Alexander Ströhle, Hannover
Wenngleich Carnitin (Abb. 1) bereits 1905 aus Muskelfleischextrakt isoliert worden war, gelang es erst Anfang der 1960er Jahre, die stereochemische Beschaffenheit des Naturstoffs vollständig aufzuklären.
Abb. 1. L-Carnitin
Wie Tabelle 1 zeigt, ist Carnitin in zahlreichen Lebensmitteln enthalten, wo es in freier und veresterter Form vorliegt. Relevante Mengen finden sich allerdings nur in Lebensmitteln tierischer Herkunft. Insbesondere Muskelfleisch weist hohe Gehalte auf − ein Umstand, der zur Namensgebung der Verbindung führte („Carnitin“ abgeleitet von lat. „carnis“: Fleisch) [20].
Tab. 1. Vorkommen von L-Carnitin in ausgewählten Lebensmitteln ([28] unter Verweis auf [20])
|
Lebensmittel |
Gehalt an L-Carnitin [mg/100 g] |
Lebensmittel |
Gehalt an L-Carnitin [mg/100 g] |
|
|
Kalbfleisch (Lende) |
132,8 |
Erbsen |
5,7 |
|
|
Rindfleisch (Lende) |
65,0 |
Vollmilch (4 % Fett) |
2,3 |
|
|
Lammfleisch |
40,5 |
Butter |
1,3 |
|
|
Putenfleisch |
21,2 |
Parmesan |
0,7 |
|
|
Schweinefleisch |
21,1 |
Zwiebeln |
0,7 |
|
|
Camembert |
14.4 |
Äpfel |
0,2 |
|
|
Joghurt |
12,2 |
Bananen |
0,2 |
|
|
Hühnerbrust |
10,4 |
Erdnüsse |
0,2 |
|
|
Kalbsleber |
6,5 |
Kartoffeln |
2,4 |
|
|
Lachs |
5,8 |
Nudeln |
0,0 |
Aufgrund der unterschiedlichen Carnitingehalte pflanzlicher und tierischer Lebensmittel variiert die Zufuhr in Abhängigkeit von der Ernährungsweise. Während Personen mit regelmäßigem Fleischkonsum täglich 1,0 bis 2,5 mg pro kg Körpergewicht (KG) zuführen, stellen vegetarische Kostformen weniger als 0,16 mg Carnitin/kg KG/Tag bereit [42]. Basierend auf Verzehrsangaben aus Frankreich wurde für eine übliche Mischkost eine durchschnittliche Aufnahme von etwa 75 mg täglich errechnet. Ovo-Lacto-Vegetarier und Veganer konsumieren mit 16 bzw. 4 mg/Tag erheblich geringere Mengen [20]. Dennoch ist die Versorgung mit Carnitin auch bei einer veganen Ernährung üblicherweise sichergestellt, da die Verbindung vom Menschen selbst in ausreichendem Maße gebildet werden kann; ein exogener Bedarf existiert somit nicht [45]. Allerdings gibt es bestimmte Lebensphasen und -umstände, wo die endogene Synthesekapazität nicht ausreicht, den Bedarf an Carnitin zu decken. Aus diesem Grund wird Carnitin als konditionell-essenzieller Nährstoff bezeichnet und der Gruppe der Vitaminoide zugerechnet [29].
Carnitin-Kinetik
Absorption
Carnitin liegt in der Nahrung in freier Form und als Carnitin-Acylester vor. Die mit der Nahrung zugeführten Carnitin-Acylester werden im Dünndarm unter Einfluss einer vom exokrinen Pankreas sezernierten Esterase hydrolytisch gespalten. Die Resorption des freien Carnitins erfolgt dann im Duodenum und Jejunum, wo Carnitin über einen aktiven, Carrier-vermittelten Mechanismus in die Enterozyten transportiert wird.
Distribution
Das resorbierte Carnitin gelangt über die Pfortader zur Leber. Von dort erreicht es zusammen mit dem in den Hepatozyten synthetisierten Carnitin den systemischen Kreislauf, wo es vorwiegend in freier, teils auch in proteingebundener Form vorliegt.
Der Import von Carnitin aus dem Blut in Organe, die auf eine exogene Versorgung angewiesen sind (u.a. Herz- und Skelettmuskulatur), erfolgt über einen Natrium-abhängigen Prozess. Verantwortlich hierfür sind zwei als OCTN1 und OCTN2 bezeichnete Carrierproteine. Im höheren Lebensalter ist die Expression von OCTN2 reduziert [31]. Dies erklärt auch, weshalb das Muskelgewebe älterer Personen einen − verglichen mit jungen Menschen − geringeren Carnitin-Gehalt aufweist [15].
Blutspiegel
Die Höhe der Carnitin-Plasmakonzentration ist alters- sowie geschlechtsabhängig und variiert in Abhängigkeit von der Zufuhr mit der Nahrung. Allerdings zeigt eine Vergleichsstudie mit Vegetariern und Mischköstlern, dass die Ernährungsweise einen nur geringen Einfluss auf die Carnitin-Versorgung erwachsener Männer ausübt. Bei Frauen und Kindern hingegen sind die nutritiven Effekte stärker ausgeprägt [33].
Exkretion
Die Ausscheidung von Carnitin erfolgt primär über die Nieren, wobei 90 bis 98 % des glomerulär filtrierten Carnitins im Tubulus reabsorbiert werden. Nur ein geringer Teil des Carnitins − vorwiegend in acetylierter Form − wird über die Galle ausgeschieden und erreicht über den enterohepatischen Kreislauf wieder die Leber. Die Verluste über die Galle sind somit gering [41].
Physiologische Bedeutung
Die physiologische Bedeutung von Carnitin basiert auf seinen Funktionen im intermediären Fett- und Energiestoffwechsel. Diese umfassen folgende Prozesse:
- Import von aktivierten, langkettigen Fettsäuren in die Mitochondrienmatrix (siehe Abb. 2)
- Transport kurz- und mittelkettiger Acyl-CoA-Formen von den Peroxisomen zu den Mitochondrien im Rahmen des Abbaus überlanger Fettsäuren
- Transport kurz- und mittelkettiger Acyl-CoA-Formen von den Peroxisomen zu den Mitochondrien im Rahmen des Abbaus methylverzweigter Fettsäuren
- Regulation des Acetyl-CoA-Pools bzw. des Anteils an freiem und gebundenem Coenzym A
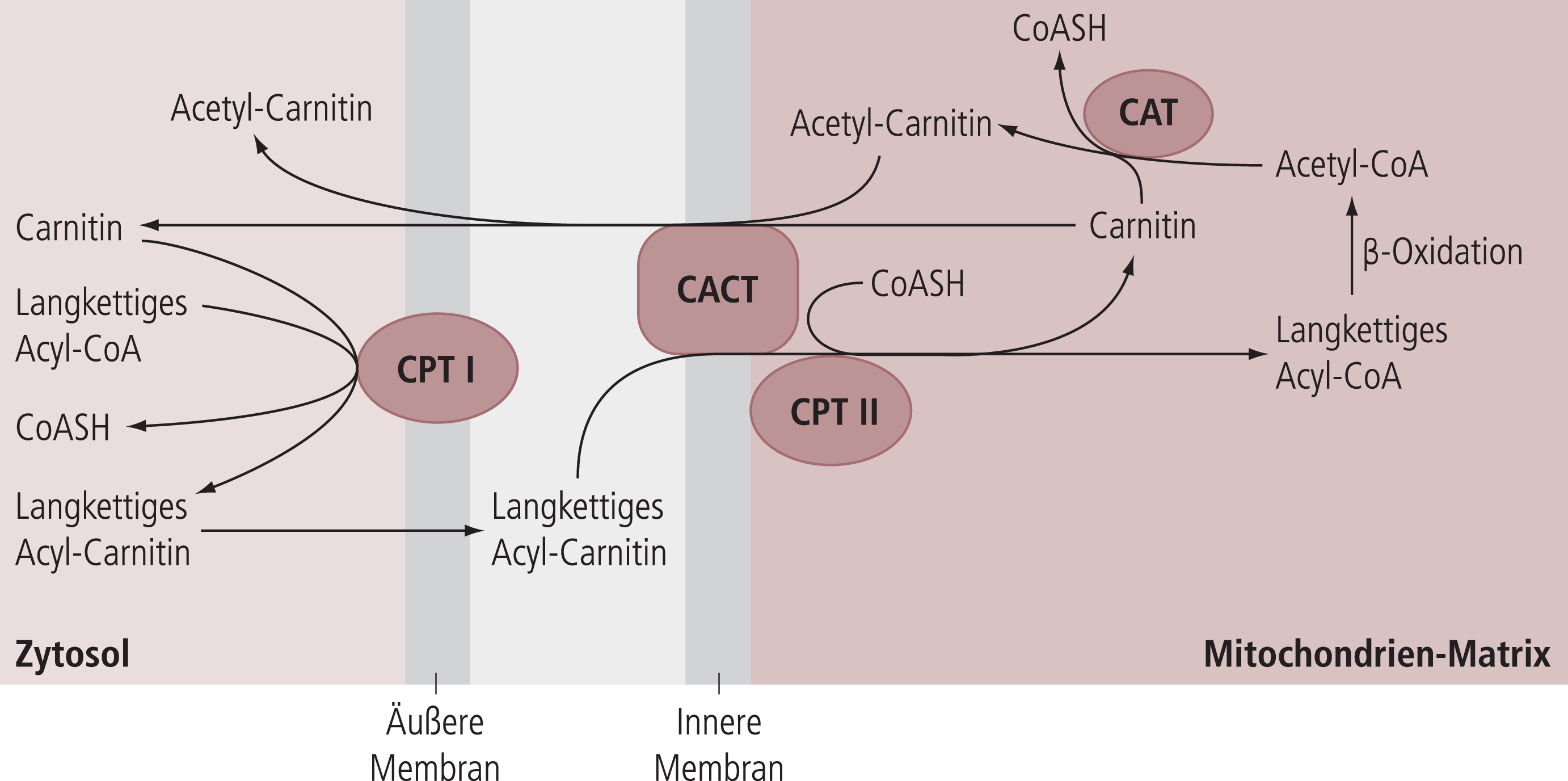
Abb. 2. Bedeutung von Carnitin für die Aufnahme aktivierter langkettiger Fettsäuren (Acyl-CoA) aus dem Zytosol in die Mitochondrien-Matrix [54]. CAT: Carnitin-Acyl-Transferase; CACT: Carnitin/Acyl-Carnitin-Carrier; CPT-1: Carnitin-Palmitoyl-Transferase I; CPT-2: Carnitin-Palmitoyl-Transferase II; CoASH: freies Coenzym A.
Einfluss von Valproinsäure auf den Carnitin-Stoffwechsel
Valproinsäure (Abb. 3; siehe Infokasten 1) interagiert in vielfältiger Weise mit dem Carnitin-Stoffwechsel. Betroffen sind die Bereiche zelluläre Aufnahme, Biosynthese, renale Ausscheidung und intrazellulärer Carnitin-Pool [32, 50, 53]:
Abb. 3. Valproinsäure
Infokasten 1: Pharmakodynamik und -kinetik von Valproinsäure [4, 8, 35, 46, 59]
Allgemeines
Valproinsäure (VPA) ist eine verzweigtkettige Monocarbonsäure (2-Propylpentansäure; Abb. 3) synthetischen Ursprungs. Aufgrund ihrer antikonvulvisen Effekte wird VPA seit den 1960er Jahren erfolgreich bei der Behandlung von zerebralen Krampfanfällen eingesetzt. VPA gilt noch heute als eines der wirksamsten Antikonvulsiva und findet insbesondere zur Therapie fokal eingeleiteter und generalisierter Anfälle (Absencen, Myoklonien, Aufwach-Grand-Mal) Verwendung. Daneben kommt VPA als Migräneprophylaktikum und zur Behandlung bipolarer Störungen zum Einsatz. Aufgrund ihrer Eigenschaft, Histon-Desacetylasen zu inhibieren, könnte VPA zukünftig in der Krebstherapie von Bedeutung sein. Vorteilhaft ist die geringe sedierende Wirkung. Neurokognitive Störungen, wie sie unter der Therapie mit anderen Antikonvulsiva häufig zu beobachten sind, bleiben meist aus.
Wirkungsmechanismus
Valproinsäure moduliert eine Reihe neurochemischer Prozesse, die für die antikonvulsive Wirkung der Substanz verantwortlich zu machen sind. Dazu zählen:
- Modulation des GABA-Systems. Valproinsäure steigert die GABA-(Gamma-Amino-Buttersäure-)vermittelte postsynaptische Hemmung und senkt so die neuronale Erregbarkeit. Dieser Effekt ist vermutlich nicht − wie früher angenommen − auf die Steigerung der Konzentration an GABA durch Valproinsäure zurückzuführen. Vielmehr legen neuere Daten eine direkte Beeinflussung des GABA-Rezeptors durch Valproinsäure nahe.
- Modulation von Ionen-Kanälen. Valproinsäure inhibiert die hochfrequenten repetitiven Entladungen depolarisierter Neuronen, indem es die Calcium-abhängigen Kaliumflüsse steigert.
Pharmakokinetische Daten
- Bioverfügbarkeit: 90–100%
- Zeit zwischen Einnahme und maximaler Serumkonzentration (tmax): 1–8 Stunden
- Zeit bis zum Erreichen einer Steady-State-Konzentration: 2 Tage
- Verteilungsvolumen: 0,09–0,17 l/kg
- Proteinbindung: 70–93%
- Eliminationshalbwertszeit: 7–17 Stunden
Metabolismus
Der Großteil der Valproinsäure wird in der Leber abgebaut; nur etwa 3% gelangen unverändert in den Urin. Hauptabbauwege sind die direkte Glucuronidierung und die oxidative Metabolisierung via Beta- und Omega-Oxidation. Während im Zuge der Beta-Oxidation atoxische und gering-toxische Produkte entstehen, geht der omega-oxidative Abbau mit der Bildung problematischer Substanzen einher. Insbesondere der Metabolit 2-Propylpenta-4-ensäure (4-en-Valproinsäure) wird für die hepatoxischen Effekte der Valproinsäure verantwortlich gemacht.
Zelluläre Aufnahme von Carnitin
Valproinsäure reagiert mit Carnitin unter Bildung von Valproyl-Carnitin-Ester (siehe Abb. 4, Reaktionsschritt 1). Sowohl Valproinsäure als auch Valproyl-Carnitin beeinträchtigen den Import von Carnitin aus dem extrazellulären Raum durch kompetitive Inhibition des OCTN2-Carriers [23, 37, 44].
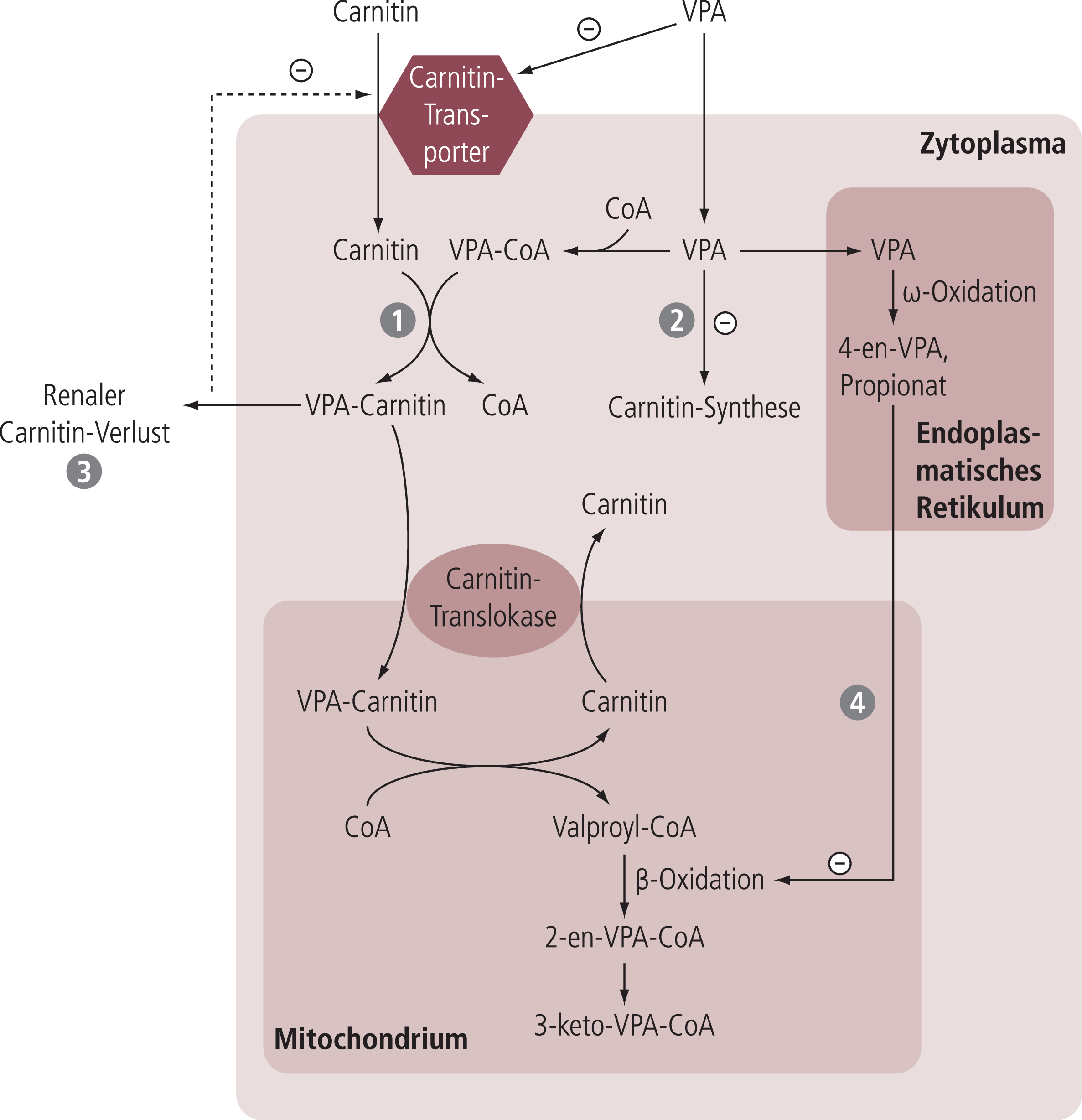
Abb. 4 Interaktionen zwischen Valproinsäure- und Carnitin-Stoffwechsel (modifiziert nach [43]). –: Inhibition; VPA: Valproinsäure; CoA: Coenzym A; weitere Erläuterungen s. Text
Biosynthese von Carnitin
Valproat vermindert die Konzentration an α-Ketoglutarat (Co-Substrat der γ-Butyro-Betain-Hydroxylase) und damit die endogene Carnitin-Synthese (siehe Abb. 4, Reaktionsschritt 2; [25]).
Ausscheidung von Carnitin
Valproyl-Carnitin-Ester werden über die Niere eliminiert, so dass es zu vermehrten Carnitin-Verlusten kommen kann (siehe Abb. 4, Reaktionsschritt 3). Quantitativ hat dieser Prozess allerdings nur eine untergeordnete Bedeutung [50].
Renale Reabsorption von Carnitin
Basierend auf den Befunden, dass (a) die tubuläre Rückresorption von Carnitin durch den OCTN2-Carrier erfolgt und (b) dieser durch Valproinsäure und Valproyl-Carnitin kompetitiv gehemmt wird, wurde postuliert, dass die Gabe von Valproat zu einer vermehrten renalen Elimination von Carnitin führt − eine Hypothese, die durch tierexperimentelle Daten untermauert wird [11]. Eine an gesunden Männern durchgeführte Kurzzeitstudie (Dauer: 34 Tage) ergab allerdings keine Hinweise für eine Valproat-induzierte Abnahme der Carnitin-Rückresorption [51].
Intrazellulärer Carnitin-Pool
Manche der beim oxidativen Abbau von Valproinsäure gebildeten Metaboliten (u.a. 2-Propylpenta-4-ensäure; syn. 4-en-Valproat) inhibieren Enzyme der Beta-Oxidation (siehe Abb. 4, Reaktionsschritt 4).
Primär hat dies einen Acyl-CoA-Stau zur Folge, wodurch Acyl-Gruppen vermehrt auf Carnitin übertragen werden; die Konzentration an freiem Carnitin nimmt damit ab. Auf der anderen Seite ist die Hemmung der Beta-Oxidation mit einer Reduktion der ATP-Bildung assoziiert, was den energieabhängigen Import von Carnitin in die Zelle beeinträchtigt [53].
Auswirkungen der Valproinsäure-Carnitin-Interaktion
Da Valproinsäure in multipler Weise die Carnitin-Homöostase des Organismus beeinträchtigt, stellt sich die Frage, welche klinischen Konsequenzen hieraus erwachsen.
Wie aus zahlreichen Untersuchungen hervorgeht, weisen Patienten unter Valproinsäure-Therapie im Vergleich mit Kontrollpersonen häufig verminderte Plasmaspiegel an Carnitin auf [Raskind/El-Chaar].
Wie die differenzierte Auswertung der Studiendaten gezeigt hat, wird das Risiko und Ausmaß einer Valproinsäure-assoziierten Hypocarnitinämie von einer Reihe von Faktoren bestimmt. Dazu zählen [16, 19]:
- Lebensalter und Gesundheitszustand: Bei Kindern und Kleinkindern mit Mehrfachbehinderung kommt es ungleich häufiger zu einer Hypocarnitinämie als bei ansonsten gesunden älteren Personen mit Epilepsie.
- Dauer der Pharmakotherapie: Das Risiko eines Carnitin-Defizits nimmt unter chronischer Valproinsäure-Therapie zu.
- Art der Pharmakotherapie: Eine Kombinationstherapie von Valproinsäure mit anderen Antikonvulsiva erhöht das Risiko für eine Hypocarnitinämie.
- Ernährungsstatus: Personen − insbesondere Heranwachsende − mit einem eingeschränkten Ernährungsstatus weisen häufig ein Carnitin-Defizit auf.
- Ernährungsweise: Das Praktizieren einer ketogenen Diät im Rahmen der Epilepsie-Behandlung erhöht das Risiko für eine Hypocarnitinämie.
Insgesamt lässt sich festhalten, dass die Ätiopathogenese des Valproinsäure-induzierten Carnitin-Defizits multifaktorieller Natur ist und eine Valproinsäure-Behandlung nicht per se mit einer Hypocarnitinämie einhergehen muss. Zu beachten ist allerdings, dass Carnitin-Plasmaspiegel im Normbereich nicht notwendigerweise eine ausreichende Versorgung der Gewebe reflektieren. So konnten in einer Studie bei Valproinsäure-behandelten Kindern trotz normaler Carnitin-Spiegel depletierte Gewebe festgestellt werden [49].
Pathophysiologische Konsequenzen des Valproinsäure-assoziierten Carnitin-Defizits
Aufgrund der zentralen Stellung von Carnitin im intermediären Stoffwechsel wurden einige der unter Valproinsäure-Therapie zu beobachtenden unerwünschten Nebenwirkungen auf ein sekundäres Carnitin-Defizit zurückgeführt. Hierzu zählen:
- Gewichtszunahme
- Hyperammonämie
- Enzephalopathie
- Hepatotoxizität
Die zugrundeliegenden Pathomechanismen und die Bewertung der Evidenz sind im Folgenden zusammengefasst.
Gewichtszunahme
Die Zunahme des Körpergewichts zählt zu den häufigsten Begleiteffekten einer Valproinsäure-Behandlung; 40 bis 70% der Patienten sind davon betroffen [55].
Ein möglicher pathophysiologischer Zusammenhang besteht in der Beeinträchtigung der Beta-Oxidation bei Carnitin-Defizit und dem dadurch verminderten Abbau von Fettsäuren. Insgesamt soll hieraus eine vermehrte Synthese von Neutralfetten resultieren [1, 9].
Nachgewiesen wurde eine Abnahme des Abbaus von (freien) Fettsäuren unter Valproinsäure-Behandlung [9, 36].
Andererseits korrelierte in einer Studie mit jungen Epilepsiepatienten die Gewichtszunahme nicht mit dem Carnitin-Plasma-Spiegel [20]. In einer Interventionsstudie zeigte sich kein signifikanter Effekt einer Carnitin-Supplementierung (15 mg/kg KG/Tag) auf das Körpergewicht bei Personen unter Valproinsäure-Behandlung [24].
Wenngleich nicht auszuschließen ist, dass ein Carnitin-Defizit zur Gewichtssteigerung unter Valproinsäure-Therapie beiträgt, scheinen hierfür primär andere Mechanismen (Valproinsäure-induzierte Leptinresistenz, Hyperleptin- und Hyperinsulinämie) verantwortlich zu sein [55].
Hyperammonämie
Unter der Behandlung mit Valproinsäure entwickeln etwa 50% der behandelten Patienten eine meist transitorische Hyperammonämie (Ammoniakspiegel >96 µg/dl) [39]. Häufig handelt es sich um asymptomatische Formen, die ohne klinische Konsequenzen bleiben [12, 32]. Als Risikofaktoren für die Entwicklung einer zum Teil schwerwiegenden Hyperammonämie gelten:
- Sehr junge Patienten (<2 Jahre)
- Polypharmakotherapie (Valproinsäure in Kombination mit Phenytoin, Phenobarbital und Carbamazepin) [13, 22, 34, 57]
- Personen mit angeborenen Defekten des Harnstoffzyklus (Ornithin-Carbamoyltransferase-Defizit) [47]
Eine pathophysiologischer Zusammenhang mit dem Carnitin-Stoffwechsel könnte auch hier in der Beeinträchtigung der Beta-Oxidation bei Carnitin-Defizit bestehen, wodurch Valproinsäure vermehrt über Omega-Oxidation abgebaut würde. Im Zuge der Omega-Oxidation von Valproinsäue werden toxische Abbauprodukte gebildet, unter anderem 4-en-Valproat. Dieses hemmt die Carbamoylphosphat-Synthase; dadurch wird die Harnstoffsynthese gehemmt und es kommt zu einem Stau von Ammoniak (Abb. 5). Die Hemmung der Harnstoffsynthese wird verstärkt, indem die Valproat-vermittelte Beeinträchtigung der Beta-Oxidation die Bereitstellung von Acetyl-CoA vermindert, wodurch die Synthese von N-Acetyl-Glutamat (NAGA), dem allosterischen Aktivator der Carbamoylphosphat-Synthase, abnimmt [32, 47] (Abb. 5).
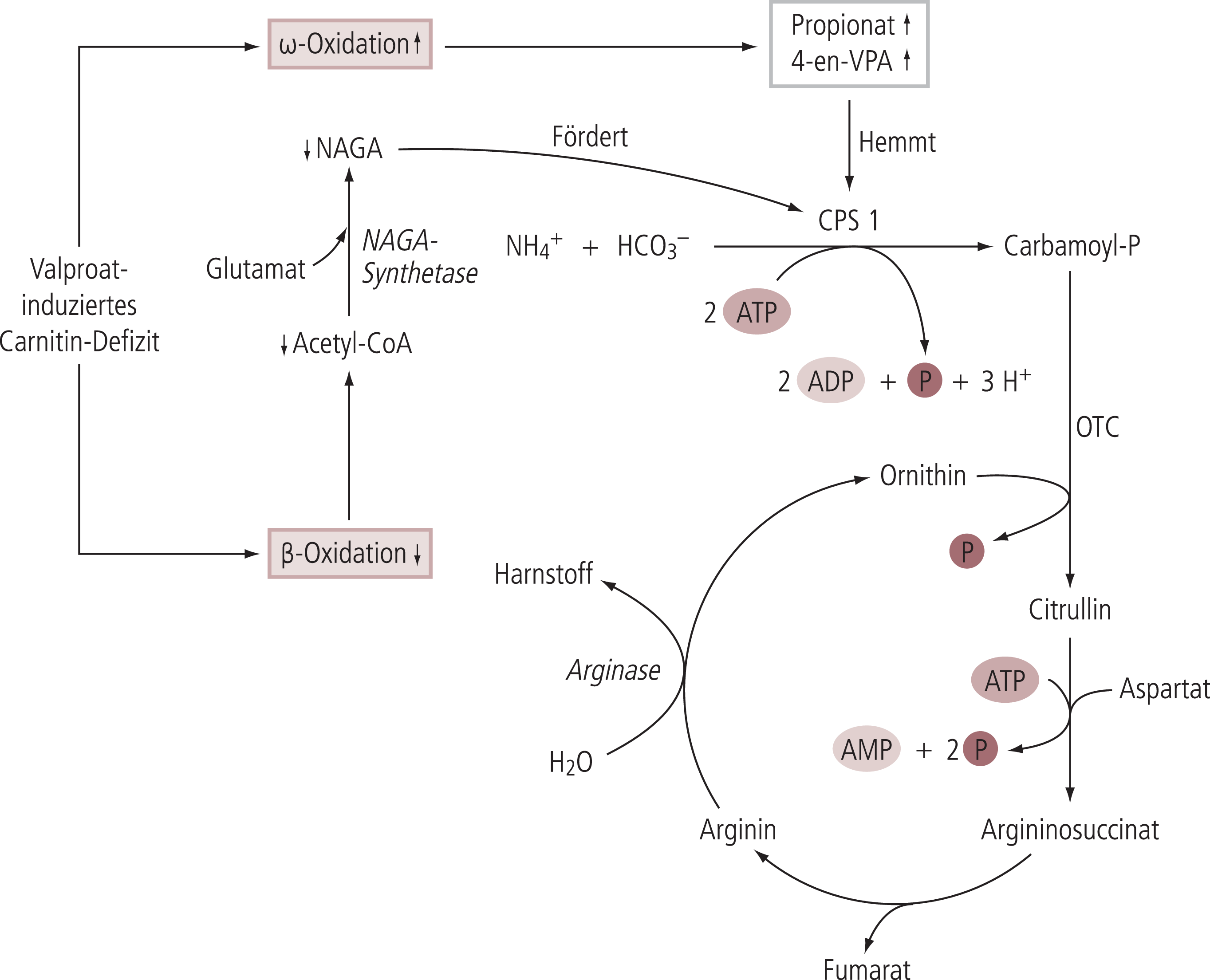
Abb. 5. Pathomechanismus der Valproinsäure-induzierten Hyperammonämie (in Anlehnung an [32]). NAGA: N-Acetylglutamat: CPS1: Carbamoylphosphat-Synthase 1; OTC: Ornithin-Carbamoyltransferase; NH4+: Ammonium; HCO3–: Hydrogencarbonat; ADP: Adenosindiphosphat; ATP: Adenosintriphosphat. ↓: Erniedrigt; ↑: Erhöht.
In Beobachtungs- und Interventionsstudien fand sich eine inverse Korrelation zwischen Gesamt-Carnitin und freiem Carnitin im Plasma und dem Ammoniakspiegel im Blut [30, 32, 56], wobei auch Studien vorliegen, die diese Befunde nicht teilen (Literatur bei [56]). Eine Carnitin-Supplementierung führte zu einer Senkung bzw. Normalisierung der Ammoniakkonzentration des Bluts [3, 7, 38].
Enzephalopathie
Die Valproinsäure-induzierte Enzephalopathie ist ein seltener, wenngleich schwerwiegender Begleiteffekt einer Valproinsäure-Behandlung [12, 47, 58].
Als Risikofaktoren für die Entwicklung einer Enzephalopathie gelten:
- Sehr junge Patienten (< 2 Jahre) [19]
- Polypharmakotherapie (Valproinsäure in Kombination mit Phenytoin, Phenobarbital und Carbamazepin) [47]
Eine mögliche Ursache ist eine Beeinträchtigung der Ammoniakentgiftung bei Valproinsäure-induziertem Carnitin-Defizit (siehe oben). Die hierdurch induzierte Hyperammonämie wird als ein Hauptauslöser der Valproinsäure-assoziierten Enzephalopathie diskutiert [32, 47, 56].
Ammoniak wirkt im Hirngewebe auf mehreren Wegen toxisch: Um die NH3-Entgiftung durch erhöhte Fixierung in Form von Glutamin zu gewährleisten, muss das Gehirn vermehrt α-Ketoglutarat und Glutamat bereitstellen. Der erhöhte Bedarf an Glutamat führt zu einem Entzug dieser Aminosäure aus dem Zytosol. Hierdurch wird die Bildung und Übertragung von Reduktionsäquivalenten (NADH) vom Zytosol in die Mitochondrien indirekt gehemmt. Somit steht weniger NADH für die Atmungskette zur Verfügung, wodurch letztlich die Energiebereitstellung durch oxidative Phosphorylierung und damit der gesamte Energiestoffwechsel der Gehirnzellen vermindert ist.
Die gesteigerte Synthese von Glutamin beeinträchtigt zudem die Funktion der Astrozyten und zieht ein zerebrales Ödem nach sich. Darüber hinaus beeinflusst Ammoniak offenbar auch direkt neuronale Membranfunktionen. So verändert es die Fluidität der synaptischen Membranen und vermindert die Dichte der postsynaptischen Serotoninrezeptoren [10].
In einigen Humanstudien konnte ein Zusammenhang zwischen der Carnitin-Versorgung, dem Ammoniakspiegel und Symptomen der Enzephalopathie aufgezeigt werden. Allerdings korreliert das Ausmaß der Symptomatik nicht notwendigerweise mit der Ammoniakkonzentration im Blut. So wurden auch bei normalen Ammoniakspiegeln Fälle einer Valproinsäure-induzierten Enzephalopathie beschrieben (Literatur bei [26, 47, 56].
Bei Personen mit Valproinsäure-induzierter Hypocarnitinämie war eine Carnitin-Supplementierung mit einer Verbesserung der neurokognitiven Funktion und des Wohlbefindens verbunden [17].
Hepatotoxizität
Unter der Medikation mit Valproinsäure kommt es häufig zu einer diskreten Erhöhung von Leberenzymen (Glutamat-Oxalacetat-Transaminase [GOT], Glutamat-Pyruvat-Transaminase [GPT]), die auf einen leberschädigenden Effekt hinweist, bei Erwachsenen aber für gewöhnlich ohne klinische Relevanz ist. Schwere Formen der Hepatotoxizität sind selten; ihre Häufigkeit wird auf 1:15000 geschätzt [5, 19, 26].
Coulter [14] postulierte 1984 die Carnitin-Hypothese der Valproinsäure-induzierten Hepatotoxizität. Ein Carnitin-Defizit, so die Vorstellung, soll die Beta-Oxidation von Valproat inhibieren, wodurch dieses vermehrt über Omega-Oxidation abgebaut wird. Im Zuge der Omega-Oxidation bilden sich Abbauprodukte, unter anderem 4-en-Valproat, die zusammen mit einem Abfall des freien CoA für die hepatotoxischen Effekte verantwortlich gemacht werden [5, 32]. Die Bildung von 4-en-Valproat-Metaboliten führt zur Glutathion-Depletion, wodurch es zu oxidativen Schäden der Leberzellen kommt [52].
Im Tiermodell konnte gezeigt werden, dass die Gabe von Carnitin einer Valproinsäure-induzierten hepatischen Steatosis und Nekrose vorbeugt [48].
In Humanstudien korreliert die Carnitin-Plasmakonzentration (Gesamt- und freies Carnitin) invers mit der Konzentration der Leberenzyme [27], und Carnitin-Supplementierung führt zur Normalisierung der Leberenzyme [19]. Die Prognose von Personen mit Valproinsäure-induzierter Leberschädigung konnte durch Carnitin-Gabe (primär i.v. appliziert) verbessert werden [6, 18, 43].
Fazit und Empfehlungen für die Praxis
Pharmaka-Nährstoff-Interaktionen sind in den letzten Jahren vermehrt ins Interesse der biomedizinischen Forschung gerückt. So existiert eine Vielzahl an Arzneistoffen, die über eine Veränderung der Distribution und Metabolisierung die Versorgung mit Mikronährstoffen beeinträchtigen. Ein in diesem Kontext relevantes Pharmakon ist Valproinsäure. Das Antikonvulsivum interferiert in multipler Weise mit der Carnitin-Homöostase, so dass eine Valproinsäure-Behandlung häufig mit einer Hypocarnitinämie assoziiert ist. Allerdings ist dieser Befund bei ansonsten gesunden Erwachsenen meist ohne klinische Relevanz. Offizielle Fachorganisationen haben sich deshalb bislang nicht für eine generelle laborchemische Überwachung der Carnitin-Versorgung (siehe Infokasten 2) beziehungsweise für eine prophylaktische Carnitin-Supplementierung unter der Behandlung mit Valproinsäure ausgesprochen.
Infokasten 2: Diagnostik des Carnitin-Mangels
Für die Bestimmung des Carnitin-Status existiert eine Reihe von Parametern. Neben der im Text erwähnten biostatistisch-relativen Definition des Carnitin-Mangels existieren auch laborchemisch ausgewiesene Cut-off-Werte. Sie ermöglichen die Diagnostik einer Hypocarnitinämie, unabhängig von einer Kontrollgruppe.
Laborchemisch ist ein Carnitin-Defizit gegeben, wenn [19]:
- die Plasmakonzentration an freiem Carnitin <20 µmol/l beträgt oder
- der Quotient aus Acyl-Carnitin und freiem Carnitin ≥ 0,4 ist.
Cave: Niedrige Plasma-Carnitin-Spiegel deuten zwar auf eine Reduktion der Carnitin-Konzentration in den Geweben hin; normale Plasmaspiegel implizieren aber nicht unbedingt, dass die Versorgung der Gewebe adäquat ist [19].
Im Gegensatz hierzu identifizierte die Paediatric Neurology Consensus Conference „L-Carnitine Supplementation in Childhood Epilepsy: Current Perspectives“ eine Reihe von Risikogruppen, die während der Valproinsäure-Behandlung ergänzend Carnitin (50–100 mg/kg KG/Tag; Maximaldosis: etwa 3g/Tag, verteilt auf 3–4 Einzeldosen) erhalten sollten [19, 32]. Hierzu zählen [19]:
- Patienten mit Valproinsäure-induzierter Hyperammonämie (mit klinischer Symptomatik)
- Sehr junge Patienten (<2 Jahre) unter Valproinsäure-haltiger Polypharmakotherapie und komplexer neurologischer Erkrankung
- Patienten, die im Zuge der ketogenen Diät eine Hypocarnitinämie entwickeln.
- Patienten unter Valproinsäure-Behandlung, die ein erhöhtes Risiko für eine toxische Schädigung der Leber aufweisen (u.a. Polypharmakotherapie, Leberfunktionsstörungen in der Vorgeschichte, reduzierter Ernährungsstatus, sonstige chronische Erkrankungen)
- Dialysepflichtige Patienten
Darüber hinaus ist eine individualisierte Carnitin-Supplementierung bei jenen Personen in Erwägung zu ziehen, die im Zuge der Valproinsäure-Behandlung eine Hypocarnitinämie mit Zeichen eines sekundären Carnitin-Defizits (Muskelschwäche, Müdigkeit) oder eine Hyperammonämie entwickeln 32]. Entsprechend wurde die Evidenz für die adjuvante Gabe von Carnitin unter der Behandlung mit Valproinsäure als möglicherweise nützlich (Evidenzgrad C) gewertet [2].
Anzumerken ist, dass Carnitin-Supplemente für gewöhnlich sehr gut toleriert werden. Nur vereinzelt beziehungsweise bei höheren (Einmal-)Dosen treten unerwünschte Begleiteffekte wie gastrointestinale Beschwerden (Diarrhö, Übelkeit) und fischartiger Körpergeruch (durch Trimethylamin) auf.
Interessenkonflikte
Der Autor erklärt, dass keine Interessenkonflikte vorliegen.
Literatur
1. Aires CC, Ijlst L, Stet F, Prip-Buus C, et al. Inhibition of hepatic carnitine palmitoyl-transferase I (CPT IA) by valproyl-CoA as a possible mechanism of valproate-induced steatosis. Biochem Pharmacol 2010;795:792–9.
2. AACE Nutrition guidelines Task Force. American Association of Clinical Endocrinologists Medical Guidelines for the clinical use of dietary supplements and nutriceuticals. Endocr Pract 2003;9:417–70.
3. Altunbaşak S, Baytok V, Tasouji M, Hergüner O, Burgut R, Kayrin L. Asymptomatic hyperammonemia in children treated with valproic acid. J Child Neurol 1997;12:461–3.
4. Arnold G, Einhäupl KM. Valproinsäure in der prophylaktischen Behandlung der Migräne. Nervenarzt 1998;6910:913–8.
5. Björnsson E. Hepatotoxicity associated with antiepileptic drugs. Acta Neurol Scand 2008;118:281–90.
6. Bohan TP, Helton E, McDonald I, König S, et al. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology 2001;56:1405–9.
7. Böhles H, Sewell AC, Wenzel D. The effect of carnitine supplementation in valproate-induced hyperammonaemia. Acta Paediatr 1996;85:446–9.
8. Bourgeois BFD. Klinische Pharmakologie der im Handel befindlichen Antiepileptika. In: Fröscher W, Vassella F, Hufnagel A. Die Epilepsien. Grundlagen, Klinik, Behandlung. 3. Aufl. Stuttgart: Schattauer, 2004:546–63.
9. Breum L, Astrup A, Gram L, Andersen T, et al. Metabolic changes during treatment with valproate in humans: implication for untoward weight gain. Metabolism 1992;41:666–70.
10. Brusilow SW. Hyperammonemic encephalopathy. Medicine (Baltimore) 2002;81: 240–9.
11. Camiña MF, Rozas I, Castro-Gago M, Paz JM, et al. Alteration of renal carnitine metabolism by anticonvulsant treatment. Neurology 1991;41:1444–8.
12. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry 2007;164:1020–7.
13. Cheung E, Wong V, Fung CW. Topiramate-valproate-induced hyperammonemic encephalopathy syndrome: case report. J Child Neurol 2005;20:157–60.
14. Coulter DL. Carnitine deficiency: a possible mechanism for valproate hepatotoxicity. Lancet 1984;1:689.
15. Crentsil V. Mechanistic contribution of carnitine deficiency to geriatric frailty. Ageing Res Rev 2010;9:265–8.
16. Cuturic M, Abramson RK, Moran RR, Hardin JW, Hall AV. Clinical correlates of low serum carnitine levels in hospitalized psychiatric patients. World J Biol Psychiatry 2011;12:73–9.
17. Cuturic M, Abramson RK, Moran RR, Hardin JW. Clinical outcomes and low-dose levocarnitine supplementation in psychiatric inpatients with documented hypocarnitinemia: a retrospective chart review. J Psychiatr Pract 2010;16:5–14.
18. DeVivo DC. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology 2002;58:507–8.
19. De Vivo DC, Bohan TP, Coulter DL, Dreifuss FE, et al. L-carnitine supplementation in childhood epilepsy: current perspectives. Epilepsia 1998;39:1216–25.
20. Demarquoy J, Georges B, Rigault C, Royer MC, et al. Radioisotopic determination of L-carnitine content in foods commonly eaten in Western countries. Food Chem 2004;86:137–42.
21. Demir E, Aysun S. Weight gain associated with valproate in childhood. Pediatr Neurol 2000;22:361–4.
22. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol 2009;32:350–2.
23. Diao L, Polli JE. Synthesis and in vitro characterization of drug conjugates of l-carnitine as potential prodrugs that target human Octn2. J Pharm Sci 2011;100:3802–16.
24. Elmslie JL, Porter RJ, Joyce PR, Hunt PJ, Mann JI. Carnitine does not improve weight loss outcomes in valproate-treated bipolar patients consuming an energy-restricted, low-fat diet. Bipolar Disord 2006;8:503–7.
25. Farkas V, Bock I, Cseko J, Sandor A. Inhibition of carnitine biosynthesis by valproic acid in rats – the biochemical mechanism of inhibition. Biochem Pharmacol 1996;52:1429–33.
26. Gerstner T, Bell N, König S. Oral valproic acid for epilepsy – long-term experience in therapy and side effects. Expert Opin Pharmacother 2008;9:285–92.
27. Goto S, Seo T, Hagiwara T, Ueda K, et al. Potential relationships between transaminase abnormality and valproic acid clearance or serum carnitine concentrations in Japanese epileptic patients. J Pharm Pharmacol 2008;60:267–72.
28. Griesheim C. Der Einfluss von l-Carnitin auf die Fettoxidation und die gesundheitsbezogene Lebensqualität von leicht übergewichtigen Senioren. Inauguraldissertation, Medizinische Fakultät der Universität Rostock, Rostock 2007 (Vefügbar unter http://rosdok.uni-rostock.de/file/rosdok_derivate_000000003429/Dissertation-Griesheim-2008.pdf; abgerufen am 26.04.2011).
29. Hahn A, Ströhle A, Wolters M. Ernährung. Physiologische Grundlagen, Prävention, Therapie. 2. Aufl. Stuttgart: Wissenschaftliche Verlagsgesellschaft, 2006.
30. Hamed SA, Abdella MM. The risk of asymptomatic hyperammonemia in children with idiopathic epilepsy treated with valproate: relationship to blood carnitine status. Epilepsy Res 2009;86:32–41.
31. Karlic H, Lohninger A, Laschan C, Lapin A, et al. Downregulation of carnitine acyltransferases and organic cation transporter OCTN2 in mononuclear cells in healthy elderly and patients with myelodysplastic syndromes. J Mol Med 2003;81:435–42.
32. Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila) 2009;47:101–11.
33. Lombard KA, Olson AL, Nelson SE, Rebouche CJ. Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am J Clin Nutr 1989;50:301–6.
34. Longin E, Teich M, Koelfen W, König S. Topiramate enhances the risk of valproate-associated side effects in three children. Epilepsia 2002;43:451–4. Erratum in: Epilepsia 2002;43:1110.
35. Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 2002;16:669–94.
36. Melegh B, Pap M, Morava E, Molnar D, et al. Carnitine-dependent changes of metabolic fuel consumption during long-term treatment with valproic acid. J Pediatr 1994;125:317–21.
37. Ohashi R, Tamai I, Inano A, Katsura M, et al. Studies on functional sites of organic cation/carnitine transporter OCTN2 (SLC22A5) using a Ser467Cys mutant protein. J Pharmacol Exp Ther 2002;302:1286–94.
38. Ohtani Y, Endo F, Matsuda I. Carnitine deficiency and hyperammonemia associated with valproic acid therapy. J Pediatr 1982;101: 782–5.
39. Raja M, Azzoni A. Valproate-induced hyperammonaemia. J Clin Psychopharmacol 2002;22:631–3.
40. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother 2000;34:630–8.
41. Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann N Y Acad Sci 2004;1033:30–41.
42. Rebouche CJ, Seim H. Carnitine metabolism and its regulation in microorganisms and mammals. Annu Rev Nutr 1998;18: 39–61.
43. Russell S. Carnitine as an antidote for acute valproate toxicity in children. Curr Opin Pediatr 2007;19:206–10.
44. Rytting E, Audus KL. Novel organic cation transporter 2-mediated carnitine uptake in placental choriocarcinoma (BeWo) cells. J Pharmacol Exp Ther 2005;312:192–8.
45. Scheck A. L-Carnitin: Sinn und Unsinn der Substitution einer körpereigenen Substanz. Teil 1: Zur Physiologie und sinnvollen Substitution. Ernahr-Umsch 1994;41:9–15.
46. Schmidt D, Elger CE. Praktische Epilepsiebehandlung. 3. Aufl. Stuttgart: Thieme, 2005.
47. Segura-Bruna N, Rodriguez-Campello A, Puente V, Roquer J. Valproate-induced hyperammonemic encephalopathy. Acta Neurol Scand 2006;114:1–7.
48. Shakoor KAK. Valproic acid induced hepatoxicity and protective role of carnitine. Pakistan J Pathol 133–4, 1997. Zitiert nach: Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila) 2009;47:101–11.
49. Shapira Y, Gutman A. Muscle carnitine deficiency in patients using valproic acid. J Pediatr 1991;118:646–9.
50. Silva MF, Aires CC, Luis PB, Ruiter JP, et al. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J Inherit Metab Dis 2008 Apr 4 [Epub ahead of print].
51. Stadler DD, Bale JF Jr, Chenard CA, Rebouche CJ. Effect of long-term valproic acid administration on the efficiency of carnitine reabsorption in humans.Metabolism 1999;48:74–9.
52. Tang W, Borel AG, Abbott FS. Conjugation of glutathione with a toxic metabolite of valproic acid, (E)-2-propyl-2,4-pentadienoic acid, catalyzed by rat hepatic glutathione-S-transferases. Drug Metab Dispos 1996;24:436–46.
53. Tein I. Carnitine transport: pathophysiology and metabolism of known molecular defects. J Inherit Metab Dis 2003;26:147–69.
54. Vaz FM, Wanders RJ. Carnitine biosynthesis in mammals. Biochem J 2002;361:417–29.
55. Verrotti A, la Torre R, Trotta D, Mohn A, Chiarelli F. Valproate-induced insulin resistance and obesity in children. Horm Res 2009;71:125–31.
56. Verrotti A, Trotta D, Morgese G, Chiarelli F. Valproate-induced hyperammonemic encephalopathy. Metab Brain Dis 2002;17:367–73.
57. Vivekanandan S, Nayak SD. Valproate-induced hyperammonemic encephalopathy enhanced by topiramate and phenobarbitone: a case report and an update. Ann Indian Acad Neurol 2010;13:145–7.
58. Wadzinski J, Franks R, Roane D, Bayard M. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med 2007; 20:499–502.
59. Wagner JM, Bug G, Jung M. Valproinsäure als Histon-Desacatylase-Hemmstoff. Neues Einsatzgebiet für einen altbekannten Arzneistoff. Pharm Unserer Zeit 2010;39:197–203.
Dr. Alexander Ströhle, Am Landwehrgraben 8, 30519 Hannover, E-Mail: stroehle@nutrition.uni-hannover.de
Valproate induced carnitine deficiency – pathobiochemistry and clinical consequences
Due to their clinical relevance drug nutrient interactions have gained increasing attention in biomedical research during recent years. Thus, there is a great number of drugs which adversely affect nutrient status by changing distribution and/or metabolism. One relevant example is valproate. The anticonvulsant interacts in several ways with the carnitine homeostasis, hence hypocarnitinemia is a common adverse effect of valproate treatment. Based on an overview of the carnitine metabolism, this article will critically examine the problem of the valproate associated carnitine deficiency.
Key words: Carnitine, valproic acid, hyperammonemia, encephalopathy, hepatotoxicity
Psychopharmakotherapie 2012; 19(01)