Christine Waldschmitt, Christoph Hiemke, Mainz, und Gabriel Eckermann, Kaufbeuren
Duloxetin hemmt selektiv die Wiederaufnahme von Serotonin und Noradrenalin und erhöht so die Konzentration der Neurotransmitter im synaptischen Spalt. Die Substanz wurde in Deutschland zunächst zur Behandlung der Belastungsharninkontinenz bei Frauen im August 2004 zugelassen, Anfang 2005 dann als Antidepressivum [9]. Seit 2006 hat sie auch die Zulassung für die Behandlung der schmerzhaften diabetischen Neuropathie [22]. Antidepressive Wirksamkeit wurde für eine Duloxetin-Tagesdosis von 60 bis 120 mg nachgewiesen. In Studien fand man eine signifikant verbesserte depressive Symptomatik bereits nach zwei Wochen im Vergleich mit Plazebo [3]. Die Untersuchung rückfallpräventiver Effekte zeigte auch bei einer Langzeitbehandlung eine gute Wirksamkeit [3–5, 19]. Von den Patienten, die 12 Wochen lang akut behandelt worden waren, erlitten in den nachfolgenden sechs Monaten unter fortgeführter Gabe von Duloxetin 17% einen Rückfall gegenüber 29% unter Plazebo. Duloxetin besitzt vergleichbare Wirksamkeit und Verträglichkeit wie selektive Serotonin-Wiederaufnahmehemmer (SSRI) oder Venlafaxin, das ebenso wie Duloxetin die Rückaufnahme von Serotonin und Noradrenalin hemmt. Im Unterschied zu Venlafaxin, welches im niedrigen Dosisbereich bis zu 150 mg/d so gut wie ausschließlich als SSRI und erst in Dosen über 150 mg „dual“ wirkt, also auch die Noradrenalin-Wiederaufnahme hemmt [8], entfaltet Duloxetin bereits bei 60 mg eine duale Wirkung [2, 12].
Unsicherheit besteht bei Duloxetin bezüglich seines pharmakokinetischen Interaktionspotenzials im Vergleich zu anderen neuen Antidepressiva. Die SSRI Paroxetin und Fluoxetin und dessen aktiver Metabolit Norfluoxetin sind potente Inhibitoren von CYP2D6, Fluvoxamin von CYP1A2, CYP2C9 und CYP2C19 [10]. Bei Kombination dieser SSRI mit Wirkstoffen, die Substrate der vorgenannten Enzyme sind, kann es wegen verminderter Clearance ohne Dosisanpassung zu einem ausgeprägten Anstieg der Plasmakonzentrationen kommen und dadurch zu schwerwiegenden unerwünschten Arzneimittelwirkungen. Gut dokumentiert ist dies für Fluoxetin und trizyklische Antidepressiva [15, 23]. Für Duloxetin wird in der Fachinformation [6] mitgeteilt, dass eine Kombination mit Inhibitoren von CYP1A2 wie Fluvoxamin oder Ciprofloxacin kontraindiziert ist, und es wird darauf verwiesen, dass es ein Inhibitor von CYP2D6 ist. Ob die pharmakokinetischen Interaktionen von und mit Duloxetin klinisch relevant sind und wie die pharmakokinetischen Interaktionen von Duloxetin im Vergleich mit anderen Antidepressiva einzuschätzen sind, wird in dieser Arbeit dargestellt.
Pharmakokinetik
Duloxetin wird nach oraler Verabreichung gut absorbiert. Die absolute Bioverfügbarkeit liegt zwischen 32% und 80%. Maximale Plasmakonzentrationen werden etwa 6 Stunden nach Einnahme der magensaftresistenten Kapseln erreicht. Die Biotransformation erfolgt in der Leber durch die Enzyme CYP1A2 und CYP2D6. Die beiden Hauptmetaboliten sind pharmakologisch inaktiv. Die Eliminationshalbwertszeit von Duloxetin beträgt zwischen 8 und 17 Stunden (im Mittel etwa 12 Stunden), nach drei Tagen ist das Steady State erreicht. Etwa 70% der Duloxetin-Metaboliten werden über den Urin, etwa 20% mit den Fäzes ausgeschieden.
CYP-Substrateigenschaften von Duloxetin – Interaktionen
Nach In-vitro-Befunden wird Duloxetin durch die Enzyme CYP1A2 und CYP2D6 metabolisiert [13, 21]. In einer neueren Arbeit [14] wurde nachgewiesen, dass CYP1A2 das am Abbau von Duloxetin bevorzugt beteiligte Enzym ist. Kombinationen mit CYP1A2-Inhibitoren wie Fluvoxamin, Ciprofloxacin oder Enoxacin sind aufgrund des zu erwartenden Anstiegs der Plasmakonzentrationen von Duloxetin kontraindiziert [6]. Fluvoxamin ist ein sehr potenter Inhibitor von CYP1A2 [9]. Es senkt in einer Dosierung von 100 mg/d die apparente Plasmaclearance von Duloxetin um 77% und steigert die Verfügbarkeit im Mittel um das Sechsfache. Die Eliminationshalbwertszeit wird durch Fluvoxamin bei einer Tagesdosis von 100 mg von 10,5 auf 26,8 Stunden verlängert [14]. In der pharmakokinetischen Untersuchung an gesunden Probanden wurden für die Kombination von Duloxetin mit Fluvoxamin keine Hinweise auf vermehrte Nebenwirkungen festgestellt [14].
Umgekehrt senkt Rauchen die Plasmakonzentrationen von Duloxetin. Aromatische Kohlenwasserstoffe im Zigarettenrauch induzieren CYP1A2. Pharmakokinetische Studien haben gezeigt, dass die Duloxetin-Plasmakonzentrationen bei Rauchern um fast 50% niedriger liegen als bei Nichtrauchern [6].
CYP2D6 scheint im Vergleich mit CYP1A2 in vivo für den Abbau von Duloxetin nach neuesten Erkenntnissen von nachgeordneter Bedeutung zu sein [14, 16]. Dies entspricht auch Befunden, bei denen die Plasmakonzentrationen von Duloxetin unter Kombination mit CYP2D6-Inhibitoren gemessen wurden [21]. Beim Vergleich von Behandlungen mit und ohne CYP2D6-Inhibitoren wurden in Kombination mit CYP2D6-Inhibitoren keine signifikant höheren Plasmaspiegel gefunden [25].
Aus den in In-vivo-Studien erkennbaren Substrateigenschaften von Duloxetin lässt sich ablesen, dass eine Behandlung mit Duloxetin in Kombination mit CYP1A2- Inhibitoren kritisch sein könnte, nicht jedoch eine Kombination mit CYP2D6-Inhibitoren. Kombinationen mit CYP1A2- Inhibitoren werden daher vom Hersteller als Kontraindikation geführt [6]. Durch begleitendes therapeutisches Drug-Monitoring (TDM), bei dem im Steady State Talspiegel im Plasma gemessen werden [11], kann das Risiko der Einstellung toxischer Wirkspiegel minimiert werden, indem die Dosis angepasst wird. Unter therapeutischen Dosen von 60 bis 120 mg sind Duloxetin-Plasmakonzentrationen zwischen 60 und 120 ng/ml zu erwarten [25]. In einer weitergehenden Untersuchung wurde gefunden, dass unter diesen Plasmakonzentrationen die Ansprechrate auf Duloxetin am höchsten war [25]. Dies spricht dafür, dass TDM insbesondere für Patienten sinnvoll ist, die auf Duloxetin nicht ausreichend ansprechen. Auch bei Rauchern ist eine Dosisanpassung unter TDM-Kontrolle angezeigt, um das Risiko der Unterdosierung mit Duloxetin zu reduzieren. Als Erhaltungsdosis scheint bei Nichtrauchern 60 mg einmal täglich auszureichen, bei Rauchern werden in der Regel 120 mg/d benötigt. Bei Rauchern, die sich entschließen, ihren Zigarettenkonsum zu reduzieren oder aufzugeben, ist es wegen der Deinduktion von CYP1A2 ebenfalls sinnvoll, den Plasmaspiegel zu kontrollieren.
CYP-Hemmeigenschaften von Duloxetin – Interaktionen
Wie beschrieben, ist Duloxetin bevorzugtes Substrat von CYP1A2, während CYP2D6 in nur geringem Umfang beteiligt ist. Bezüglich des Hemmpotenzials von Duloxetin verhält es sich umgekehrt. Bei Einsatz therapeutischer Dosen wird CYP1A2 durch Duloxetin nicht gehemmt [14], wohl aber CYP2D6 [16, 21]. In einer Studie, die 2007 von Preskorn und Mitarbeitern publiziert wurde [16], wurde ein erster direkter Vergleich der Hemmeffekte von Duloxetin, Escitalopram und Sertralin auf die funktionelle Aktivität von CYP2D6 vorgenommen. Untersucht wurde die veränderte Pharmakokinetik von Metoprolol. Metoprolol ist ausschließlich Substrat von CYP2D6. Die pharmakokinetischen Parameter von Metoprolol wurden vor (Tag –7) und nach einer 17-tägigen (Tag 17) Kombination mit 20 mg/d Escitalopram, 60 mg/d Duloxetin oder 100 mg/d Sertralin bei gesunden Probanden gemessen. Alle drei Antidepressiva hatten einen statistisch signifikanten (p<0,05) Einfluss auf die Pharmakokinetik von Metoprolol. Im Vergleich mit Sertralin und Escitalopram hemmte Duloxetin die Metoprolol-Elimination stärker (Tab. 1). Gemessen am Anstieg der Plasmakonzentrations-Zeit-Kurve ergab sich für Metoprolol folgende Rangfolge des CYP2D6-hemmenden Potenzials: Duloxetin (+49%) > Escitalopram (+29%) > Sertralin (+28%). Für den Anstieg der Spitzenplasmaspiegel von Metoprolol ergab sich nach Zugabe der drei Antidepressiva die gleiche Reihenfolge: Duloxetin (+40%) > Escitalopram (+30%) > Sertralin (+26%).
Tab. 1. Pharmakokinetische Parameter (Cmax: maximale Plasmakonzentration; AUC: Fläche unter der Plasmakonzentrations-Zeit-Kurve; t1/2: Eliminationshalbwertszeit; Cl: Clearance) von Metoprolol im Steady State vor (Tag –7) und nach (Tag 17) zusätzlicher Gabe von Escitalopram, Sertralin oder Duloxetin [Daten nach 16]
|
Pharmakokinetik |
Escitalopram (n=15) |
Sertralin (n=16) |
Duloxetin (n=16) |
|||
|
Tag –7 |
Tag 17 |
Tag –7 |
Tag 17 |
Tag –7 |
Tag 17 |
|
|
Cmax [ng/ml] |
168±33 |
240±33* |
175±12 |
235±15* |
147±91 |
247±82* |
|
AUC [h·ng/ml] |
1217±293 |
1721±339* |
1120±136 |
1559±150* |
831±927 |
1616±830* |
|
t½ [h] |
3,82±0,22 |
3,86±0,27* |
3,26±0,23 |
3,96±0,15* |
3,03±1,23 |
4,22±1,23* |
|
Cl [ml/min] |
2577±293 |
1299±258* |
1378±139 |
968±11* |
2169±1616 |
743±309* |
* p<0,01, Tag –7 = keine Antidepressiva-Behandlung, Tag 17 = Antidepressiva-Behandlung
Bei TDM werden in der Regel Minimalspiegel (Cmin) gemessen und bewertet. Berechnet man aus den Ergebnissen von Preskorn und Mitarbeitern [16] die Talspiegel von Metoprolol, dann ergibt sich durch Escitalopram keine, durch Sertralin eine 1,7fache und durch Duloxetin eine 2,2fache Erhöhung der Metoprolol-Spiegel (Abb. 1). Die potenten CYP2D6-Inhibitoren Fluoxetin und Paroxetin steigern die Blutspiegel wesentlich ausgeprägter, nämlich um das 4fache beziehungsweise 6fache [17]. Bezogen auf Metoprolol als In-vivo-Substrat von CYP2D6 stellt sich Duloxetin als moderater Inhibitor von CYP2D6 dar, schwächer als Fluoxetin oder Paroxetin, aber stärker als Sertralin oder Escitalopram.
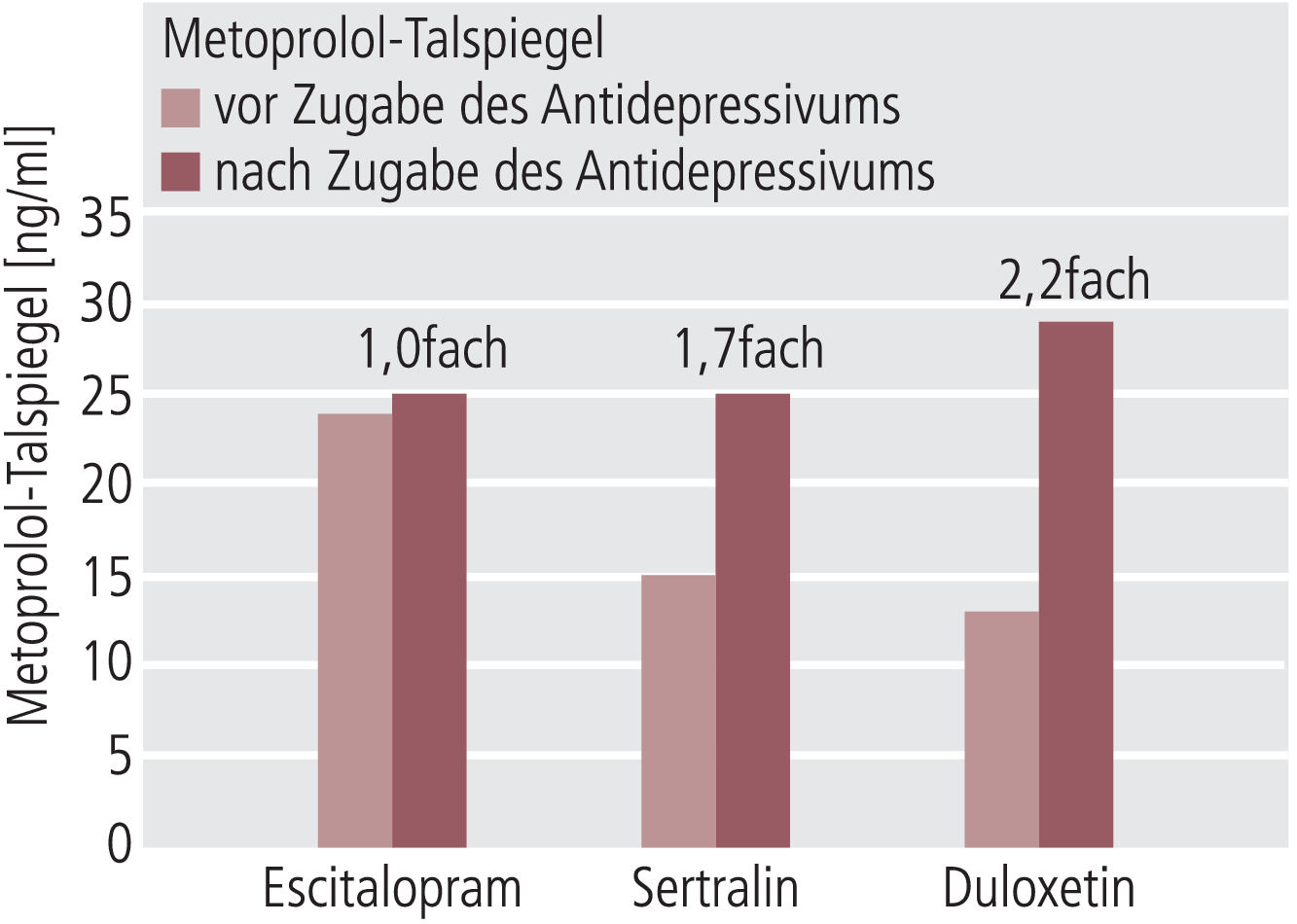
Abb. 1. Veränderung der Talspiegel im Steady State von Metoprolol (Substrat von CYP2D6) vor und nach Zugabe von Escitalopram, Sertralin oder Duloxetin (berechnet aus Daten von Preskorn et al. [16]) zur Darstellung der unterschiedlichen In-vivo-Hemmpotenziale der drei Antidepressiva auf CYP2D6
In einer Untersuchung von Skinner et al. [21], die ebenfalls an gesunden Probanden durchgeführt wurde, wurde Desipramin als CYP2D6-Substrat benutzt. Desipramin wird im Wesentlichen durch CYP2D6 zu 2-Hydroxydesipramin hydroxyliert. Im Unterschied zu Metoprolol ist Desipramin kein Inhibitor von CYP2D6. Es entfällt daher ein bei Metoprolol anzunehmender Hemmeffekt (Abb. 2). Bei gleichzeitiger Gabe von täglich 120 mg Duloxetin und Desipramin 50 mg/d änderten sich sämtliche pharmakokinetischen Parameter von Desipramin. Die Desipramin-Minimalspiegel stiegen um das 3,3fache (Abb. 3). Nebenwirkungen traten bei den gesunden Probanden unter der Kombination beider Antidepressiva nicht häufiger auf als unter Desipramin-Monotherapie, und klinisch relevante Parameter wie Herzfrequenz oder Blutdruck änderten sich nicht unter der Kombination.
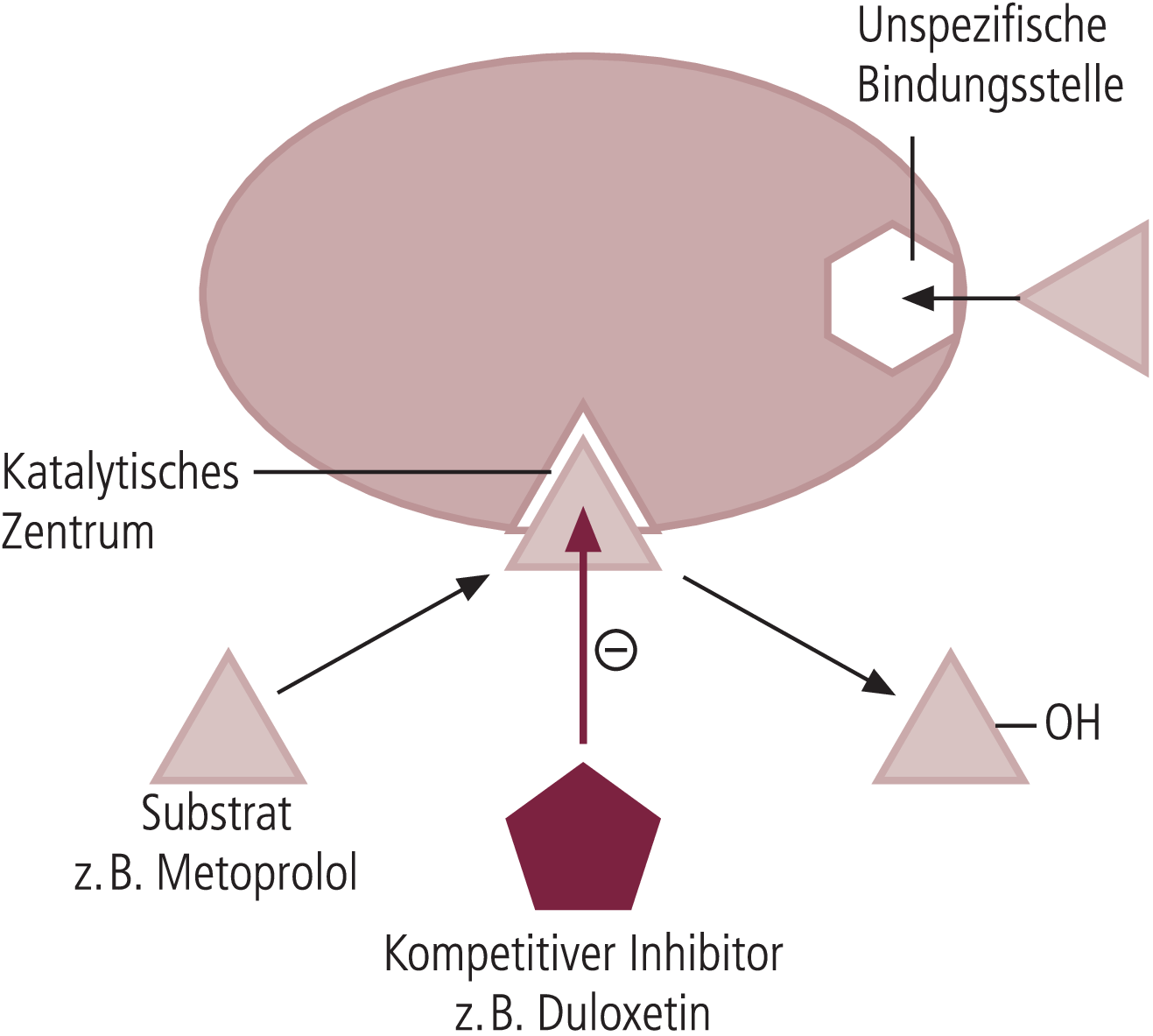
Abb. 2. Schematische Darstellung des durch Cytochrom P450-2D6 katalysierten Abbaus von Metoprolol, das sowohl Substrat als auch Inhibitor von CYP2D6 ist.
Bei hohen Substratkonzentrationen besetzt Metoprolol eine unspezifische Bindungsstelle. Dadurch kommt es zu einer Hemmung des Enzyms (Substrathemmung durch allosterischen Effekt). Duloxetin ist Substrat von CYP2D6 und hemmt dadurch die Hydroxylierung von Metoprolol wahrscheinlich kompetitiv.
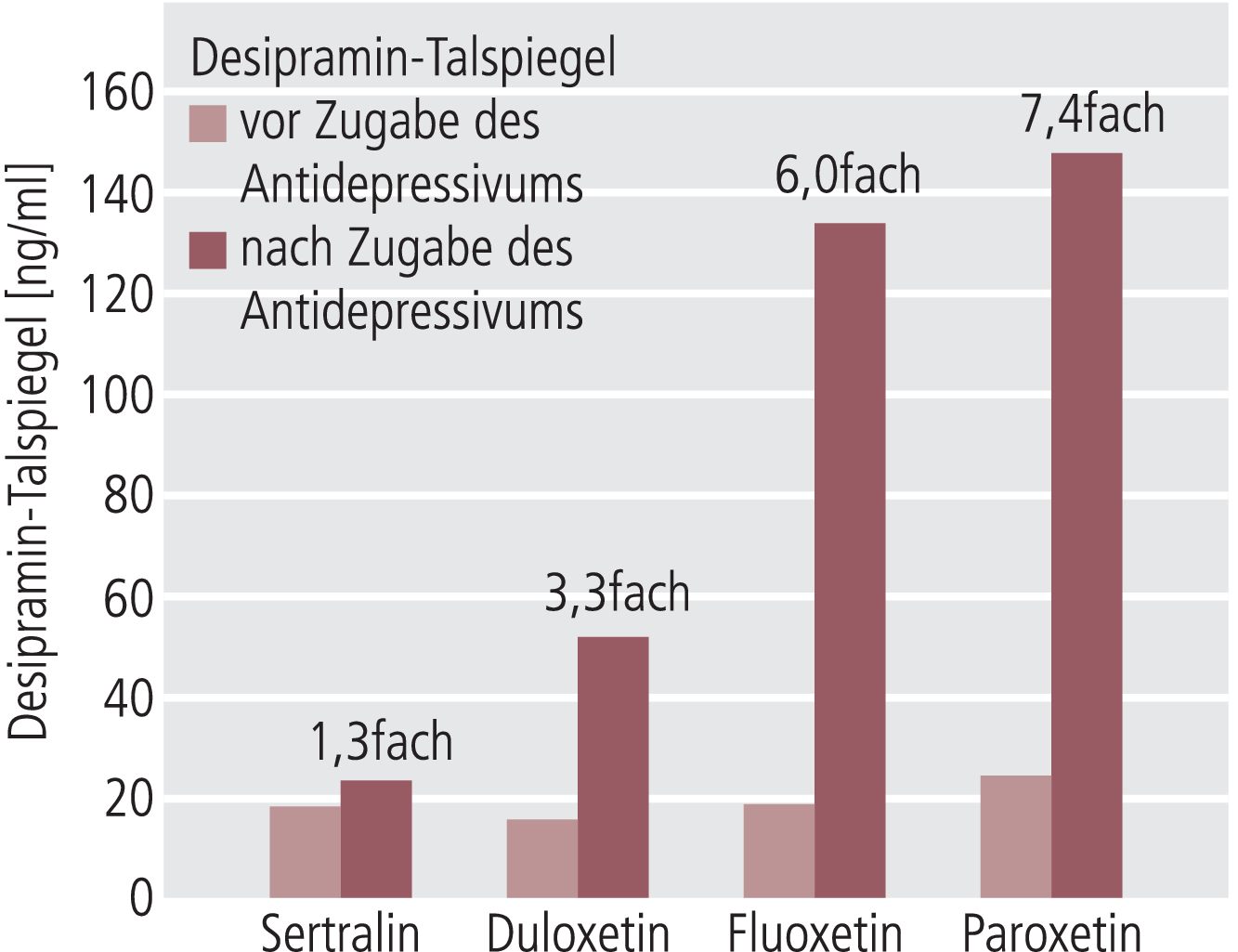
Abb. 3. Veränderung der Talspiegel (Co) im Steady State von Desipramin (Substrat von CYP2D6) vor und nach Zugabe von Sertralin, Duloxetin, Fluoxetin oder Paroxetin (berechnet aus Daten von Preskorn et al. [16]) zur Darstellung der unterschiedlichen In-vivo-Hemmpotenziale der vier Antidepressiva auf CYP2D6) [mod. nach 1, 18, 21]
Um das CYP2D6-inhibitorische Potenzial von Sertralin und Fluoxetin zu vergleichen, benutzte Preskorn in einer Studie ebenfalls den Einfluss auf die Pharmakokinetik von Desipramin [18]. Nach 21 Tagen Behandlung mit Sertralin 50 mg/d stieg der maximale Plasmaspiegel von Desipramin um nur 31%, während nach Komedikation mit Fluoxetin 20 mg/d ein 4facher Anstieg zu beobachten war (Tab. 2).
Tab. 2. Pharmakokinetische Parameter (Cmax: maximale Plasmakonzentration; AUC: Fläche unter der Plasmakonzentrations-Zeit-Kurve; t1/2: Eliminationshalbwertszeit) von Desipramin im Steady State vor (Tag 7) und nach (Tag 28) Gabe von Sertralin oder Fluoxetin [Daten nach 18]
|
Pharmakokinetik |
Sertralin (n=9) |
Fluoxetin (n=9) |
||
|
Tag 7 |
Tag 28 |
Tag 7 |
Tag 28 |
|
|
Cmax [ng/ml] |
24,5±2,6 |
32,8±4,5* |
51,2±6,4 |
193,3±19,7* |
|
AUC [h·ng/ml] |
387±47 |
486±74 |
881±144 |
3890±384* |
|
t½ [h] |
7,3±0,3 |
5,8±0,6 |
7,1±0,5 |
5,2±0,8 |
* p<0,01, Tag 7 = nur Desipramin-Behandlung, Tag 28 = 21 Tage Behandlung mit Sertralin bzw. Fluoxetin
Skinner untersuchte in einer weiteren Studie den Effekt von Paroxetin, dem bekanntermaßen potentesten Inhibitor von CYP2D6 [10], auf die Pharmakokinetik von Duloxetin. Die gleichzeitige Gabe von Duloxetin 40 mg/d bei anhaltender Paroxetin-Therapie mit einer Dosierung von 20 mg/d führte zu einer um 60% erhöhten Duloxetin-Plasmakonzentration (Tab. 3).
Tab. 3. Pharmakokinetische Parameter (Cmax: maximale Plasmakonzentration; AUC: Fläche unter der Plasmakonzentrations-Zeit-Kurve; t1/2: Eliminationshalbwertszeit; Cl: Clearance; Mittelwerte±SD) von Duloxetin unter Monotherapie und in Kombination mit dem CYP2D6-Inhibitor Paroxetin [Daten nach 21]
|
Pharmako- |
Duloxetin 40 mg/d |
|
|
kinetik |
allein (n=10) |
+ Paroxetin 20 mg/d (n=9) |
|
Cmax [ng/ml] |
35,2±8,4 |
55,7±10,0* |
|
AUC [h·ng/ml] |
497±165 |
780±204* |
|
t½ [h] |
10 |
13* |
|
Cl [ml/min] |
1472±462 |
913±260* |
* p<0,01
Trotz signifikanten Anstiegs der pharmakokinetischen Parameter von Desipramin durch Duloxetin ist das Ausmaß der Änderungen infolge der CYP2D6-Hemmung verglichen mit Paroxetin relativ gering. Abbildung 3 stellt in einer Übersicht den Einfluss der jeweiligen Komedikation auf die Talspiegel von Desipramin im Steady State dar.
Nach der Untersuchung von Skinner und Mitarbeitern [21] wird durch eine CYP2D6-Inhibition mit Paroxetin die Duloxetin-Pharmakokinetik signifikant verändert. Aber der moderate Effekt von Paroxetin auf den Duloxetin-Plasmaspiegel war mit keiner signifikanten Änderung in der Verträglichkeit verbunden.
Das neuerdings als Antidepressivum zugelassene Bupropion ist ebenfalls ein Inhibitor von CYP2D6 [20]. Der Hemmeffekt ist in vitro allerdings nur schwach ausgeprägt, wohl aber deutlich in vivo. Der Abbau von Dextromethorphan wird durch Bupropion deutlich verlangsamt. Wahrscheinlich geht der Hemmeffekt primär nicht von Bupropion selbst, sondern von Metaboliten aus [20]. Es ist allerdings unklar, ob das pharmakokinetische Interaktionspotenzial von Bupropion riskanter als das von Duloxetin einzuschätzen ist. Da es für Bupropion Fallberichte über Intoxikationen gibt (siehe www.psiac.de/), wurde der Hemmeffekt in Tabelle 4 so eingeordnet wie der von Fluoxetin oder Paroxetin. Nach Einschätzung von Spina und Mitarbeitern [24] wird Bupropion bezüglich seines CYP2D6-Hemmpotenzials mit Duloxetin gleichgesetzt.
Tab. 4. Hemmpotenzial von neuen Antidepressiva auf Cytochrom-P450-Enzyme bei therapeutischen Dosen der jeweiligen Antidepressiva
|
Cytochrom-P450-Isoenzym (CYP) |
||||||
|
1A2 |
2B6 |
2C9 |
2C19 |
2D6 |
3A4 |
|
|
Bupropion |
0 |
+ |
0 |
0 |
++ |
0 |
|
Citalopram Desmethylcitalopram |
0 0 |
0 0 |
0 0 |
+ 0 |
+ + |
0 + |
|
Duloxetin |
0 |
0 |
0 |
0 |
++ |
0 |
|
Escitalopram |
0 |
0 |
0 |
+ |
+ |
0 |
|
Fluoxetin |
0 |
0 |
+ |
+ |
+++ |
+ |
|
Fluvoxamin |
+++ |
+ |
++ |
+++ |
+ |
+ |
|
Mirtazapin |
+ |
0 |
0 |
0 |
+ |
+ |
|
Paroxetin |
+ |
+ |
0 |
+ |
+++ |
+ |
|
Reboxetin |
0 |
– |
0 |
0 |
0 |
+ |
|
Sertralin Desmethylsertralin |
0 + |
0 0 |
0 + |
0 + |
+ + |
0 + |
|
Venlafaxin |
0 |
0 |
0 |
0 |
+ |
+ |
0 keine Hemmung; – keine Daten verfügbar; + geringfügige Hemmung ohne klinische Bedeutung; ++ moderate Hemmung, unter Umständen bedeutsam, Dosisanpassung empfohlen; +++ deutlicher Anstieg der Plasmaspiegel, Dosisanpassung erforderlich, wenn möglich mit Kontrolle der Plasmaspiegel [Zusammenstellung nach 1, 7, 10, 16, 18, 21, 25]
Die Ergebnisse der pharmakokinetischen Untersuchungen mit kleinen Fallzahlen lassen allerdings nur bedingt einen Rückschluss auf die klinische Relevanz der Hemmeffekte von Duloxetin bei Kombinationsbehandlungen zu. Eigene Analysen von TDM-Daten zur Abschätzung des Interaktionspotenzials von Duloxetin haben an über 100 Patienten ergeben, dass eine Kombination mit den CYP2D6-Substraten Mirtazapin, Risperidon, Amitriptylin oder Aripiprazol zu keinen signifikanten Änderungen der Plasmaspiegel führte, weder für die Muttersubstanzen noch für die Metaboliten. Alle gemessenen Konzentrationen lagen im therapeutischen Bereich und es wurden bei diesen Anforderungen keine Nebenwirkungen berichtet [25]. Trotzdem sollte man bei Kombination von Duloxetin und Substraten von CYP2D6 die Hemmwirkung von Duloxetin beachten und mögliche kritische Interaktionseffekte unbedingt bedenken, beispielsweise bei einer Kombination mit den Betablockern Metoprolol, Nebivolol oder Propranolol (Bisoprolol scheint diesbezüglich nicht problematisch) oder auch mit dem Schmerzmittel Tramadol.
Fazit
Duloxetin wird bevorzugt über CYP1A2 und nachgeordnet durch CYP2D6 abgebaut. Deshalb steigt bei Kombination von Duloxetin mit Fluvoxamin oder anderen Inhibitoren von CYP1A2 die Duloxetin-Plasmakonzentration deutlich und ist bei Rauchern niedriger als bei Nichtrauchern. Eine Kombination von Duloxetin mit CYP2D6-Inhibitoren hat nach bisheriger Datenlage keine klinisch relevanten Auswirkungen. Duloxetin hat keine eigene Hemmwirkung auf CYP1A2, ist aber ein moderater Inhibitor von CYP2D6, potenter als Sertralin, Citalopram oder Escitalopram und schwächer als Fluoxetin oder Paroxetin (Tab. 4). Bisher gibt es keinen Fallbericht, aus dem sich für Duloxetin eine klinische Relevanz pharmakokinetischer Interaktionen ableiten ließe. In Situationen jedoch, in denen mit veränderten Aktivitäten von CYP1A2 oder CYP2D6 zu rechnen ist, etwa bei Kombinationsbehandlung mit CYP1A2-Inhibitoren, bei Rauchern [6a] oder bei Kombinationsbehandlung mit CYP2D6-Substraten, gibt es gute Evidenz, die Behandlung klinisch zu überwachen, möglichst mit Kontrolle der Blutspiegel, um eine maximale Sicherheit und Wirksamkeit zu erzielen.
Literatur
1. Alderman J, Preskorn SH, Greenblatt DJ, Harrison W, et al. Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 1997;17:284–91.
2. Bymaster FP, Dreshfiled-Ahmad LJ, Threlkeld PG, Shaw JL, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology 2001;25:871–80.
3. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine, 60 mg once daily, for major depression disorder: A randomized double-blind placebo-controlled trial. J Clin Psychiatry 2002;63:308–15.
4. Detke MJ, Wiltse CG, Mallinckrodt CH. Duloxetine in the acute and long-term treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur Neuropsychopharmacol 2004;14:457–70.
5. Detke MJ, Gilaberte I, Perahia DG, Wang F, et al. Duloxetine vs. placebo in the prevention of relapse of major depression disorder. Eur Psychiatry 2004;19(Suppl 1):214s.
6. Fachinformation Cymbalta, Stand August 2007.
6a.Fric M, Pfuhlmann B, Laux G, Riederer P, et al. The influence of smoking on the serum level of duloxetine. Pharmacopsychiatry 2008;41:151–5.
7. Gillmann PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updates. Br J Pharmacol 2007;151:737–48.
8. Harvey AT, Rudolph RL, Preskorn SH. Evidence of the dual mechanism of action of venlafaxine. Arch Gen Psychiatry 2000;57: 503–9.
9. Hiemke C, Hampel C, Weigmann H. Pharmakologie von Duloxetin. Psychopharmakotherapie 2006;13:12–8.
10. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther 2000;85:11–28.
11. Hiemke C, Baumann P, Laux G, Kuss HJ, et al. Therapeutisches Drug-Monitoring in der Psychiatrie. Konsensus-Leitlinie der AGNP. Psychopharmakotherapie 2005;12: 166–82.
12. Karpa KD, Cavanaugh JE, Lakoski JM. Duloxetine pharmacology: profile of a dual monoamine modulator. CNS Drug Rev 2002;8: 361–76.
13. Lantz RJ, Gillespie TA, Rash TJ, Kuo F, et al. Metabolism, excretion, and pharmacokinetics of duloxetine in healthy human subjects. Drug Metab Dispos 2003;31:1142–50.
14. Lobo ED, Bergstrom RF, Reddy S, Quinlan T, et al. In vitro and in vivo evaluations of cytochrome P450-1A2 interactions with duloxetine. Clin Pharmacokinet 2008;47:191–202.
15. Preskorn SH, Shah R, Neff M, Golbeck AL, et al. The potential for clinically significant drug-drug interactions involving the CYP2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract 2007;13: 5–12.
16. Preskorn SH, Greenblatt DJ, Flockhart D, Luo Y, et al. Comparison of duloxetine, escitalopram, and sertraline effects on cytochrome P450-2D6 function in healthy volunteers. J Clin Psychopharmacol 2007;27:28–34.
17. Preskorn SH. Reproducibility of the in vivo effect of the selective serotonin reuptake inhibitors on the in vivo function of cytochrome P450-2D6: An update (part I). J Psychiatr Pract 2003;2:150–8.
18. Preskorn SH, Alderman J, Chung M, Harrison W, et al. Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J Clin Psychopharmacol 1994;14:90–8.
19. Raskin J, Goldstein DJ, Mallinchrodt CH, Ferguson MB. Duloxetine in the long-term treatment of major depressive disorder. J Clin Psychiatry 2003;64:1237–44.
20. Reese MJ, Wurm RM, Muir KT, Generaux GT, et al. An in vitro mechanistic study to elucidate the desipramine/bupropion clinical drug-drug interaction. Drug Metab Dispos 2008;36:1198–201.
21. Skinner MH, Kuan HY, Pan A, Sathirakul K, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P450-2D6 in healthy volunteers. Clin Pharmacol Ther 2003;73: 170–7.
22. Smith TR. Duloxetine in diabetic neuropathy. Expert Opin Pharmacother 2006;7:215–23.
23. Spina E, Avenoso A, Scordi MG, Ancione M, et al. Inhibition of risperidone metabolism by fluoxetine in patients with schizophrenia: a clinically relevant pharmakokinetic drug interaction. J Clin Psychopharmacol 2002;22: 419–23.
24. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther 2008;30:1206–27.
25. Waldschmitt C, Vogel F, Pfuhlmann B, Hiemke C. Duloxetine serum concentrations and clinical effects. Data from a therapeutic drug monitoring (TDM) survey. Int J Neuropsychopharmacol 2008;11(Suppl 1):191.
Prof. Dr. Christoph Hiemke, Christine Waldschmitt, Klinik für Psychiatrie und Psychotherapie der Universität Mainz, Untere Zahlbacher Str. 8, 55101 Mainz, E-Mail: hiemke@mail.uni-mainz.de Dr. med. Gabriel Eckermann, Bezirkskrankenhaus Kaufbeuren, Kemnather Straße 16, 87600 Kaufbeuren
Pharmacokinetic interaction potential of duloxetine
Duloxetine is a balanced selective serotonin and noradrenaline reuptake inhibitor which is indicated for the treatment of depression and in addition of stress-induced urinary incontinence in females and diabetogenic neuropathic pain. The antidepressant potential of duloxetine and its tolerability are similar to those of venlafaxine and of selective serotonin reuptake inhibitors. In vivo, duloxetine is primarily metabolized by CYP1A2. Combinations with inhibitors of CYP1A2 like fluvoxamine or ciprofloxacine must therefore be monitored by analysis of plasma concentrations for dose correction (recommended target plasma concentration 60 to 120 ng/ml). CYP2D6 is of minor importance for the in vivo degradation of duloxetine, and combinations with CYP2D6 inhibitors do not lead to clinically relevant elevations of duloxetine plasma concentrations. However, duloxetine by itself is an inhibitor of CYP2D6. The inhibitory potential is less pronounced than those of paroxetine or fluoxetine. Under steady state conditions, trough plasma levels of desipramine or metoprolol have been shown to increase by about threefold under therapeutic doses of duloxetine (60 mg) compared with a mean increase of sixfold by fluoxetine or paroxetine. So far, case reports on adverse events due to inhibition of CYP2D6 by duloxetine are lacking. Nevertheless, the CYP2D6 inhibitory potential of duloxetine may be clinically relevant when the combined drug is a preferred substrate of CYP2D6 and exhibits a narrow therapeutic range.
Keywords: Duloxetine, antidepressants, drug interactions, CYP2D6, CYP1A2
Psychopharmakotherapie 2009; 16(01)