. Nikolaus S, Larisch R, Beu M, et al. In vivo measurement of D2 receptor density and affinity for 18F-(3-N-methyl)benperidol in the rat striatum with a PET system for small laboratory animals. J Nucl Med 2003;44:618–624.
473. Nilsen OG, Dale O. Single dose pharmacokinetics of trazodone in healthy subjects. Pharmacol Toxicol 1992;71:150–153.
474. Nilsson MI, Meresaar U, Ånggard E. Clinical pharmacokinetics of methadone. Acta Anaesthesiol Scand Suppl 1982;74:66–69.
475. Nishikage H, Nakanishi T, Takamitsu Y, et al. Sequential changes in the plasma concentration of risperidone following intentional overdose. Clin Neuropharmacol 2002;25:307–309.
476. Nnadi CU, Malhotra AK. Clinical and pharmacogenetic studies of iloperidone. Per Med 2008;5:367–375.
477. Norman T, Chiu E, James RH, et al. Single oral dose pharmacokinetics of tiapride in patients with Huntington‘s disease. Eur J Clin Pharmacol 1987;32:583–586.
478. Nozawa M, Ohnuma T, Matsubara Y, et al. The relationship between the response of clinical symptoms and plasma olanzapine concentration, based on pharmacogenetics. Ther Drug Monit 2008;30:35–40.
479. Nutt JG, Fellman JH. Pharmacokinetics of levodopa. Clin Neuropharmacol 1984;7:35–49.
480. Nyberg S, Nordström AL, Halldin C, et al. Positron emission tomography studies on D2 dopamine receptor occupancy and plasma antipsychotic drug levels in man. Int Clin Psychopharmacol 1995 Sep;10 Suppl 3:81–85.
481. Nyholm D. Pharmacokinetic optimisation in the treatment of Parkinson‘s disease: an update. Clin Pharmacokinet 2006;45:109–136.
482. Obach RS, Cox LM, Tremaine LM. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: an in vitro study. Drug Metab Dispos 2005;33:262–270 .
483. Öhman D, Cherma MD, Norlander B, et al. Determination of serum reboxetine enantiomers in patients on chronic medication with racemic reboxetine. Ther Drug Monit 2003;25:174–182 .
484. Öhman D, Norlander B, Peterson C, et al. Bioanalysis of racemic reboxetine and its desethylated metabolite in a therapeutic drug monitoring setting using solid phase extraction and HPLC. Ther Drug Monit 2001;23:27–34.
485. Olesen OV, Linnet K. Hydroxylation and demethylation of the tricyclic antidepressant nortriptyline by cDNA-expressed human cytochrome P-450 isozymes. Drug Metab Dispos. 1997;25:740–744.
486. Olesen OV, Linnet K. Identification of the human cytochrome P450 isoforms mediating in vitro N-dealkylation of perphenazine. Br J Clin Pharmacol 2000;50:563–571.
487. Olesen OV, Linnet K. Contributions of five human cytochrome P450 isoforms to the N-demethylation of clozapine in vitro at low and high concentrations. J Clin Pharmacol 2001;41:823–832.
488. Orsulak PJ. Therapeutic monitoring of antidepressant drugs: current methodology and applications. J Clin Psychiatry 1986;47 Suppl: 39–52 .
489. Orsulak PJ. Therapeutic monitoring of antidepressant drugs: guidelines updated. Ther Drug Monit 1989;11:497–507.
490. Orsulak PJ, Schildkraut JJ. Guidelines for therapeutic monitoring of tricyclic antidepressant plasma levels. Ther Drug Monit 1979;1:199–208.
491. Ostad Haji E, Tadić A, Wagner S, et al. Association between citalopram serum levels and clinical improvement of patients with major depression. J Clin Psychopharmacol 2011;31:281–286.
492. Ota T, Shinotoh H, Fukushi K, et al. Estimation of plasma IC50 of donepezil for cerebral acetylcholinesterase inhibition in patients with Alzheimer’s disease using positron emission tomography. Clin Neuropharmacol 2010;33:74–78.
493. Palego L, Biondi L, Giannaccini G, et al. Clozapine, norclozapine plasma levels, their sum and ratio in 50 psychotic patients: influence of patient-related variables. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:473–480.
494. Panagiotidis G, Arthur HW, Lindh JD, et al. Depot haloperidol treatment in outpatients with schizophrenia on monotherapy: impact of CYP2D6 polymorphism on pharmacokinetics and treatment outcome. Ther Drug Monit 2007;29:417–422.
495. Paris BL, Ogilvie BW, Scheinkoenig JA, et al. In vitro inhibition and induction of human liver cytochrome p450 enzymes by milnacipran. Drug Metab Dispos 2009;37:2045–2054.
496. Park JY, Kim KA, Park PW, et al. Effect of CYP3A5*3 genotype on the pharmacokinetics and pharmacodynamics of alprazolam in healthy subjects.Clin Pharmacol Ther 2006;79:590–599.
497. Park PW, Seo YH, Ahn JY, et al. Effect of CYP3A5*3 genotype on serum carbamazepine concentrations at Steady-state in Korean epileptic patients. J Clin Pharm Ther 2009;34:569–574.
498. Parker DR, McIntyre IM. Case studies of post–mortem quetiapine: therapeutic or toxic concentrations? J Analyt Toxicol 2005;29:407–412.
499. Patsalos PN, Berry DJ, Bourgeois BF, et al. Antiepileptic drugs–best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008;49:1239–1276.
500. Paz E, Bouzas L, Hermida J, et al. Evaluation of three dosing models for the prediction of Steady-state trough clozapine concentrations. Clin Biochem 2008;41:603–606.
501. Pearce GA, Day RO. Compliance with criteria necessary for effective drug concentration monitoring. Ther Drug Monit 1990;12:250–257.
502. Pedersen OL, Gram LF, Kristensen CB, et al. Overdosage of Antidepressants: Clinical and Pharmacokinetic Aspects. Eur J Clin Pharmacol 1982;23:513–521.
503. Perez J, Chiron C, Musial C, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia 1999;40:1618–1626.
504. Perry PJ. The relationship between antidepressant response and tricyclic antidepressant plasma concentrations: a retrospective analysis of the literature using logistic regression analysis. Clin Pharmacokinet 1987;13:381–392.
505. Perry PJ. Therapeutic drug monitoring of antipsychotics. Psychopharmacol Bull 2001;35:19–29.
506. Perry PJ, Browne JL, Alexander B, et al. Relationship of free nortriptyline levels to therapeutic response. Acta Psychiatr Scand 1985;72:120–125.
507. Perry PJ, Miller DD, Arndt SV, et al. Clozapine and norclozapine plasma concentrations and clinical response of treatment-refractory schizophrenic patients. Am J Psychiatry 1991;148:231–235.
508. Perry PJ, Miller DD, Arndt SV, et al. Haloperidol dosing requirements: the contribution of smoking and nonlinear pharmacokinetics. J Clin Psychopharmacol 1993;13: 46–51.
509. Perry PJ, Sanger T, Beasley C. Olanzapine plasma concentrations and clinical response in acutely III schizophrenic patients. J Clin Psychopharmacol 1997;17:472–477.
510. Perry PJ, Zeilmann C, Arndt S. Tricyclic antidepressant concentrations in plasma: an estimate of their sensitivity and specificity as a predictor of response. J Clin Psychopharmacol 1994;14:230–240.
511. Perucca E, Cloyd J, Critchley D, et al. Rufinamide: clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia 2008;49: 1123–1141.
512. Petit P, Lonjon R, Cociglio M, et al. Carbamazepine and its 10,11–epoxide metabolite in acute mania: clinical and pharmacokinetic correlates. Eur J Clin Pharmacol 1991;41:541–546.
513. Peyronneau MA, Delaforge M, Riviere R, et al. High affinity of ergopeptides for cytochromes P450 3A. Importance of their peptide moiety for P450 recognition and hydroxylation of bromocriptine. Eur J Biochem 1994;223:947–956.
514. Pichini S, Papaseit E, Joya X, et al. Pharmacokinetics and therapeutic drug monitoring of psychotropic drugs in pediatrics. Ther Drug Monit 2009;31:283–318.
515. Pierce DM, Franklin RA, Harry TV, et al. Pharmacodynamic correlates of modified absorption: studies with lormetazepam. Br J Clin Pharmacol 1984;18:31–35.
516. Poggesi I, Benedetti MS, Whomsley R, et al. Pharmacokinetics in special populations. Drug Metab Rev 2009;41:422–454..
517. Potgieter GE, Groenewoud G, Jordaan PJ, et al. Pharmacokinetics of pipamperone from three different tablet formulations. Arzneimittelforschung 2002;52:430–444.
518. Pounder DJ, Jones GR. Post-mortem drug redistribution – a toxicological nightmare. Forensic Sci Int 1990;45:253–263.
519. Prakash C, Kamel A, Cui D, et al. Identification of the major human liver cytochrome P450 isoform(s) responsible for the formation of the primary metabolites of ziprasidone and prediction of possible drug interactions. Br J Clin Pharmacol 2000;49 Suppl 1:35S–42S.
520. Preskorn S, Patroneva A, Silman H, et al. Comparison of the pharmacokinetics of venlafaxine extended release and desvenafaxine in extensive and poor cytochrome P450 metabolizers. J Clin Psychopharmacol 2009;29:39–43.
521. Preskorn SH. CNS drug development: part I: the early period of CNS drugs. J Psychiatr Pract 2010;16:334–339.
522. Preskorn SH. CNS drug development: Part II: Advances from the 1960s to the 1990s. J Psychiatr Pract 2010;16:413–415.
523. Preskorn SH. Patients who do not respond to the „usual“ dose: why Terry fell off the dose–response curve. J Psychiatr Pract 2009;15:460–466.
524. Preskorn SH. Practical application of therapeutic drug monitoring: a tale of two patients. J Psychiatr Pract 2008;14:301–306.
525. Preskorn SH. Tricyclic antidepressant plasma level monitoring: an improvement over the dose–response approach. J Clin Psychiatry 1986;47 Suppl1:24–30.
526. Preskorn SH, Burke MJ, Fast GA. Therapeutic drug monitoring: Principles and practice. Ther Drug Monit 1993;16:611–641.
527. Preskorn SH, Fast GA. Therapeutic drug monitoring for antidepressants: efficacy, safety, and cost effectiveness. J Clin Psychiat 1991;52 Suppl: 23–33.
528. Preskorn SH, Fast GA. Tricyclic antidepressant–induced seizures and plasma drug concentration. J Clin Psychiatry 1992;53:160–162.
529. Preskorn SH, Fleck RJ, Schroeder DH. Therapeutic drug monitoring of bupropion. Am J Psychiatry 1990;147:1690–1691.
530. Preskorn SH, Jerkovich GS. Central nervous system toxicity of tricyclic antidepressants: phenomenology, course, risk factors, and role of therapeutic drug monitoring. J Clin Psychopharmacol 1990;10:88–95.
531. Puech A, Fleurot O, Rein W. Amisulpride, an atypical antipsychotic, in the treatment of acute episodes of schizophrenia: a dose-ranging study vs. haloperidol. Acta Psychiatr Scand 1998;98:65–72.
532. Pumariega AJ, Nelson R, Rotenberg L. Varenicline-induced mixed mood and psychotic episode in a patient with a past history of depression. CNS Spectr 2008;13:511–514.
533. Puozzo C, Albin H, Vinçon G, et al. Pharmacokinetics of milnacipran in liver impairment. Eur J Drug Metab Pharmacokinet 1998;23:273–279.
534. Raaflaub J. On the pharmacokinetics of chlorprothixene in man. Experientia 1975;31:557–558.
535. Radtke RA. Pharmacokinetics of levetiracetam. Epilepsia 2001;42 Suppl 4:24–27.
536. Rao VA, Bishop M, Coppen A. Clinical state, plasma levels of haloperidol and prolactin: a correlation study in chronic schizophrenia. Br J Psychiatry 1980;137:518–521.
537. Rasmussen BB, Brøsen K. Is therapeutic drug monitoring a case for optimizing clinical outcome and avoiding interactions of the selective serotonin reuptake inhibitors? Ther Drug Monit 2000;22:143–154.
538. Rasmussen PV, Jensen TS, Sindrup SH, et al. TDM–based imipramine treatment in neuropathic pain. Ther Drug Monit 2004;26:352–360.
539. Rauschenbach R, Gieschen H, Husemann M, et al. Stable expression of human cytochrome P450 3A4 in V79 cells and its application for metabolic profiling of ergot derivatives. Eur J Pharmacol 1995;293:183–190.
540. Ravva P, Gastonguay MR, Tensfeldt TG, et al. Population pharmacokinetic analysis of varenicline in adult smokers. Br J Clin Pharmacol 2009;68:669–681.
541. Rees JA. Clinical interpretation of pharmacokinetic data on dothiepine hydrochloride (Dosulepin, Prothiaden). J Int Med Res 1981;9:98–102.
542. Regenthal R, Krueger M, Koeppel C, et al. Drug levels: therapeutic and toxic serum/plasma concentrations of common drugs. J Clin Monit Comput 1999;15:529–544.
543. Reimold M, Solbach C, Noda S, et al. Occupancy of dopamine D(1), D (2) and serotonin (2A) receptors in schizophrenic patients treated with flupentixol in comparison with risperidone and haloperidol. Psychopharmacology (Berl) 2007;190:241–249.
544. Reis M, Aamo T, Ahlner J, et al. Reference concentrations of antidepressants. A compilation of post-mortem and therapeutic levels. J Analyt Toxicol 2007;31:254–264.
545. Reis M, Aamo T, Spigset O, et al. Serum concentrations of antidepressant drugs in a naturalistic setting: compilation based on a large therapeutic drug monitoring database. Ther Drug Monit 2009;31:42–56.
546. Reis M, Åberg–Wistedt A, Ågren H, et al. Compliance with SSRI medication during 6 months of treatment for major depression: an evaluation by determination of repeated serum drug concentrations. J Affect Disorders 2004;82:443–446.
547. Reis M, Akerblad AC, Ekselius L, et al. Partial compliance as determined from plasma levels of sertraline and its metabolite in depressed patients in primary care. J Clin Psychopharmacol 2010;30:746–748.
548. Reis M, Chermá MD, Carlsson B, et al. On behalf of the task force for TDM of escitalopram in Sweden. Therapeutic drug monitoring of escitalopram in an outpatient setting. Ther Drug Monit 2007;29:758–766.
549. Reis M, Lundmark J, Bengtsson F. Therapeutic drug monitoring of racemic citalopram: a 5-year experience in Sweden, 1992–1997. Ther Drug Monit 2003;25:183–191.
550. Reis M, Lundmark J, Björk H, et al. Therapeutic drug monitoring of racemic venlafaxine and its main metabolites in an everyday clinical setting. Ther Drug Monit 2002;24:545–553.
551. Reis M, Olsson G, Carlsson B, et al. Serum levels of citalopram and its main metabolites in adolescent patients treated in a naturalistic clinical setting. J Clin Psychopharmacol 2002;22:406–413.
552. Reis M, Prochazka J, Sitsen A, et al. Inter- and intraindividual pharmacokinetic variations of mirtazapine and its N-demethyl metabolite in patients treated for major depressive disorder: a 6-month therapeutic drug monitoring study. Ther Drug Monit 2005;27:469–477.
553. Remington G, Mamo D, Labelle A, et al. A PET study evaluating dopamine D2 receptor occupancy for long-acting injectable risperidone. Am J Psychiatry 2006;163:396–401.
554. Renwick AB, Mistry H, Ball SE, et al. Metabolism of Zaleplon by human hepatic microsomal cytochrome P450 isoforms. Xenobiotica 1998;28:337–348.
555. Riant P, Urien S, Albengres E, et al. Effects of the binding of imipramine to erythrocytes and plasma proteins on its transport through the rat blood–brain barrier. J Neurochem 1988;51:421–425.
556. Richens, A. Banfield CR, Salfi M, et al. Single and multiple dose pharmacokinetics of felbamate in the elderly. Br J Clin Pharmacol 1997;44:129–134.
557. Riedel M, Schwarz MJ, Strassnig M, et al. Risperidone plasma levels, clinical response and side–effects. Eur Arch Psychiatry Clin Neurosi 2005;255:261–268.
558. Rivas N, Buelga DS, Elger CE, et al. Population pharmacokinetics of lamotrigine with data from therapeutic drug monitoring in German and Spanish patients with epilepsy. Ther Drug Monit 2008;30:483–489.
559. Rivera–Calimlim L, Castañeda L, Lasagna L. Effects of mode of management on plasma chlorpromazine in psychiatric patients. Clin Pharmacol Ther 1973;14:978–986.
560. Roberts RL, Joyce PR, Mulder RT, et al. A common P–glycoprotein polymorphism is associated with nortriptyline-induced postural hypotension in patients treated for major depression. Pharmacogenomics J 2002;2:191–196.
561. Robertson P JR, Hellriegel ET. Clinical pharmacokinetic profile of modafinil. Clin Pharmacokinet 2003;42:123–137.
562. Rochat B, Kosel M, Boss G, et al. Stereoselective biotransformation of the selective serotonin reuptake inhibitor, citalopram, and its demethylated metabolites by monoamine oxidases in human liver. Biochem Pharmacol 1998;56:15–23.
563. Rogers SL, Friedhoff LT. The efficacy and safety of donepezil in patients with Alzheimer‘s disease: results of a US multicentre, randomized, double-blind, placebo-controlled trial. The Donepezil Study Group. Dementia 1996;7:293–303.
564. Roman M, Kronstrand R, Lindstedt D, et al. Quantitation of seven low-dosage antipsychotic drugs in human postmortem blood using LC-MS-MS. J Anal Toxicol 2008;32:147–155 .
565. Ronfeld RA, Tremaine LM, Wilner KD. Pharmacokinetics of sertraline and its N-demethyl metabolite in elderly and young male and female volunteers. Clin Pharmacokinet 1997;32 Suppl 1:22–30 .
566. Rosenzweig P, Canal M, Patat A, et al. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum Psychopharmacol 2002;17:1–13.
567. Rotzinger S, Fang J, Baker GB. Trazodone is metabolized to m-chlorophenylpiperazine by CYP3A4 from human sources. Drug Metab Dispos 1998;26:572–575.
568. Rougemont M, Ulrich S, Hiemke C, et al. French summaries of product characteristics: content in relation to therapeutic monitoring of psychotropic drugs. Fundam Clin Pharmacol 2010;24:377–384 .
569. Rudorfer V, Potter WZ. The role of metabolites of antidepressants in the treatment of depression. CNS Drugs 1997;7:273–312.
570. Ruottinen HM, Rinne UK. Effect of one month‘s treatment with peripherally acting catechol-O-methyltransferase inhibitor, entacapone, on pharmacokinetics and motor response to levodopa in advanced parkinsonian patients. Clin Neuropharmacol 1996;19:222–233.
571. Sachse J, Härtter S, Hiemke C. Automated determination of ziprasidone by HPLC with column switching and spectrophotometric detection. Ther Drug Monit 2005;27:158–162.
572. Sachse J, Härtter S, Weigmann H, et al. Automated determination of amisulpride by liquid chromatography with column switching and spectrophotometric detection. J Chromatogr B Analyt Technol Biomed Life Sci 2003;784:405–410.
573. Sachse J, Köller J, Härtter S, et al. Automated analysis of quetiapine and other antipsychotic drugs in human blood by high performance-liquid chromatography with column-switching and spectrophotometric detection. J Chromatogr B Analyt Technol Biomed Life Sci 2006;830:342–348.
574. Sagar KA, Smyth MR. Bioavailability studies of oral dosage forms containing levodopa and carbidopa using column–switching chromatography followed by electrochemical detection. Analyst 2000;125: 439–445.
575. Sage JI, Mark MH. Pharmacokinetics of continuous–release carbidopa/levodopa. Clin Neuropharmacol 1994;17 Suppl 2:1–6.
576. Sainati SM, Hubbard JW, Chi E, et al. Safety, tolerability, and effect of food on the pharmacokinetics of iloperidone (HP 873), a potential atypical antipsychotic. J Clin Pharmacol 1995;35:713–720.
577. Saint–Marcoux F, Sauvage FL, Marquet P. Current role of LC–MS in therapeutic drug monitoring. Anal Bioanal Chem 2007;388:1327–1349.
578. Saivin S, Hulot T, Chabac S, et al. Clinical pharmacokinetics of acamprosate. Clin Pharmacokinet 1998;35:331–345.
579. Sajatovic M, Velligan DI, Weiden PJ, et al. Measurement of psychiatric treatment adherence. J Psychosom Res 2010;69:591–599.
580. Salazar DE, Frackiewicz EJ, Dockens R, et al. Pharmacokinetics and tolerability of buspirone during oral administration to children and adolescents with anxiety disorder and normal healthy adults. J Clin Pharmacol 2001;41:1351–1358.
581. Sallee FR, Pollock BG, Stiller RL, et al. Pharmacokinetics of pimozide in adults and children with Tourette‘s syndrome. J Clin Pharmacol 1987;27:776–781.
582. Sartorius N, Baghai TC, Baldwin DS, et al. Antidepressant medications and other treatments of depressive disorders: a CINP Task Force report based on a review of evidence. Int J Neuropsychopharmacol 2007;10 Suppl 1:S1–S207.
583. Sauer JM, Ring BJ, Witcher JW. Clinical Pharmacokinetics of atomoxetine. Clin Pharmacokinet 2005;44:571–590.
584. Sauvage FL, Gaulier JM, Lachâtre G, et al. Pitfalls and prevention strategies for liquid chromatography–tandem mass spectrometry in the selected reaction–monitoring mode for drug analysis. Clin Chem 2008;54:1519–1527.
585. Schulberg HC, Katon W, Simon GE, et al. Treating major depression in primary care practice: an update of the Agency for Health Care Policy and Research Practice Guidelines. Arch Gen Psychiatry 1998;55:1121–1127.
586. Schulz M, Schmoldt A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie 2003;58:447–474.
587. Schwarzenbach F, Netillard C, Demoly P, et al. Antidepressant response and fluvoxamine plasma concentrations: a pilot study. Pharm World Sci 2003;25:27–29.
588. Scordo MG, Spina E, Dahl ML, et al. Influence of CYP2C9, 2C19 and 2D6 genetic polymorphisms on the Steady-state plasma concentrations of the enantiomers of fluoxetine and norfluoxetine. Basic Clin Pharmacol Toxicol 2005;97:296–301.
589. Seiler W, Wetzel H, Hillert A, et al. Pharmacokinetics and bioavailability of benperidol in schizophrenic patients after intravenous and two different kinds of oral application. Psychopharmacology (Berl) 1994;116:457–463.
590. Service JA, Waring WS. QT Prolongation and delayed atrioventricular conduction caused by acute ingestion of trazodone. Clin Toxicol (Phila) 2008;46:71–73.
591. Shams M, Hiemke C, Härtter S. Therapeutic drug monitoring of antidepressant mirtazapine and its N-demethylated metabolite in human serum. Ther Drug Monit 2004;26:78–84
592. Shams ME, Arneth B, Hiemke C, et al. CYP2D6 polymorphism and clinical effect of the antidepressant venlafaxine. J Clin Pharm Ther 2006;31:493–502.
593. Sharma S, Joshi S, Chadda RK. Therapeutic drug monitoring of lithium in patients with bipolar affective disorder: experiences from a tertiary care hospital in India. Am J Ther 2009;16:393–397.
594. Shin JG, Soukova N, Flockart DA. Effect of antipsychotic drugs on human liver cytochrome P–450 (CYP) isoforms in vitro: Preferential inhibition of CYP2D6. Drug Metab Dispos 1999;27:1078–1084.
595. Shinderman M, Maxwell S, Brawand–Amey M, et al. Cytochrome P4503A4 metabolic activity, methadone blood concentrations, and methadone doses. Drug Alcohol Depend 2003;69:205–211.
596. Shiraga T, Kaneko H, Iwasaki K, et al. Identification of cytochrome P450 enzymes involved in the metabolism of zotepine, an antipsychotic drug, in human liver microsomes. Xenobiotica 1999;29: 217–229.
597. Shua–Haim J, Smith J, Picard F, et al. Steady-state pharmacokinetics of rivastigmine in patients with mild to moderate Alzheimer‘s disease not affected by co-administration of memantine: an open-label, crossover, single–centre study. Clin Drug Investig 2008;28:361–374.
598. Sidhu J, Priskorn M, Poulsen M, et al. Steady-state pharmacokinetics of the enantiomers of citalopram and its metabolites in human. Chirality 1997;9:686–692.
599. Siva N. Tackling the booming trade in counterfeit drugs. Lancet 2010;376:1725–1726.
600. Skinner MH, Kuan HY, Skerjanec A, et al. Effect of age on the pharmacokinetics of duloxetine in women. Br J Clin Pharmacol 2004;57:54–61 .
601. Skogh E, Bengtsson F, Nordin C. Could discontinuing smoking be hazardous for patients administered clozapine medication? A case report. Ther Drug Monit 1999;21:580–582.
602. Skogh E, Reis M, Dahl ML, et al. Therapeutic drug monitoring data on olanzapine and its N–demethyl metabolite in the naturalistic clinical setting. Ther Drug Monit 2002;24:518–526.
603. Small JG, Hirsch SR, Arvanitis LA, et al. Quetiapine in patients with schizophrenia – a high- and low-dose double-blind comparison with placebo. Seroquel study group. Arch Gen Psychiatry 1997;54:549–557.
604. Smith RB, Kroboth PD, Vanderlugt JT, et al. Pharmacokinetics and pharmacodynamics of alprazolam after oral and IV administration. Psychopharmacology (Berl) 1984;84:452–456.
605. Smith SW. Chiral toxicology: it‘s the same thing … only different. Toxicol Sci 2009;110:4–30.
606. Snoeck E, Van Peer A, Sack M, et al. Influence of age, renal and liver impairment on the pharmacokinetics of risperidone in man. Psychopharmacology (Berl) 1995;122:223–229.
607. Søgaard B, Mengel H, Rao N, et al. The pharmacokinetics of escitalopram after oral and intravenous administration of single and multiple doses to healthy subjects. J Clin Pharmacol 2005;45:1400–1406.
608. Soldin P, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet 2009;48:143–157.
609. Someya T, Muratake T, Hirokane G, et al. Interindividual variation in bromperidl metabolism and relationship to therapeutic effects. J Clin Psychopharmacol 2000;20:175–180.
610. Søndergaard Khinchi M, Nielsen KA, Dahl M, et al. Lamotrigine therapeutic thresholds. Seizure 2008;17:391–395.
611. Soni SD. Fluspirilene in the treatment of non-hospitalized schizophrenic patients. Curr Med Res Opin 1977;4:645–649.
612. Sparshatt A, Taylor D, Patel MX, et al. A systematic review of aripiprazole–dose, plasma concentration, receptor occupancy, and response: implications for therapeutic drug monitoring. J Clin Psychiatry 2010;71:1447–1456.
613. Sparshatt A, Taylor D, Patel MX, et al. Amisulpride – dose, plasma concentration, occupancy and response: implications for therapeutic drug monitoring. Acta Psychiatr Scand 2009, 120:416–428.
614. Spencer TJ, Biederman J, Ciccone PE, et al. PET study examining pharmacokinetics, detection and likeability, and dopamine transporter receptor occupancy of short- and long–acting oral methylphenidate. Am J Psychiatry 2006;163:387–395 .
615. Spencer TJ, Bonab AA, Dougherty DD, et al. A PET study examining pharmacokinetics and dopamine transporter occupancy of two long–acting formulations of methylphenidate in adults. Int J Mol Med 2010;25:261–265.
616. Spigset O, Hägg S, Stegmayr B, et al. Citalopram pharmacokinetics in patients with chronic renal failure and the effect of haemodialysis. Eur J Clin Pharmacol 2000;59:699–703.
617. Spina E, Avenoso A, Facciolà G, et al. Relationship between plasma risperidone and 9-hydroxyrisperidone concentrations and clinical response in patients with schizophrenia. Psychopharmacology (Berl) 2001;153:238–243.
618. Spina E, Birgersson C, von Bahr Ö, et al. Phenotypic consistency in hydroxylation of desmethylimipramine and debrisoquine in healthy subjects and in human liver microsomes. Clin Pharmacol Ther 1984;36:677–682.
619. Sproule BA, Hardy BG, Shulman KI. Differential pharmacokinetics of lithium in elderly patients. Drugs Aging 2000;16:165–177.
620. Stassen HH, Anghelescu IG, Angst J, et al. Predicting response to psychopharmacological treatment. Survey of recent results. Pharmacopsychiatry 2011 this issue.
621. Stassen HH, Angst J, Hell D, et al. Is there a common resilience mechanism underlying antidepressant drug response? Evidence from 2848 patients. J Clin Psychiatry 2007;68:1195–1205.
622. Stead AH, Moffat AC. A collection of therapeutic, toxic and fatal blood drug concentrations in man. Hum Exp Toxicol 1983;3:437–464.
623. Steimer W. Pharmacogenetics and Psychoactive Drug Therapy: Ready for the Patient? Ther Drug Monit 2010;32:381–386.
624. Steimer W, Potter JM. Pharmacogenetic screening and therapeutic drugs. Clin Chim Acta 2002;315:137–155.
625. Steimer W, Zöpf K, von Amelunxen S, et al. Amitriptyline or not, that is the question: pharmacogenetic testing of CYP2D6 and CYP2C19 identifies patients with low or high risk for side effects in amitriptyline therapy. Clin Chem 2005;51:376–385.
626. Stieffenhofer V, Hiemke C. Pharmacogenetics, therapeutic drug monitoring and non compliance. Ther Umsch 2010;67:309–315.
627. Stieffenhofer V, Saglam H, Schmidtmann I, et al. Clozapine plasma level monitoring for prediction of rehospitalization schizophrenic outpatients. Pharmacopsychiatry 2011;44:55–59.
628. Stock B, Spiteller G. Metabolism of antiparkinson drugs. An example of competitive hydroxylation. Arzneimittelforschung 1979;29:610–615.
629. Störmer E, Brockmöller J, Roots I, et al. Cytochrome P-450 enzymes and FMO3 contribute to the disposition of the antipsychotic drug in vitro. Psychopharmacology (Berl) 2000;151:312–320.
630. Störmer E, von Moltke LL, Shader RI, et al. Metabolism of the antidepressant mirtazapine in vitro: contribution of cytochromes P-450 1A2, 2D6, and 3A4. Drug Metab Dispos 2000;28:1168–1175.
631. Suhara T, Takano A, Sudo Y, et al. High levels of serotonin transporter occupancy with low–dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch Gen Psychiatry 2003;60:386–391.
632. Sutfin TA, Perini GI, Molnar G, et al. Multiple–dose pharmacokinetics of imipramine and its major active and conjugated metabolites in depressed patients. J Clin Psychopharmacol 1988;8:48–53.
633. Suzuki A, Otani K, Ishida M, et al. Increased plasma concentrations of bromperidol and its reduced metabolite with levomepromazine, but not with thioridazine. Ther Drug Monit 1997;19:261–264.
634. Suzuki Y, Fukui N, Sawamura K, et al. Concentration–response relationship for fluvoxamine using remission as an endpoint: a receiver operating characteristics curve analysis in major depression. J Clin Psychopharmacol 2008;28:325–328.
635. Svirbely JR, Speicher CE. The importance of request and report forms in the interpretation of therapeutic drug monitoring data. Ther Drug Monit 1980;2:211–216.
636. Sweet RA, Pollock BG, Kirshner M, et al. Pharmacokinetics of single- and multiple-dose bupropion in elderly patients with depression. J Clin Pharmacol 1995;35:876–884.
637. Sweet RA, Pollock BG, Mulsant BH, et al. Pharmacologic profile of perphenazine‘s metabolites. J Clin Psychopharmacol 2000;20:181–187.
638. Szegedi A, Jansen WT, van Willigenburg AP, et al. Early improvement in the first 2 weeks as a predictor of treatment outcome in patients with major depressive disorder: a meta-analysis including 6562 patients. J Clin Psychiatry 2009;70:344–353.
639. Takano A, Suhara T, Ichimiya T, et al. Time course of in vivo 5–HTT transporter occupancy by fluvoxamine. J Clin Psychopharmacol 2006;26:188–191.
640. Takano A, Suzuki K, Kosaka J, et al. A dose-finding study of duloxetine based on serotonin transporter occupancy. Psychopharmacology 2006;185:395–399.
641. Tan L, Yu JT, Sun YP, et al. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. Clin Neurol Neurosurg 2010;112:320–323.
642. Tanaka O, Kondo T, Otani K, et al. Single oral dose kinetics of zotepine and its relationship to prolactin response and side effects. Ther Drug Monit 1998;20:117–119.
643. Tanaka E, Kurata N, Yasuhara H. How useful is the „cocktail approach“ for evaluating human hepatic drug metabolizing capacity using cytochrome P450 phenotyping probes in vivo? J Clin Pharm Ther 2003;28:157–165.
644. Tasker TCG, Kaye CM, Zussman BD, et al. Paroxetine plasma levels: lack of correlation with efficacy or adverse events. Acta Psychiatr Scand 1989;80 Suppl 350:152–155.
645. Tateishi T, Watanabe M, Kumai T, et al. CYP3A is responsible for N-dealkylation of haloperidol and bromperidol and oxidation of their reduced forms by human liver microsomes. Life Sci 2000;67:2913–2920.
646. Taylor D. Antidepressant drugs and cardiovascular pathology: a clinical overview of effectiveness and safety. Acta Psychiatr Scand 2008;118:434–442.
647. Taylor D. Psychopharmacology and adverse effects of antipsychotic long–acting injections: a review. Br J Psychiatry Suppl 2009;52:S13–S19.
648. Thanacoody RH, Daly AK, Reilly JG, et al. Factors affecting drug concentrations and QT interval during thioridazine therapy. Clin Pharmacol Ther 2007;82:555–565.
649. The Scottish Schizophrenia Research Group. The Scottish first episode Schizophrenia study II. Treatment: pimozide versus flupenthixol. Br J Psychiatry 1987;150:334–338.
650. Thieme D, Rolf B, Sachs H, et al. Correlation of inter-individual variations of amitriptyline metabolism examined in hairs with CYP2C19 and CYP2D6 polymorphisms. Int J Legal Med 2008;122:149–155.
651. Timmer CJ, Sitsen JM, Delbressine LP. Clinical pharmacokinetics of mirtazapine. Clin Pharmacokinet 2000;38:461–474.
652. Tiseo PJ, Rogers SL, Friedhoff LT. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following evening administration. Br J Clin Pharmacol 1998;46 Suppl 1:13–18.
653. Titier K, Canal M, Déridet E, et al. Determination of myocardium to plasma concentration ratios of five antipsychotic drugs: comparison with their ability to induce arrhythmia and sudden death in clinical practice. Toxicol Appl Pharmacol 2004;199:52–60.
654. Toennes SW, Maurer HH. Microsoft Excel in pharmacokinetics – an easy way to solve kinetic problems in clinical toxicology, legal medicine or doping control. In Sachs H, Bernhard W, Jeger A, editors. Proceedings of the 34th International TIAFT Meeting, Interlaken, 11–15 August 1996. Leipzig: Molina, 1997. p. 201–204.
655. Tokairin T, Fukasawa T, Yasui-Furukori N, et al. Inhibition of the metabolism of brotizolam by erythromycin in humans: in vivo evidence for the involvement of CYP3A4 in brotizolam metabolism. Br J Clin Pharmacol 2005;60:172–175.
656. Tokunaga H, Kudo K, Imamura T, et al. Plasma concentrations of antipsychotic drugs in psychiatric inpatients. Nippon Hoigaku Zasshi 1997;51:417–422.
657. Tompson DJ, Vearer D. Steady-state pharmacokinetic properties of a 24–hour prolonged-release formulation of ropinirole: results of two randomized studies in patients with Parkinson‘s disease. Clin Ther 2007;29:2654–2666.
658. Topiwala A, Fazel S. The pharmacological management of violence in schizophrenia: a structured review. Expert Rev Neurother 2011;11:53–63.
659. Tornio A, Neuvonen PJ, Backman JT. The CYP2C8 inhibitor gemfibrozil does not increase the plasma concentrations of zopiclone. Eur J Clin Pharmacol 2006;62: 645–651.
660. Touw DJ, Neef C, Thomson AH, et al. Cost-effectiveness of therapeutic drug monitoring: a systematic review. Ther Drug Monit 2005;27:10–17.
661. Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR(*)D measurement–based care. Neuropsychopharmacol 2007;32:2479–2489.
662. Tsai MH, Lin KM, Hsiao MC et al Genetic polymorphisms of cytochrome P450 enzymes influence metabolism of the antidepressant escitalopram and treatment response. Pharmacogenomics 2010;11:537–546.
663. Tuerck D, Wang Y, Maboudian M, et al. Similar bioavailability of dexmethylphenidate extended (bimodal) release, dexmethyl-phenidate immediate release and racemic methylphenidate extended (bimodal) release formulations in man. Int J Clin Pharmacol Ther 2007;45:662–668.
664. Turbott J, Norman TR;Burrows GD, et al. Pharmacokinetics of nortriptyline in elderly volunteers. Commun Psychopharmacol 1980;4:225–231.
665. Turpeinen M, Koivuviita N, Tolonen A, et al. Effect of renal impairment on the pharmacokinetics of bupropion and its metabolites. Br J Clin Pharmacol 2007;64:165–173.
666. Uchida H, Mamo DC, Mulsant BH, et al. Increased antipsychotic sensitivity in elderly patients: evidence and mechanisms. J Clin Psychiatry 2009;70:397–405.
667. Uhr M, Steckler T, Yassouridis A, et al. Penetration of amitriptyline, but not of fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to mdr1a P-glycoprotein gene disruption. Neuropsychopharmacology 2000;22:380–387.
668. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 2008;57:203–239.
669. Ujiie Y, Fukasawa T, Yasui–Furukori N, et al. Rifampicin markedly decreases plasma concentration and hypnotic effect of brotizolam. Ther Drug Monit 2006;28: 299–302.
670. Ulrich S, Baumann B, Wolf R, et al.Therapeutic drug monitoring of clozapine and relapse–a retrospective study of routine clinical data. Int J Clin Pharmacol Ther 2003;41:3–13.
671. Ulrich S, Hiemke C, Laux G, et al. TDM group of the Arbeitsgemeinschaft Neuropsychopharmakologie und Pharmakopsychiatrie (AGNP). Value and actuality of the prescription information for therapeutic drug monitoring of psychopharmaceuticals: a comparison with the medico–scientific evidence. Pharmacopsychiatry 2007;40:121–127.
672. Ulrich S, Läuter J. Comprehensive survey of the relationship between serum concentration and therapeutic effect of amitriptyline in depression. Clin Pharmacokinet 2002;41:853–876 .
673. Ulrich S, Sandmann U, Genz A. Serum concentrations of haloperidol pyridinium metablites and the relationship with tardive dyskinesia and parkonsism: a cross-section study in psychiatric patients. Pharmacopsychiatry 2005;38:171–177.
674. Ulrich S, Wurthmann C, Brosz M, et al. The relationship between serum concentration and therapeutic effect of haloperidol in patients with acute schizophrenia. Clin Pharmacokinet 1998;34:227–263.
675. Valdes R Jr, Payne DA, Linder MW, editors. Laboratory medicine practice guidelines and recommendations for laboratory analysis and application of pharmacogenetics to clinical practice. Washington, DC: National Academy of Clinical Biochemistry;2010.
676. Van der Weide J, Steijns LS, van Weelden MJ. The effect of smoking and cytochrome P450 CYP1A2 genetic polymorphism on clozapine clearance and dose requirement. Pharmacogenetics 2003;13:169–172.
677. Van der Weide J, van Baalen–Benedek EH, Kootstra–Ros JE. Metabolic ratios of psychotropics as indication of cytochrome P450 2D6/2C19 genotype. Ther Drug Monit 2005;27:478–483.
678. Van der Zwaag C, McGee M, McEvoy JP, et al. Response of patients with treatment-refractory schizophrenia to clozapine within three serum level ranges. Am J Psychiatry 1996;153:1579–1584.
679. Van Gorp F, Whyte IM, Isbister GK. Clinical and ECG effects of Escitalopram overdose. Ann Emerg Med 2009;54:4–408.
680. Van Putten T, Marder SR, Wirshing WC, et al. Neuroleptic plasma levels. Schizophr Bull 1991;17:197–216.
681. Varsaldi F, Miglio G, Scordo MG, et al. Impact of the CYP2D6 polymorphism on Steady-state plasma concentrations and clinical outcome of donepezil in Alzheimer‘s disease patients. Eur J Clin Pharmacol 2006;62:721–726.
682. Vasudev K, Das S, Goswami U, et al. Pharmacokinetics of valproic acid in patients with bipolar disorder. J Psychopharmacol 2001;15:187–190.
683. Vasudev K, Goswami U, Kohli K. Carbamazepine and valproate monotherapy: feasibility, relative safety and efficacy, and therapeutic drug monitoring in manic disorder. Psychopharmacology (Berl) 2000;150:15–23.
684. Veefkind AH, Haffmans PMJ, Hoencamp E. Venlafaxine serum levels and CYP2D6 genotype. Ther Drug Monit 2000;22:202–208.
685. Velligan DI, Lam YW, Glahn DC, et al. Defining and assessing adherence to oral antipsychotics: a review of the literature. Schizophr Bull 2006;32:724–742.
686. Venkatakrishnan K, Culm KE, Ehrenberg BL, et al. Kinetics and dynamics of intravenous adinazolam, N-desmethyl adinazolam, and alprazolam in healthy volunteers. J Clin Pharmacol 2005;45:529–537.
687. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): implications for interactions with enzyme-inducing drugs. J Clin Pharmacol 1999;39:567–577.
688. Vernaleken I, Fellows C, Janouschek H, et al. Striatal and extrastriatal D2/D3-receptor-binding properties of ziprasidone: a positron emission tomography study with [18F]Fallypride and [11C]raclopride (D2/D3-receptor occupancy of ziprasidone). J Clin Psychopharmacol 2008;28:608–617.
689. Vernaleken I, Janouschek H, Raptis M, et al. Dopamine D2/3 receptor occupancy by quetiapine in striatal and extrastriatal areas. Int J Neuropsychopharmacol 2010;13:951–960.
690. Vernaleken I, Siessmeier T, Buchholz HG, et al. High striatal occupancy of D2-like dopamine receptors by amisulpride in the brain of patients with schizophrenia. Int J Neuropsychopharmacol 2004;7:421–430.
691. Ververs FF, Voorbij HA, Zwarts P, et al. Effect of cytochrome P450 2D6 genotype on maternal paroxetine plasma concentrations during pregnancy. Clin Pharmacokinet 2009;48:677–683.
692. Viala A, Ba B, Durand A, et al. Comparative study of the pharmacokinetics of zuclopenthixol decanoate and fluphenazine decanoate. Psychopharmacology (Berl) 1988;94:293–297.
693. Vine W, Bowers LD. Cyclosporine: structure, pharmacokinetics, and therapeutic drug monitoring. Crit Rev Clin Lab Sci 1987;25:275–311.
694. Viola MS, Bercellini MA, Saidon P, et al. Pharmacokinetic variability of oxcarbazepine in epileptic patients. Medicina (B Aires) 2000;60:914–918.
695. Vogel F, Gansmüller R, Leiblein T, et al. The use of ziprasidone in clinical practice: Analysis of pharmacokinetic and pharmacodynamic aspects from data of a drug monitoring survey. Eur Psychiatry 2009;24:143–148.
696. Voineskos AN, Wilson AA, Boovariwala A, et al. Serotonin transporter occupancy of high-dose selective serotonin reuptake inhibitors during major depressive disorder measured with [11C]DASB positron emission tomography.Psychopharmacology (Berl) 2007;193:539–545.
697. Von Moltke LL, Greenblatt DJ, Giancarlo GM, et al. Escitalopram (S–citalopram) and its metabolites in vitro: cytochromes mediating biotransformation, inhibitory effects, and comparison to R–citalopram. Drug Metab Dispos 2001;29:1102–1109.
698. Von Moltke LL, Greenblatt DJ, Granda BW, et al. Zolpidem metabolism in vitro: responsible cytochromes, chemical inhibitors, and in vivo correlations. Br J Clin Pharmacol 1999;48:89–97.
699. Vormfelde SV, Bitsch A, Meineke I, et al. Non-response to maprotiline caused by ultra-rapid metabolism that is different from CYP2D6? Eur J Clin Pharmacol 1997;52:387–390.
700. Vuille F, Amey M, Baumann P. Use of plasma level monitoring of antidepressants in clinical practice. Towards an analysis of clinical utility. Pharmacopsychiatry 1991;24:190–195.
701. Waade RB, Christensen H, Rudberg I, et al. Influence of comedication on serum concentrations of aripiprazole and dehydroaripiprazole. Ther Drug Monit 2009;31:233–238.
702. Waldschmitt C, Vogel F, Maurer C, et al. Measurement of duloxetine in blood using high–performance liquid chromatography with spectrophotometric detection and column switching. Ther Drug Monit 2007;29:767–772.
703. Waldschmitt C, Vogel F, Pfuhlmann B, et al. Duloxetine serum concentrations and clinical effects. Data from a therapeutic drug monitoring (TDM) survey Pharmacopsychiatry 2009;42:189–193.
704. Wan J, Xia H, He N, et al. The elimination of diazepam in Chinese subjects is dependent on the mephenytoin oxidation phenotype. Br J Clin Pharmacol 1996;42:471–474.
705. Wang JH, Liu ZQ, Wang W, et al. Pharmacokinetics of sertraline in relation to genetic polymorphism of CYP2C19. Clin Pharmacol Ther 2001;70:42–47.
706. Ward E, Musa MN, Bailey LG. Clinical pharmacokinetics of lithium. J Clin Pharmacol 1994;34:280–285 .
707. Weber J, McCormack PL. Asenapine. CNS Drugs. 2009;23:781–792.
708. Weiden PJ, Kozma C, Grogg A, et al. Partial compliance and risk of rehospitalization among California Medicaid patients with schizophrenia. Psychiatr Serv 2004;55:886–891.
709. Weigmann H, Bierbrauer J, Härtter S, et al. Automated determination of clozapine and major metabolites in serum and urine. Ther Drug Monit 1997;19:480–488.
710. Weigmann H, Härtter S, Hiemke C. Automated determination of clomipramine and its major metabolites in human and rat serum by high-performance liquid chromatography with on-line column-switching. J Chromatogr B Biomed Sci Appl 1998;710: 227–233 .
711. Weigmann H, Härtter S, Maehrlein S, et al. Simultaneous determination of olanzapine, clozapine and demethylated metabolites in serum by on-line column-switching high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 2001;759:63–71.
712. Weiss U, Marksteiner J, Kemmler G, et al. Effects of age and sex on olanzapine plasma concentrations. J Clin Psychopharmacol 2005;25:570–574.
713. Wen B, Ma L, Zhu M. Bioactivation of the tricyclic antidepressant amitriptyline and its metabolite nortriptyline to arene oxide intermediates in human liver microsomes and recombinant P450s. Chem Biol Interact 2008;173:59–67.
714. Wen B, Zhou M. Metabolic activation of the phenothiazine antipsychotics chlorpromazine and thioridazine to electrophilic iminoquinone species in human liver microsomes and recombinant P450s. Chem Biol Interact 2009;181:220–226.
715. White NC, Litovitz T, Clancy C. Suicidal antidepressant overdoses: a comparative analysis by antidepressant type. J Med Toxicol 2008;4:238–250.
716. Wienkers LC, Allievi C, Hauer MJ, et al. Cytochrome P-450-mediated metabolism of the individual enantiomers of the antidepressant agent reboxetine in human liver microsomes. Drug Metab Dispos 1999;27:1334–1340.
717. Wiesel FA, Alfredsson G, Ehrnebo M, et al. The pharmacokinetics of intravenous and oral sulpiride in healthy human subjects. Eur J Clin Pharmacol 1980;17:385–391.
718. Wille SM, Cooreman SG, Neels HM, et al. Relevant issues in the monitoring and the toxicology of antidepressants. Crit Rev Clin Lab Sci 2008;45:25–89.
719. Willmore LJ, Abelson MB, Ben-Menachem E, et al. Vigabatrin: 2008 update. Epilepsia 2009;50:163–173.
720. Wilson JF. Survey of reference ranges and clinical measurements for psychoactive drugs in serum. Ther Drug Monit 2003;25:243–247.
721. Wilting I, Heerdink ER, Mersch PP, et al. Association between lithium serum level, mood state, and patient-reported adverse drug reactions during long-term lithium treatment: a naturalistic follow-up study. Bipolar Disord 2009;11:434–440.
722. Wincor MZ, Munjack DJ, Palmer R. Alprazolam levels and response in panic disorder: preliminary results. J Clin Psychopharmacol 1991;11:48–51.
723. Winter HR, Earley WR, Hamer-Maansson JE, et al. Steady-state pharmacokinetic, safety, and tolerability profiles of quetiapine, Norquetiapine, and other quetiapine metabolites in peidatric and adult patients with psychotic disorders. J Child Adolesc Psychopharmacol 2008;18:81–98.
724. Wójcikowski J, Boksa J, Daniel WA. Main contribution of the cytochrome P450 isoenzyme 1A2 (CYP1A2) to N-demethylation and 5-sulfoxidation of the phenothiazine neuroleptic chlorpromazine in human liver - a comparison with other phenothiazines. Biochem Pharmacol 2010;80:1252–1259.
725. Wójcikowski J, Daniel WA. Perazine at therapeutic drug concentrations inhibits human cytochrome P450 isoenzyme 1A2 (CYP1A2) and caffeine metabolism – an in vitro study. Pharmacol Rep 2009;61:851–858.
726. Wójcikowski J, Pichard–Garcia L, Maurel P, et al. Contribution of human cytochrome p–450 isoforms to the metabolism of the simplest phenothiazine neuroleptic promazine. Br J Pharmacol 2003;138:1465–1474.
727. Wolff K, Hay AW, Rasitrick D, et al. Steady-state pharmacokinetics of methadone in opioid addicts. Eur J Clin Pharmacol 1993;44:189–194.
728. Wong SL, Granneman GR. Modeling of sertindole pharmacokinetic dispostions in healthy volunteers in short term dose–escalation studies. J Pharmaceut Sci 1998;87:1629–1631.
729. Wong SL, Menacherry S, Mulford D, et al. Pharmacokinetics of sertindole and dehydrosertindole in volunteers with normal or impaired renal function. Eur J Clin Pharmacol 1997;52:223–227.
730. Wright CE, Sisson TL, Ichhpurani AK, et al. Steady-state pharmacokinetic properties of pramipexole in healthy volunteers. J Clin Pharmacol 1997;37:520–525.
731. Wynalda MA, Wienkers LC. Assessment of potential interactions between dopamine receptor agonists and various human cytochrome P450 enzymes using a simple in vitro inhibition screen. Drug Metab Dispos 1997;25:1211–1214.
732. Xiang Q, Zhao X, Zhou Y, et al. Effect of CYP2D6, CYP3A5, and MDR1 genetic polymorphisms on the pharmacokinetics of risperidone and its active moiety. J Clin Pharmacol 2010;50:659–666.
733. Xu P, Li HD, Zhang BK, et al. Pharmacokinetics and tolerability of modafinil tablets in Chinese subjects. J Clin Pharm Ther 2008;33:429–437.
734. Yao C, Raoufinia A, Gold M, et al. Steady-state pharmacokinetics of galantamine are not affected by addition of memantine in healthy subjects. J Clin Pharmacol 2005;45:519–528.
735. Yasui-Furukori N, Kondo T, Ishida M, et al. The characteristics of side–effects of bromperidol in schizophrenic patients. Psychiatry Clin Neurosci 2002;56:103–106.
736. Yasui-Furukori N, Saito M, Nakagami T, et al. Association between multidrug resistance 1 (MDR1) gene polymorphisms and therapeutic response to bromperidol in schizophrenic patients: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:286–291.
737. Yasui-Furukori N, Saito M, Nakagami T, et al. Clinical response to risperidone in relation to plasma drug concentrations in acutely exacerbated schizophrenic patients. J Psychopharmacol 2010;24:987–994.
738. Yeung PK, Hubbard JW, Korschinski ED, et al. Pharmacokinetics of chlorpromazine and key metabolites. Eur J Clin Pharmacol 1993;45:563–569.
739. Yin OQ, Wing YK, Cheung Y, et al. Phenotype-genotype relationship and clinical effects of citalopram in Chinese patients. J Clin Psychopharmacol. 2006;26:367–372.
740. Yu DK, Dimmitt DC, Lanman RC, et al. Pharmacokinetics of dothiepin in humans: a single dose dose-proportionality study. J Pharm Sci 1986;75:582–585.
741. Zernig G, Hiemke C, Havemann-Reinecke U., et al. Empfehlungen für die gutachterliche Bewertung von Medikamentenspiegeln in der Psychiatrie im gerichtsanhängigen Schadensfall. Psychopharmakotherapie 2009;16:57–64.
742. Zernig G, Lechner T, Kramer-Reinstadler K, et al. What the clinician still has to be reminded of. Ther Drug Monit 2004;26:582.
743. Zernig G, Ng K, Hiemke C, et al. Therapeutic drug monitoring–based clozapine dosing recommendations. Ther Drug Monit 2007;29:130–131.
744. Zhao Q, Iyer GR, Verhaeghe T, et al. Pharmacokinetics and safety of galantamine in subjects with hepatic impairment and healthy volunteers. J Clin Pharmacol 2002;42:428–436.
745. Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin Pharmacokinet 2009;48:689–723.
746. Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part II. Clin Pharmacokinet 2009;48:761–804.
747. Zhou SF, Liu JP, Chowbay B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev 2009c;41:89–295.
748. Zimmerman NP, Hickie IB, McGorry PD. Guidelines for youth depression: time to incorporate new perspectives. Med J Aust 2010;193:557.
749. Zullino DF, Delessert D, Eap CB, et al. Tobacco and cannabis smoking cessation can lead to intoxication with clozapine or olanzapine. Int Clin Psychopharmacol 2002;17:141–143.
*Diese Arbeit ist eine Übersetzung von „Hiemke C, et al. AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2011. Pharmacopsychiatry 2011;44:195–235”, © Georg Thieme Verlag, Stuttgart, 2011.
Prof. Dr. Christoph Hiemke für die TDM-Gruppe der AGNP, Klinik für Psychiatrie und Psychotherapie, Universitätsmedizin Mainz, Untere Zahlbacher Straße 8, 55128 Mainz, E-Mail: hiemke@uni-mainz.de
Psychopharmakotherapie 2012; 19(03)