Torsten Reum, Bonn
Jeder Anwendung eines neuen Arzneimittels am Menschen gehen präklinische Untersuchungen voraus, um Informationen über dessen Wirkungen und Sicherheit zu erhalten. Die präklinische Prüfung und Entwicklung beinhaltet die Durchführung experimenteller Studien an verschiedenen ausgewählten biologischen Systemen zur Untersuchung der pharmakologischen und toxikologischen Eigenschaften des Arzneimittels. Nationale und internationale Gesetze und Regelungen legen fest, dass für jedes neue Arzneimittel vor der Testung am Menschen im Rahmen klinischer Studien der Phasen I bis III und vor einer Marktzulassung jeweils in bestimmtem Umfang präklinische Studien durchzuführen sind. Diese Untersuchungen sollen einerseits das pharmakologische Wirkungsprofil beschreiben und die beabsichtigte pharmakodynamische Wirkung nachweisen, andererseits sollen sie anhand weitgehend standardisierter Prüfverfahren valide Informationen zur Sicherheit und Toxizität des Wirkstoffs liefern. Das Arzneimittelgesetz (AMG, 14. Novelle, Stand 21.09.2005) sieht vor, dass der pharmakodynamische Wirkungsnachweis für eine Substanz unter Berücksichtigung des jeweils gesicherten Standes der wissenschaftlichen Erkenntnisse erfolgen soll und insbesondere Tierversuche durch andere Prüfverfahren zu ersetzen sind, wenn dies im Hinblick auf den Prüfzweck wissenschaftlich vertretbar ist (AMG, §26: Arzneimittelprüfrichtlinien). Nach Möglichkeit soll anhand entsprechender experimenteller Verfahren beziehungsweise unter Verwendung geeigneter Krankheitsmodelle auch ein Vergleich mit anderen Wirkstoffen, die bei der entsprechenden Krankheit wirksam sind, angestellt werden.
So klar diese gesetzlichen Rahmenbedingungen zum Wirkungsnachweis für Arzneimittel allgemein erscheinen, so problematisch erweisen sie sich im konkreten Fall der präklinischen Entwicklung und Testung von neuroprotektiv wirkenden Substanzen. Es mangelt an validen präklinischen Testsystemen, die neben dem pharmakodynamischen Wirkungsnachweis beispielsweise auch eine zuverlässige Voraussage von nachhaltigen und klinisch relevanten neuroprotektiven Effekten beim betroffenen Patienten ermöglichen oder ein latent neurodegeneratives Potenzial beim Menschen anzeigen können. Eine wirksame Pharmakotherapie, die nachweislich neuroprotektive Wirkungen beim Patienten hat, also zu einer Verzögerung oder gar zum Aufhalten der fortschreitenden Neurodegeneration führt, ist bislang nicht verfügbar. Da geeignete Krankheitsmodelle und wirksame Vergleichssubstanzen zur präklinischen Testung von potenziell neuroprotektiv wirkenden Substanzen nicht zur Verfügung stehen, ist eine zuverlässige Prognose der klinischen Wirksamkeit bei Neurodegenerationen im Rahmen der präklinischen Entwicklung gegenwärtig kaum möglich.
Neurodegenerative Erkrankungen verlaufen chronisch progredient. Bei jedem Arzneimittel, das der Patient über einen längeren Zeitraum einnimmt, besteht die Möglichkeit, dass es den Krankheitsverlauf nachhaltig beeinflusst und gegebenenfalls auch verschlechtert. Es muss daher auch gefragt werden, ob die etablierten und weitgehend standardisierten präklinischen Teststrategien zur Sicherheits- und Toxizitätsprüfung geeignet sind, mögliche neurodegenerative Potenziale und Prozesse adäquat zu erkennen und zu charakterisieren. Diese spezifischen Probleme bei der präklinischen Testung von Arzneimitteln sollen in der vorliegenden Arbeit weiter analysiert werden, dafür sollten zunächst aber einige Aspekte der neurodegenerativen Erkrankungen und der neuroprotektiven Therapieansätze erläutert werden.
Neurodegenerative Erkrankungen
Unter einer Neurodegeneration versteht man einen chronisch fortschreitenden Schädigungsprozess an Nervenzellen, der zum Verlust neuronaler Funktionen und/oder zum Absterben der Neuronen führt. Dieser Prozess ist in der Regel auf definierte Zelltypen und/oder bestimmte Regionen des Zentralnervensystems (ZNS) begrenzt. Die Neuroprotektion ist analog dazu jeder substanzielle Effekt zur Verhinderung oder Verlangsamung beziehungsweise Abschwächung von neurodegenerativen Vorgängen. Unter den zahlreichen mit einer solchen progredienten Neurodegeneration einhergehenden Syndromen kommen die Alzheimer-Krankheit und der Morbus Parkinson in den westlichen Industrienationen am häufigsten vor. Die Pathogenese der Alzheimer-Krankheit ist gekennzeichnet durch die vermehrte Entstehung und Ablagerung von Amyloid-Plaques und Neurofibrillen, nachfolgend kommt es zu zahlreichen zerebralen Funktionsstörungen und letztlich zur Desintegration neuronaler Schaltkreise und zum fortschreitenden Absterben von Nervenzellen [35]. Das pathophysiologische Korrelat des Morbus Parkinson ist die selektive Degeneration neuromelaninhaltiger dopaminerger Nervenzellen in der Substantia nigra pars compacta. Als Ursache wird eine mitochondriale Komplex-I-Dysfunktion in diesen Neuronen angenommen, die zahlreiche weitere Funktionsstörungen wie zellulären Energiemangel, oxidativen Stress, Exzitotoxizität und Entzündungen hervorruft, die den Krankheitsprozess weiter verstärken [9, 38]. Die Prävalenz neurodegenerativer Erkrankungen steigt mit zunehmendem Lebensalter [19] und wird daher für eine älter werdende Gesellschaft immer mehr zum Problem. Abbildung 1 zeigt den typischen Verlauf einer neurodegenerativen Erkrankung und verdeutlicht die daraus erwachsenden Probleme.
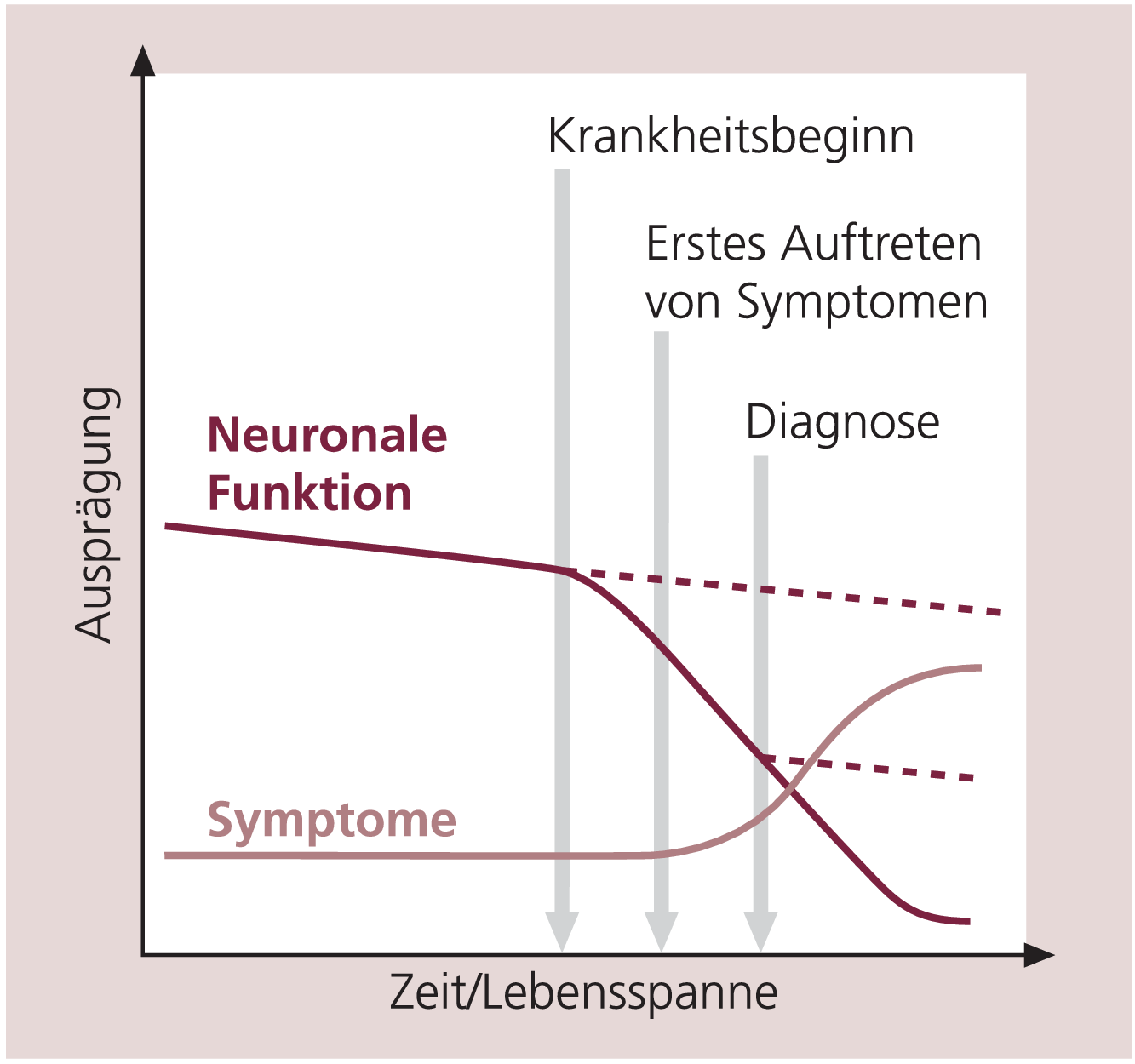
Abb. 1. Zeitlicher Verlauf einer neurodegenerativen Erkrankung Der beschleunigte Verlust neuronaler Funktionen (dunkelrote Linie) markiert den Krankheitsbeginn, führt jedoch aufgrund von Kompensationsmechanismen nicht unmittelbar zum Auftreten von Krankheitssymptomen. Zwischen dem Einsetzen der ersten Symptome (hellrote Linie) und der Diagnosestellung liegt in der Regel eine weitere Zeitspanne. Der Krankheitsprozess läuft auch nach der Diagnosestellung unvermindert weiter, weil durch die Pharmakotherapie gegenwärtig nur die Symptomatik behandelt werden kann. Neuroprotektiva, die zur Abschwächung oder Aufhebung des fortschreitenden neuronalen Funktionsverlustes führen könnten (untere Strichlinie), sind noch nicht verfügbar.
Obwohl der krankheitsbedingte Zell- und Funktionsverlust im ZNS kontinuierlich zunimmt, treten beispielsweise bei Parkinson-Patienten die typischen Krankheitssymptome erst dann auf, wenn bereits 50 bis 60% der dopaminergen Zellen abgestorben sind [1]. Die etablierte Pharmakotherapie des Morbus Parkinson kann den Degenerationsprozess nicht aufhalten, gegenwärtig ist damit nur eine funktionelle Verbesserung der Symptomatik möglich. Auch wenn zukünftig zum Zeitpunkt der Diagnose bereits eine wirksame neuroprotektive Arzneimittelbehandlung eingeleitet werden könnte, wäre damit allenfalls das Absterben weiterer Nervenzellen zu verhindern, nicht aber der stattgefundene Neuronenverlust auszugleichen.
Warum ist eine wirksame neuroprotektive Therapie auf pharmakologischer Basis bis heute nicht verfügbar, obwohl die wesentlichen Mechanismen der Pathogenese des Morbus Parkinson hinreichend bekannt sind? Die Kenntnis dieser Mechanismen führte zur Testung zahlreicher Wirkstoffe aus verschiedenen Substanzgruppen, wie Antioxidanzien, Dopaminrezeptor-Agonisten, Glutamatrezeptor-Antagonisten oder Monoaminoxidase-(MAO-)B-Hemmer in etablierten Krankheitsmodellen des Morbus Parkinson und in anderen präklinischen Experimenten – mit überwiegend positiven Ergebnissen [15, 25]. Trotzdem haben diese Wirkstoffe die klinischen Erwartungen nicht erfüllen können [30]. Kann eine vertiefte Analyse konkreter Untersuchungsbedingungen diesen Widerspruch auflösen? Das ist im Einzelfall sicherlich möglich. Retrospektiv lassen sich vielfach Ursachen benennen, die entweder in den Substanzeigenschaften, in den experimentellen Bedingungen oder auch im Design der klinischen Studien zu finden sind. Eine solche Analyse konkreter Einzelbeispiele ist jedoch für den prospektiven Ansatz eines präklinischen Experiments und für die generelle Beantwortung der Frage, ob präklinische Untersuchungen und Tiermodelle zur Voraussage und Bewertung neuroprotektiver Arzneimittelwirkungen am Menschen prinzipiell geeignet sind, wenig hilfreich. Allgemein gültige Schlussfolgerungen zur Eignung von präklinischen Testmethoden und zur Übertragbarkeit experimenteller Daten auf den Menschen erfordern daher eher einen induktiven Ansatz.
Präklinische Arzneimittelentwicklung
Die präklinische Arzneimittelentwicklung dient der umfassenden Charakterisierung der Wirkungen eines Arzneimittels vor dessen entsprechender Anwendung am Menschen. Bei den Wirkungen, die ein Arzneimittel auf den Organismus ausübt, wird zwischen den pharmakodynamischen und den toxischen Effekten unterschieden. Zu den pharmakodynamischen Effekten werden in erster Linie die therapeutisch gewünschten Wirkungen gezählt, aber auch weitere unerwünschte Reaktionen, die auf diesem Wirkungsmechanismus beruhen. Andere schädliche Effekte einer Substanz wären als toxische Arzneimittelwirkungen zu bezeichnen. Pharmakodynamische und toxische Arzneimittelwirkungen sind in den meisten Fällen konzentrations- beziehungsweise dosisabhängig, erste Hinweise zur Sicherheit eines Arzneimittels können daher bereits aus dem Abstand von effektiver und toxischer Konzentration am jeweiligen Wirkort abgeleitet werden. Der Konzentrationsverlauf am Wirkort lässt sich jedoch kaum direkt erfassen, zur Extrapolation sind daher Untersuchungen zur Pharmako- beziehungsweise Toxikokinetik notwendig. Schlüsselvorgänge wie Resorption, Verteilung, Metabolismus und Exkretion eines Wirkstoffs sollten daher möglichst frühzeitig an einer relevanten, das heißt dem Menschen in dieser Beziehung vergleichbaren Versuchstierspezies untersucht worden sein.
Die präklinische Arzneimittelentwicklung schafft die Voraussetzungen für die notwendigen klinischen Studien der Phasen I bis III, und die größte Bedeutung der Ergebnisse von präklinischen Studien liegt in der adäquaten Sicherheitsbewertung für die eingeschlossenen Probanden und Patienten, insbesondere bei der Erstanwendung am Menschen. Die zur Genehmigung von klinischen Studien erforderlichen präklinischen Untersuchungen sind international harmonisiert und im ICH-M3-Dokument [11] dargelegt (ICH = International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use).
Die gewonnenen präklinischen Daten zur Pharmakodynamik einer Substanz sind für die Nutzen-Risiko-Bewertung der frühen klinischen Prüfungen von großer Bedeutung, denn erste Aussagen zur Wirksamkeit des Arzneimittels am Menschen sind in der Regel erst während der Phase II möglich. Der Wirksamkeitsnachweis ist eine unabdingbare Voraussetzung zur Erlangung der Marktzulassung eines Arzneimittels und kann nur anhand von klinischen Ergebnissen erbracht werden. Aufgrund der erwähnten Diskrepanz von präklinischen und klinischen Ergebnissen zur neuroprotektiven Arzneimittelwirkung müssten die präklinischen Test- und Krankheitsmodelle so optimiert werden, dass zukünftig eine wesentlich verbesserte Vorhersagbarkeit für die klinische Wirksamkeit möglichst schon anhand dieser Experimente und gegebenenfalls anhand der Ergebnisse der frühen klinischen Testphasen möglich ist, denn eine aufwendige, teure und zeitintensive klinische Testung von Neuroprotektiva, deren Wirksamkeit nicht belegt werden kann, sollte möglichst unterbleiben oder rechtzeitig abgebrochen werden.
Übertragbarkeit präklinischer Daten auf den Menschen
Von zahlreichen Wissenschaftlern wird zur Erklärung von Diskrepanzen zwischen präklinischen Daten und klinischen Ergebnissen immer wieder betont, dass die Komplexität des Krankheitsgeschehens beim Menschen durch die präklinischen Modelle nicht oder nicht adäquat wiedergegeben werden kann. Aussagen zur prinzipiellen Verwendbarkeit präklinischer Testmethoden und zu deren Übertragungsmöglichkeiten auf das komplexe Geschehen beim gesunden Menschen und beim Patienten mit einer bestimmten Krankheit sind nur möglich, wenn die physiologischen Wechselbeziehungen und die konkreten pathophysiologischen Mechanismen im experimentellen Ansatz angemessen widergespiegelt werden können. Daher muss die Pathophysiologie und Pathobiochemie der menschlichen Erkrankung durch alle verfügbaren Untersuchungsverfahren umfassend aufgeklärt und für ein präklinisches Krankheitsmodell möglichst weitgehend berücksichtigt sein.
Um entscheiden zu können, welchen Stellenwert man präklinischen Daten beimessen kann und welche Möglichkeiten und Grenzen konkrete präklinische Untersuchungen aufweisen, müsste die Komplexität der untersuchten Krankheit und des verwendeten Modells gezielt untersucht und nach systematischen Gesichtspunkten analysiert werden. Für beide Systeme (Krankheit und Modell) müssen daher die relevanten strukturellen und funktionellen Hierarchieebenen erkennbar sein (Abb. 2). Der Begriff Hierarchie wird meist auf die starr fixierte vertikale Rangordnung in einem System eingegrenzt, also auf die klar ausgerichteten und festgelegten Beziehungen zwischen oberen und unteren Systemebenen. Es wird dabei leicht übersehen, dass für jede Struktur und jeden Prozess mit der Einordnung in eine bestimmte Hierarchieebene eine Vielzahl von Wechselbeziehungen zu anderen Strukturen und Prozessen entstehen, die stets in drei Grundrichtungen verlaufen. Nach oben hin ist der jeweilige Prozess der „abhängige“ Teil eines übergeordneten Systems, in das eine Eingliederung erfolgen muss, nach unten integriert und „kontrolliert“ derselbe Prozess aber gleichzeitig verschiedene Subprozesse, ist also selbst ein übergeordnetes System. Neben diesen vertikal ausgerichteten „Abhängigkeiten bzw. Kontroll- oder Integrationsfunktionen“ sind jedoch auf der jeweils gleichen Organisationsebene vielfältige „gleichberechtigte“ Wechselbeziehungen von Bedeutung. Aufgrund dieser differenzierteren Betrachtung soll der Begriff der Hierarchie in Analogie zu Wilber [45] durch den Begriff Holarchie ersetzt werden, denn auf jeder hier dargestellten Ebene finden wir sowohl das Phänomen der Differenzierung, nämlich das Vorhandensein oder Erzeugen einer Vielfalt verschiedener zum Teil sehr unterschiedlicher Prozesse oder Strukturen wie beispielsweise Moleküle, Organellen, Zelltypen und Organe, als auch das der Integration, das heißt, die Zellen, Gewebe, Organe usw. sind in einer bestimmten einzigartigen strukturellen und funktionellen Wechselbeziehung organisiert. Dieses Wechselspiel von eigenständiger Funktion als Zelle und Funktion im Zell- oder Gewebeverband erzeugt so (und nur so!) das entsprechende Organ. Im sprichwörtlichen Sinn ergibt das Ganze aufgrund seiner Komplexität mehr als die Summe seiner Teile.
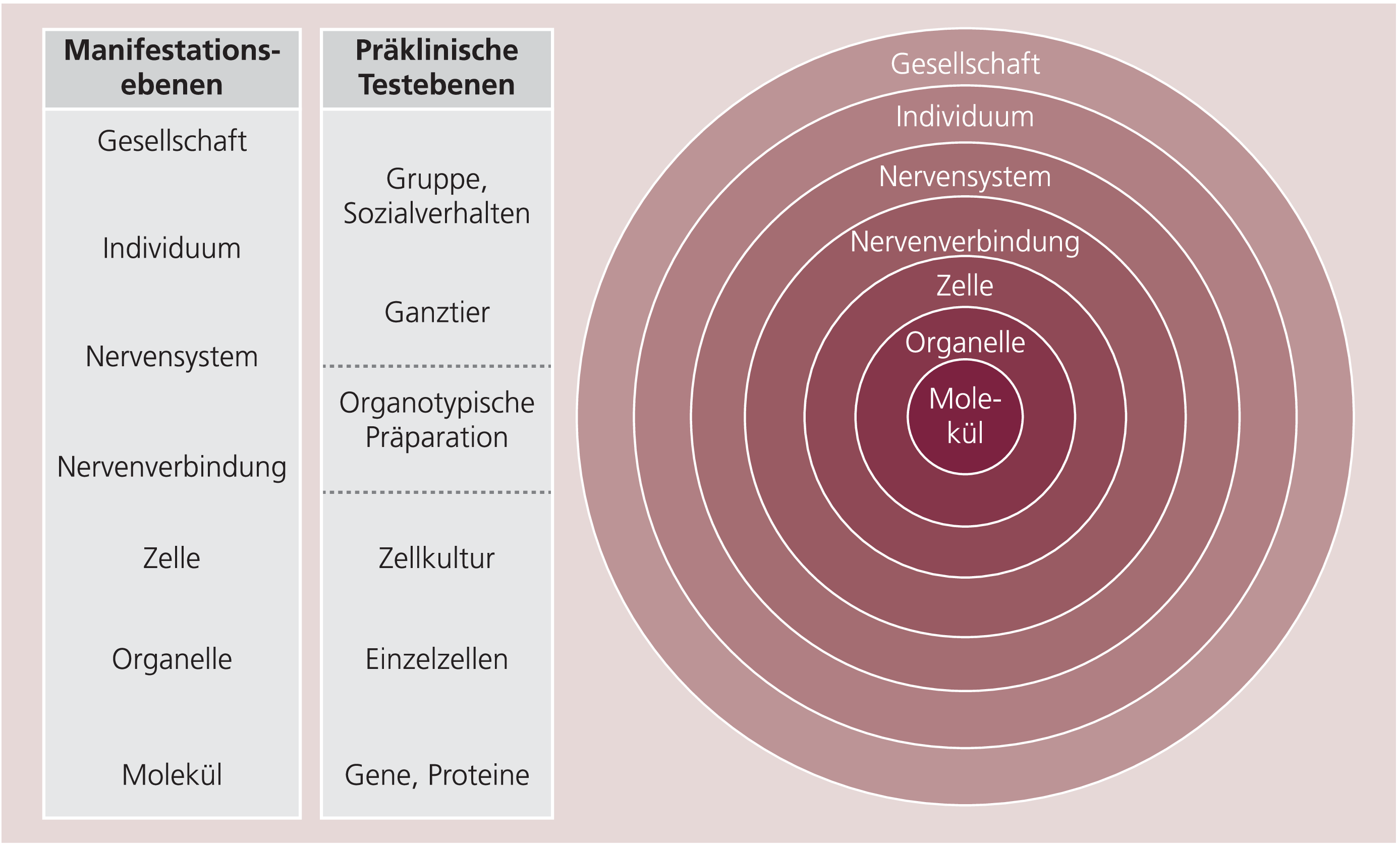
Abb. 2. Darstellung und Auflistung der Holarchieebenen, auf denen sich Krankheiten des ZNS manifestieren können (rechter Abschnitt und linke Spalte), und Auflistung der präklinischen Testebenen, die den Manifestationsebenen einer Krankheit zugeordnet werden können (rechte Spalte). Die Zunahme der Komplexität wird durch die konzentrischen Kreise in der rechten Bildhälfte symbolisiert. Weitere Erläuterungen siehe Text
Je komplexer die Strukturen auf den höheren Holarchieebenen werden, umso mannigfaltiger werden theoretisch die Kombinationsmöglichkeiten der „abhängigen“ Elemente der niederen Ebenen, und umso komplexer wird das System selbst, was in Abbildung 2 rechts durch die Zunahme des jeweiligen Kreisumfangs symbolisiert wird. Mit der Zunahme der Komplexität steigt einerseits die Störanfälligkeit des Systems, andererseits wachsen aber auch die Möglichkeiten der Kompensation. Wird das System auf einer bestimmten Komplexitäts- oder Holarchieebene gestört, kann diese Störung auf der gleichen Ebene kompensiert werden (indem z.B. nach Apoptose einer Zelle die Nachbarzelle die ausgefallene Funktion mit übernimmt) und/oder die Kompensation wird auf eine höhere Ebene verlagert, so wie beispielsweise die komplexe Regulation der Hormonfreisetzung beim Mangel von Steroidhormonen in der Peripherie vonstatten geht.
Störungen, die auf den unterschiedlichen Ebenen infolge einer Krankheit auftreten, sind in vielen Fällen als pathophysiologische Korrelate erkennbar. Sie treten ab einer bestimmten Ebene – der Manifestationsebene – in Erscheinung und sind dann auch auf den höheren Ebenen erkennbar, solange dort keine Kompensation erfolgt. Bei genetischer Determinierung einer Krankheit ist die „primäre“ „Störung“ auf molekularer Ebene zu finden, aber es existieren durchaus zahlreiche Kompensationsmöglichkeiten auf den höheren Holarchieebenen, die den Ausbruch und den Verlauf der Krankheit beeinflussen und teilweise sehr variabel gestalten können. Ein interessantes Beispiel für komplexe Wechselwirkungen auf zahlreichen Ebenen, die das Auftreten und den Verlauf einer Krankheit bestimmen, stellt die Schizophrenie dar. Obwohl eine Heredität der Schizophrenie nicht ernsthaft in Zweifel gezogen werden kann, liegen selbst bei eineiigen Zwillingen mit identischem Genom die Konkordanzraten für das Auftreten der Krankheit nur bei etwa 50% [40]. Die Diagnose muss anhand von psychopathologischen Symptomen gestellt werden, denn somatisch determinierte pathophysiologische Korrelate konnten bisher weder durch gezielte Laboruntersuchungen noch durch die bildgebende Diagnostik identifiziert werden [2], obwohl diese Methoden in den letzten Jahren immens verbessert wurden. Die Manifestationsebenen der Schizophrenie könnten daher durchaus erst im Bereich des Individuums beziehungsweise der Gesellschaft zu finden sein, denn die Krankheitseinsicht ist beim Patienten selbst oft nicht vorhanden, und das jeweilige soziale beziehungsweise gesellschaftliche Umfeld des Patienten kann den Ausbruch und den Verlauf der Schizophrenie sehr wesentlich beeinflussen [29].
Systematische Charakterisierung der präklinischen Testsysteme
Im Rahmen des Holarchiekonzepts können Komplexitätsgrad und Manifestationsebene einer Krankheit oder eines pathophysiologischen Prozesses zur Klassifizierung von präklinischen Testsystemen herangezogen werden. In Abbildung 2 sind in der Spalte der Bildmitte verschiedene präklinische Testsysteme aufgelistet, deren Reihenfolge dem Komplexitätsgrad entspricht und deren Lokalisierung mit der entsprechenden Manifestationsebene in der linken Spalte übereinstimmt. Je nachdem, auf welcher Manifestationsebene eine bestimmte funktionelle Störung oder Krankheit auftritt beziehungsweise untersucht werden soll, kann ein Testsystem auf der entsprechenden Holarchieebene verwendet werden.
Beispielsweise kann die Wirkung spezifisch wirkender Neurotoxine an einer Zellkultur untersucht werden, sofern die verwendeten Zellen die jeweils notwendigen Eigenschaften oder Zielstrukturen aufweisen, die zur intrazellulären Aufnahme und zur Wirkung dieses Neurotoxins erforderlich sind. Im Falle von selektiv neurodegenerativ wirkenden Toxinen, wie 6-Hydroxydopamin (6-OHDA), müssen die Zellen beispielsweise dopaminerge Transmissionseigenschaften aufweisen und über funktionsfähige membranständige Dopamintransporter verfügen. Mit dem Testsystem Zellkultur sind aber keine direkten Rückschlüsse von der beobachteten Toxinwirkung auf die höheren Manifestationsebenen möglich, also beispielsweise auf die Beeinflussung der neuronalen Erregungsausbreitung, die Störung motorischer und sensorischer Körperfunktionen und die damit verbundenen Auswirkungen auf den Patienten als Individuum, oder auch auf die Gesellschaft (z.B. familiäre Belastung, Arbeitsausfall, Invalidisierung). Diese Rückschlüsse aus präklinischen Daten sind nur durch Extrapolation möglich, und die Genauigkeit der Extrapolation hängt davon ab, wie gut das experimentelle Modell im Zusammenspiel der jeweiligen Ebenen nach Einwirkung definierter Störgrößen verstanden wird.
Natürlich kann auch primär ein präklinisches Modell, das höhere Testebenen einschließt, verwendet werden. Die Wirkung von Neurotoxinen wird beispielsweise sehr oft direkt am Versuchstier untersucht, wobei hier die Kompensationsmöglichkeiten höherer Ebenen unbedingt in Betracht zu ziehen sind. Eine partielle Läsion dopaminerger ZNS-Strukturen durch 6-OHDA ist beispielsweise durch Analyse des Spontanverhaltens der Ratte kaum erkennbar [7].
Die Auflistung der präklinischen Testsysteme in Abbildung 2 ist durch zwei Strichlinien unterbrochen. Die untere Linie markiert die Grenze für die Verwendung von Modellen beziehungsweise Testmaterial menschlichen Ursprungs. Von wenigen Ausnahmen abgesehen, bei denen neurochirurgisch gewonnenes organotypisches Gewebe für präklinische Untersuchungen verwendet werden kann, ist für die präklinische Routinetestung die Verwendung von menschlichen neuronalen Zelllinien als obere Grenze anzusehen, komplexere Ebenen des menschlichen Nervensystems müssen klinisch untersucht werden. Die zweite Linie oberhalb der organotypischen Gewebepräparationen markiert die Grenze für die Verwendbarkeit von so genannten alternativen präklinischen Testmethoden oder In-vitro-Modellen. Entsprechend dem 3-R-Konzept (Reduction, Refinement and Replacement of animal studies) sollen präklinische Tierversuche nach Möglichkeit durch alternative Testmethoden ersetzt werden [13] (s.a. §26 AMG: Arzneimittelprüfrichtlinien). Auch hierbei gilt, dass die Ergebnisse aus alternativen Testverfahren für eine Prüfsubstanz nur indirekte Rückschlüsse auf deren Wechselwirkungen oberhalb der getesteten Komplexitätsebene zulassen. Alternative Testmethoden schließen jedoch die Verwendung von Tierversuchen (außer denen an Säugetieren) nicht grundsätzlich aus, daher ist auch hier die Linie unterbrochen.
Gerade bei der Verwendung alternativer Testmodelle oder Tierspezies, beispielsweise von Fischen oder Insekten, stellt sich die Frage nach der Validität der Methode und nach der Übertragbarkeit der damit erzielten Ergebnisse auf den Menschen, denn im Vergleich zum Menschen weist das Nervensystem der eben genannten Tierklassen wesentlich größere Unterschiede auf als das der üblicherweise verwendeten Säugetierspezies. Damit kommt generell ein zweiter wichtiger Aspekt für die Eignung und Übertragbarkeit von präklinischen Daten hinzu, die Vergleichbarkeit der an einer bestimmten Spezies gewonnenen Ergebnisse mit der jeweiligen physiologischen oder/und pathophysiologischen Situation beim Menschen. Dieses Kriterium „Vergleichbarkeit“ ist aber nicht zwangsläufig mit dem Grad der genetischen Verwandtschaft gleichzusetzen sondern entsprechend den verwendeten Krankheitsmodellen und Untersuchungsparametern zu bewerten. Maßgebend ist hier eindeutig die konkrete experimentelle Simulation der Bedingungen im menschlichen Organismus beziehungsweise der entsprechenden Krankheit. Unter Umständen können ganz bestimmte funktionelle oder „phänotypische“ Eigenschaften untersucht werden, die eine genetisch weiter entfernte Spezies für ein konkretes Experiment vergleichsweise besser geeignet erscheinen lassen. Diese Eigenschaften können, beispielsweise in der Frage der Extrapolation von Pharmakokinetikdaten, dazu führen, dass sich das Nagetier und nicht der ebenfalls untersuchte Primat in diesem Fall als die relevantere, das heißt „menschenähnlichere“ Spezies herausstellt. Auch hinsichtlich der Ausprägung krankheitsspezifischer Symptome und Merkmale stellt sich nach eingehender Prüfung heraus, dass die häufig verwendete Versuchstierspezies Ratte für die Untersuchung von neurodegenerativen oder neuroprotektiven Wirkungen beispielsweise im Vergleich zum Primaten unter mehreren Aspekten sehr gut geeignet ist [8].
Ein drittes Kriterium, das für die Interpretation und Übertragbarkeit präklinischer Ergebnisse auf den Menschen nicht vernachlässigt werden darf, ist die Berücksichtigung der Ontogenese. Im Zusammenhang mit der Manifestation von neurodegenerativen Prozessen sind der ontogenetische Entwicklungsstand und der Zeitrahmen pathophysiologischer Reaktionen von entscheidender Bedeutung. Selbst bei einer eindeutig genetisch bedingten Krankheitsprädisposition, beim Morbus Huntington beispielsweise, kommt es erst in der Lebensmitte zum allmählichen Ausbruch der Krankheit. Entsprechende präklinische Modelle, die analog zur Situationen im Menschen die Altersabhängigkeit degenerativer Effekte berücksichtigen und einen chronisch progredienten Verlauf der Erkrankung simulieren, sind in ihrer Aussagekraft den etablierten Ansätzen mit schnell wirkenden Neurotoxinen sicherlich klar überlegen. In anderen Fällen sind die so genannten vulnerablen Zeitfenster, in denen Wirkstoffe und Toxine neurodegenerative Veränderungen hervorrufen können, in die frühe Individualentwicklung verlagert. Substanzen wie Ethylalkohol, Benzodiazepine, bestimmte Narkotika und Antikonvulsiva führen beispielsweise bei Ratten zur umfangreichen Apoptose von Nervenzellen während der Synaptogenese in einer sehr frühen Entwicklungsperiode [17]. Dem Aspekt der Ontogenese, insbesondere der Berücksichtigung der frühen Entwicklungsperioden einschließlich der Adoleszenz und der gezielten präklinischen Testung für Arzneimittelanwendungen bei Kindern und Jugendlichen, wird in der präklinischen Testung noch zu wenig Rechnung getragen. Die Notwendigkeit, Arzneimittel gezielt für die Anwendung in diesen Altersgruppen zu entwickeln und zuzulassen, ist international erkannt worden, und die gezielte klinische Entwicklung von Arzneimittel zur Anwendung bei Kindern und Jugendlichen ist erklärtes Ziel der EMEA. 2007 ist deshalb eine entsprechende Verordnung auf europäischer Ebene in Kraft getreten [43].
Die drei soeben dargestellten Bewertungskriterien für präklinische Testmethoden – der Komplexitätsgrad des verwendeten Testsystems, der Grad der Vergleichbarkeit von Tier und Mensch hinsichtlich der zu untersuchenden Funktionen und Prozesse sowie die Berücksichtigung der Ontogenese – könnten in einem dreidimensionalen Koordinatensystem darstellt werden (Abb. 3). Der Komplexitätsgrad wird durch die y-Achse bestimmt und nimmt nach oben zu. Die Speziesunterschiede beziehungsweise die Vergleichbarkeit mit dem Menschen sind durch den jeweiligen Abstand auf der x-Achse gekennzeichnet. Die z-Achse markiert die speziesspezifische Ontogenese und umfasst jeweils die gesamte Lebensspanne der entsprechenden Spezies. Auf dieser Achse ist der Abstand zwischen der verwendeten Tierspezies und dem Menschen dann am geringsten, wenn der entsprechende ontogenetische Entwicklungsstand des Tiers dem jeweiligen Lebensabschnitt des Menschen (z.B. uterine Entwicklung, Kindheit, Adoleszenz und Altern) entspricht, in dem die zu untersuchenden Störungen und Krankheiten auftreten, und der Zeitverlauf des Krankheitsprozesses adäquat berücksichtigt ist. Dieser Umstand wird beispielsweise bei Untersuchungen des reproduktionstoxikologischen oder des karzinogenen Potenzials eines Arzneimittels berücksichtigt. Bei diesen Untersuchungen werden die Tiere kontinuierlich über bestimmte ontogenetische Entwicklungsabschnitte hinweg mit der Testsubstanz behandelt, um damit mögliche arzneimittelbedingte Einflüsse auf die embryofetale und postpartale Entwicklung beziehungsweise auf die Entstehung und Entwicklung von Tumoren erfassen und bewerten zu können. Der Mensch soll in diesem System nicht als Punkt dargestellt werden, der sich entsprechend dem Lebensalter auf der z-Achse bewegt, sondern er ist aufgrund der interindividuell unterschiedlichen Prädisposition für bestimmte Krankheiten und deren Langzeiteffekte eher als Punktwolke mit einem Schweif parallel zur z-Achse anzusehen.
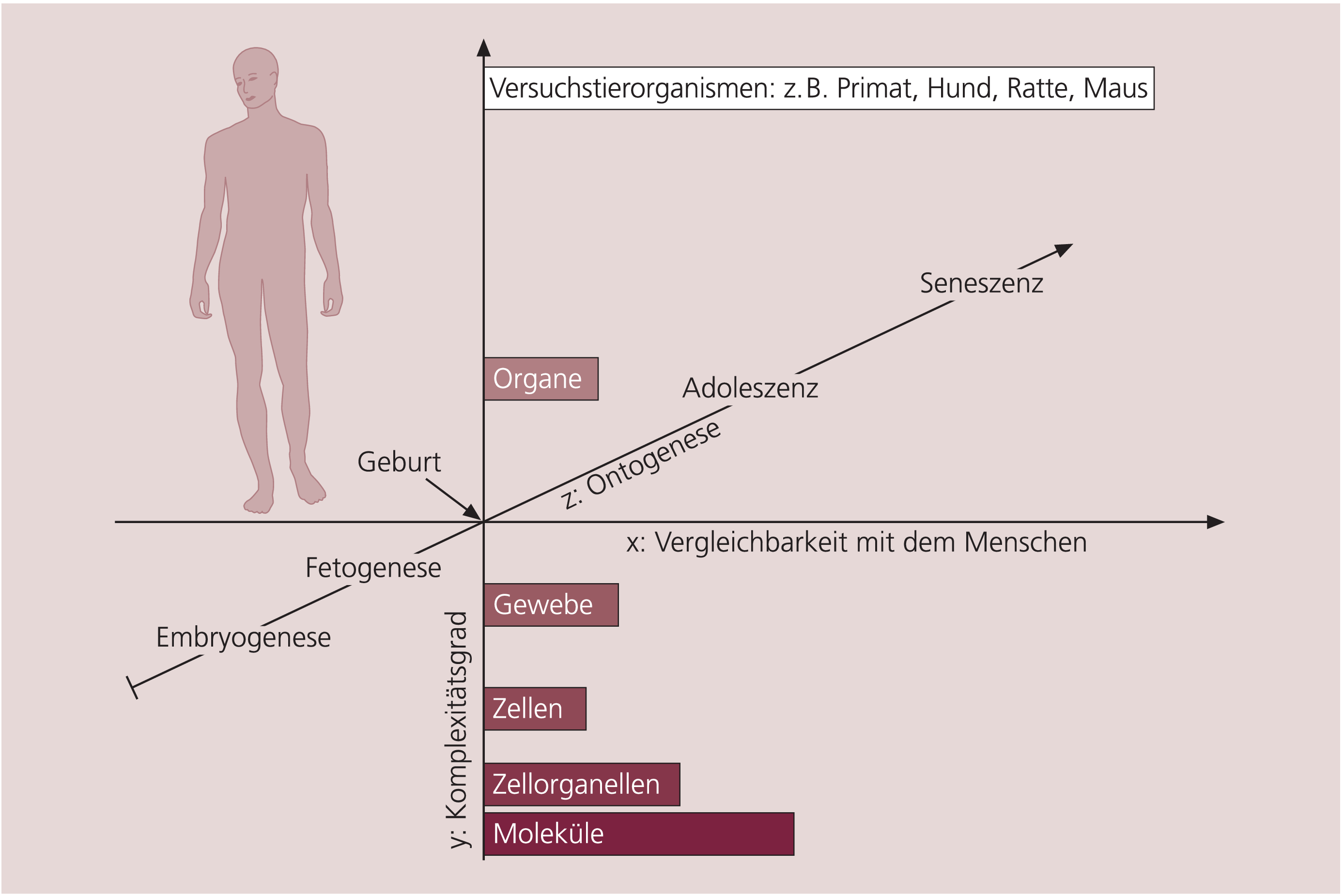
Abb. 3. Dreidimensionales Koordinatensystem zur Einstufung und vergleichenden Bewertung von präklinischen Testmethoden Die drei Kriterien, d.h. der Grad der Vergleichbarkeit der untersuchten Vorgänge in der verwendeten Spezies und im Menschen, der durch die Methode gewährleistete Komplexitätsgrad und die Berücksichtigung der Ontogenese für krankheitsspezifische Untersuchungen, sind durch die Achsen x, y und z repräsentiert. Für jede Testmethode könnte in diesem System eine Koordinate zugeordnet werden, die einen bestimmten Abstand zur Koordinate des Patientenkollektivs mit der entsprechenden Krankheit aufweist. Je kürzer dieser Abstand wäre, desto besser wäre die Übertragbarkeit der präklinisch gewonnenen Daten auf die jeweilige Situation beim Menschen. Weitere Erläuterungen siehe Text
Für eine präklinische Methode können durch die Positionsbestimmungen auf diesen drei Achsen Koordinaten bestimmt werden, die eine Zuordnung und Vergleichbarkeit verschiedener Methoden möglich machen. Der jeweilige räumliche Abstand, den ein bestimmtes Testsystem mit seiner spezifischen Koordinate im Vergleich zur „Koordinate Mensch“ einnimmt, kann als relatives Maß für die Extrapolierbarkeit beziehungsweise Übertragbarkeit der von dieser Methode gewonnenen Daten herangezogen werden. Die Skalierung der einzelnen Achsen, die letztendlich als Maßstab für den gesuchten Abstand zur „Koordinate Mensch“ anzusehen ist, kann jedoch nicht quantifiziert werden, was einen direkten Methodenvergleich erschwert. Dennoch scheint eine generelle Einstufung der Aussagekraft und Relevanz von präklinischen Modellen und Methoden sowie deren Vergleich prinzipiell möglich und mit diesem Ansatz anschaulich darstellbar zu sein.
Für die Übertragbarkeit und Relevanz der präklinischen Daten aus bestimmten Modellen und methodischen Ansätzen sind außerdem die dafür ausgewählten Biomarker von entscheidender Bedeutung. Für eine konkrete Fragestellung müssen die jeweils am besten geeigneten Biomarker oder Endpunkte herangezogen und validiert werden. Sollten sich die aus präklinischen Experimenten gezogenen Schlussfolgerungen im Nachhinein als nicht zutreffend herausstellen, wie beispielsweise nach den schwerwiegenden und sehr bedauerlichen Zwischenfällen bei sechs Probanden infolge der Erstanwendung des Präparats TGN1412 der Firma TeGenero im März 2006, dann müssen sowohl die untersuchten Biomarker als auch die verwendeten Methoden hinsichtlich ihrer Eignung reevaluiert werden [39]. Welche Bedeutung die Auswahl der präklinischen Modelle und der verwendeten Biomarker für die Bewertung neurodegenerativer Effekte haben kann, soll im nächsten Kapitel an einem konkreten Beispiel verdeutlicht werden.
Neurodegeneration durch Methylphenidat – ein falsch interpretierter präklinischer Befund?
Methylphenidat ist ein Standardtherapeutikum zur Langzeitbehandlung der Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS) bei Kindern und Jugendlichen, das in Deutschland sehr häufig verordnet wird. Die Anzahl der definierten Tagesdosen stieg in den vergangenen Jahren kontinuierlich und erreichte im Jahr 2005 eine Anzahl von 33 Mio. [24]. Ein Rückgang dieser Entwicklung ist bislang nicht zu erkennen.
Vor einigen Jahren erschien eine wissenschaftliche Arbeit, in der juvenile und adulte Ratten für zwei Wochen mit Methylphenidat (2 mg/kg/d per os) behandelt wurden [27]. Die anschließend in vitro durchgeführten Radioligand-Bindungsassays zeigten bei den in der juvenilen Phase behandelten Tieren eine Abnahme der Dichte (Bmax) des hoch affinen Dopamin-Transportproteins (DAT) im Striatum, wenn das Gehirn etwa eine oder sechs Wochen nach Abschluss der Behandlung untersucht wurde, während bei den im adulten Stadium behandelten Ratten keine Veränderungen der DAT-Bindung festgestellt wurden. Dieser Befund wurde von den Autoren als begünstigender Faktor für neurodegenerative Veränderungen, wie sie beim Morbus Parkinson beobachtet werden, gedeutet. Diese Daten haben in Deutschland unter den Wissenschaftlern und in der Öffentlichkeit eine breite Diskussion um mögliche irreversible Schäden einer Langzeittherapie mit Methylphenidat, insbesondere bei Anwendung im Kindes- und Jugendalter, entfacht. Selbst in Medien für medizinische Fachkreise wurden nach Erscheinen dieser Arbeit starke Zweifel an der Sicherheit dieser wirksamen und über Jahrzehnte bewährten Behandlung geäußert [3].
Obwohl weiterhin keine gezielten Untersuchungen von neurodegenerativen Langzeiteffekten durch Methylphenidat am Versuchstier vorliegen, sind bis heute zahlreiche relevante Daten zum Wirkstoff und zur postulierten Schädigung publiziert worden. Anhand dieser Ergebnisse soll versucht werden, die erhobenen Befunde angemessen zu bewerten und die Frage nach möglichen neurodegenerativen Wirkungen durch eine Behandlung mit Methylphenidat indirekt zu beantworten.
Der Wirkstoff Methylphenidat gehört zur Klasse der Psychostimulanzien. Diese historisch bedingte Zuordnung beruht auf einem pharmakologischen Wirkungsspektrum, das durch eine allgemeine Erhöhung des ZNS-Aktivitätszustands gekennzeichnet ist und beim Menschen unter anderem zur Steigerung von lokomotorischer Aktivität und psychischer Reaktivität führt. Die bekanntesten Vertreter der Psychostimulanzien sind die Phenylethylamin-Derivate Amphetamin und Metamphetamin, im weiteren Textverlauf als Amphetamine bezeichnet. Beide Substanzen sind neurotoxisch und führen bei entsprechend hoher Dosierung zur Degeneration dopaminerger Nervenzellen [32, 33]. Es liegt nahe, diese Neurotoxizität im Analogieschluss auch dem Methylphenidat zuzuschreiben. Falls eine latente Neurotoxizität tatsächlich vorhanden ist, wäre das bei einer Methylphenidat-Langzeitbehandlung von zahlreichen Kindern und Jugendlichen in Deutschland äußerst bedenklich.
Vergleicht man die pharmakodynamischen Wirkungen der Amphetamine mit denen des Methylphenidats, dann treten auf zellulärer und subzellulärer Ebene die wesentlichen Unterschiede im Wirkungsmechanismus zutage, die in Abbildung 4 im Detail skizziert sind. Amphetamine und Methylphenidat beeinflussen unter anderem die Funktion des DAT an dopaminergen Neuronen, jedoch auf sehr unterschiedliche Weise. Methylphenidat bindet an den DAT und hemmt den Rücktransport von Dopamin in die Präsynapse. Die dadurch bedingte längere Verweilzeit des Transmitters an postsynaptischen Dopaminrezeptoren verstärkt die dopaminerge Transmission. Bei ADHS-Patienten wurde im Striatum eine primär erhöhte DAT-Dichte festgestellt [20], eine Hemmung dieses essenziellen Transportproteins durch Methylphenidat würde die funktionellen Störungen bei ADHS ausgleichen und damit die klinische Wirksamkeit zumindest teilweise erklären. Die Amphetamine und einige weitere strukturverwandte Analoga interagieren ebenfalls mit dem DAT. Sie führen jedoch, da sie als DAT-Substrat in die Nervenzelle eingeschleust werden, zu einer vermehrten Freisetzung von Dopamin aus dem zytosolischen Pool in den Extrazellulärraum (Abb. 4).
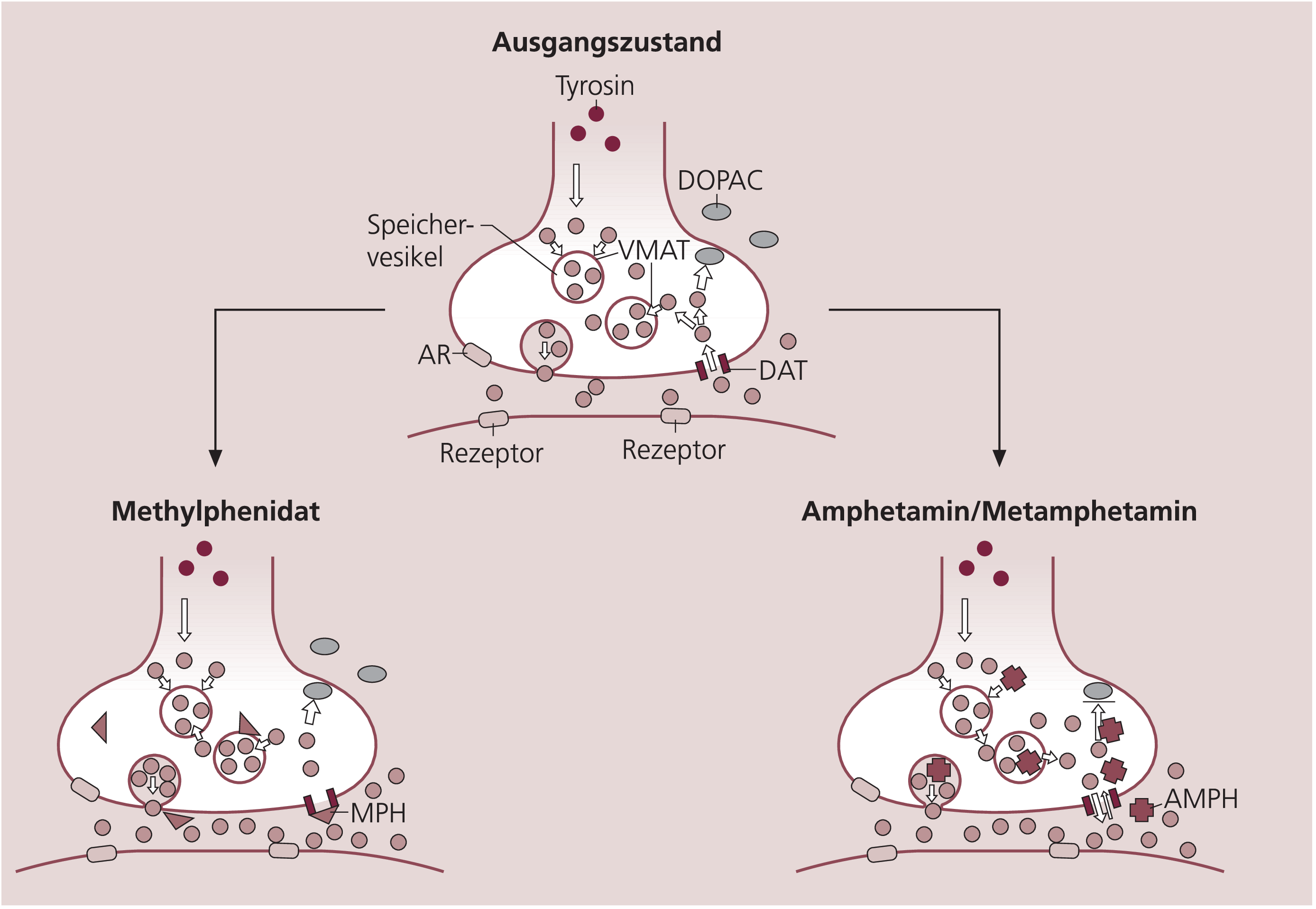
Abb. 4. Schematische Darstellung der Angriffspunkte und pharmakologischen Wirkungen von Methylphenidat und von Amphetamin/Metamphetamin auf die Transmission von dopaminergen Nervenendigungen Neben dem extrazellulären Konzentrationsanstieg von Dopamin, der sowohl durch Methylphenidat als auch durch Amphetamin und Metamphetamin ausgelöst wird, ergeben sich aufgrund der verschiedenen Angriffspunkte beider Wirkstoffe zahlreiche Differenzen in der Wirkung. Hervorzuheben sind insbesondere die unterschiedlichen Effekte auf die intrazelluläre Dopaminverteilung, die erhebliche Konsequenzen für das neurodegenerative Potenzial dieser Substanzen haben. Detaillierte Erläuterungen sind im laufenden Text zu finden. Die kleinen geschlossenen Kreise stellen Dopaminmoleküle dar, aus deren Häufigkeit die Konzentration im entsprechenden Kompartiment abzuleiten ist. AR = Autorezeptor, AMPH = Amphetamin/Metamphetamin, DAT = Dopamintransporter, DOPAC = Dihydroxyphenylessigsäure, MPH = Methylphenidat, VMAT = vesikulärer Monoamintransporter
Methylphenidat und Amphetamine rufen am Versuchstier vergleichbare Wirkungen und Verhaltensänderungen hervor [5], die im Wesentlichen auf den extrazellulären Anstieg der Dopaminkonzentration im Striatum und im Nucleus accumbens zurückzuführen sind [21]. Diese akuten Wirkungen haben jedoch nichts mit den neurotoxischen Effekten der Amphetamine zu tun, die nach heutigem Kenntnisstand eindeutig auf die Beeinflussung der intrazellulären Kompartimentierung von Dopamin zurückgeführt werden. Experimentell wurde nachgewiesen, dass die extrazelluläre Dopaminkonzentration für die Zellschädigung keine Rolle spielt [23]. Wie bereits erläutert und in Abbildung 4 dargestellt, binden Amphetamine als Substrat an den DAT, werden auf diesem Weg in das Neuron eingeschleust und intrazellulär über den vesikulären Monoamintransporter (VMAT) in den Dopamin-Speichervesikeln angereichert. Das dort gespeicherte Dopamin wird im Gegenzug in das Zytosol zurückverteilt [42]. Die zytosolische Transmitterkonzentration steigt, und Dopamin wird durch Amphetamine via DAT retrograd aus den Nervenendigungen in den extrazellulären Raum ausgeschleust [4, 42].
Amphetamine wirken selektiv neurotoxisch auf das dopaminerge Neuron. Es kommt zu Schädigungen am Axon und an den Nervenendigungen, die jedoch nicht zwangsläufig zum Zelltod führen [32, 33]. Die Mechanismen sind hinreichend bekannt, und der Erhöhung der intrazellulären Dopaminkonzentration wird dabei eine zentrale Rolle zugeschrieben. Es kommt zur verstärkten Autooxidation von Dopamin mit Bildung von Dopaminchinon und anderen reaktiven Verbindungen [41], die intrazelluläre Schäden durch oxidativen Stress hervorrufen [14, 23] und zusätzlich im Bereich der geschädigten Nervenendigungen eine anhaltende Glutamatfreisetzung erzeugen [46]. Eine dadurch bedingte Aktivierung von NMDA-Rezeptoren mit exzitotoxischer Wirkung wird als weiterer wesentlicher Pathogenitätsfaktor angesehen [25].
Die neurotoxische Wirkung der Amphetamine auf dopaminerge Zellen und die pathophysiologische Schädigungskaskade beim Morbus Parkinson weisen zahlreiche Parallelen auf, angefangen von ähnlichen Genexpressionsmustern, die auf die Aktivierung zellschädigender und proapoptotischer Mechanismen schließen lassen [6, 18, 47], über reaktive Metaboliten und die Beeinflussung der Mitochondrienfunktion [9], bis hin zur gegenseitigen Verstärkung spezifisch neurotoxischer Effekte bei kombinierter Gabe von Amphetaminen und Substanzen, die ein experimentelles Parkinson-Syndrom auslösen [28].
Für Methylphenidat gibt es bislang keinen bedeutsamen Hinweis, dass dadurch vergleichbare toxische Wirkungen wie durch Amphetamine oder andere zur Erzeugung eines experimentellen Morbus Parkinson genutzte Toxine ausgelöst werden. Im Gegenteil, durch Ex-vivo-Studien wurde nachgewiesen, dass Methylphenidat zusätzlich zur DAT-Blockade durch Aktivierung des VMAT intrazellulär zur Umverteilung von Dopamin aus dem Zytosol in die vesikulären Speicher beiträgt [36]. Der VMAT hat eine sehr wichtige Schutzfunktion. In neuronalen Kulturen von VMAT-Knock-out-Mäusen wurde aufgrund der höheren zytosolischen Dopaminkonzentration eine extrem gesteigerte Empfindlichkeit gegenüber Metamphetamin festgestellt [22]. Die Aktivität des VMAT als „Dopamin-Sequestrierer“ ist ein wichtiger neuroprotektiver Faktor. Methylphenidat erhöht die Aktivität des VMAT-2-Proteins, verstärkt die Dopaminspeicherung und schützt die Zelle vor dem instabilen Transmittermolekül [16, 37], während Metamphetamin im gleichen Experiment den diametral entgegengesetzten Effekt hervorruft, der zur Degeneration dopaminerger Nervenendigungen führt [16]. Diese Befunde werden durch In-vivo-Studien an Mäusen gestützt, bei denen die wiederholte Gabe von Methylphenidat über einige Tage eine Verminderung im striatalen Gehalt von Dopamin und DOPAC bewirkt, die jedoch reversibel ist und nicht auf ein spezifisches neurotoxisches Potenzial hindeutet [48]. Demgegenüber führt Amphetamin bei Primaten in therapeutisch relevanten Expositionen zur Schädigung von dopaminergen Nervenendigungen [34].
Die in der Studie von Moll et al. [27] beschriebene Abnahme der striatalen DAT-Dichte ist angesichts der pharmakologischen Wirkungen zu erwarten, denn bei einer selektiven Bindung und Hemmung des DAT durch Methylphenidat muss mit adaptiven Veränderungen dieses Transportproteins gerechnet werden. Sowohl in klinischen als auch in tierexperimentellen Studien ruft Methylphenidat eine Reduktion der DAT-Bindungsdichte hervor [31, 44], die als kompensatorische Down-Regulation anzusehen ist, durch die Aktivierung der Proteinkinase C initiiert wird und als endozytotische Internalisierung des membranständigen Transporters anzusehen ist [26]. Dieser Vorgang geht per se nicht mit neurodegenerativen Veränderungen einher. Die DAT-Bindungsdichte ist damit als alleiniger Biomarker zur Erfassung einer dopaminergen Neurodegeneration, insbesondere nach pharmakologisch bedingter DAT-Blockade, völlig ungeeignet. Zum Nachweis Morbus-Parkinson-ähnlicher Veränderungen am Tier sollte eine gezielte histologische Untersuchung der mesenzephalen dopaminergen Neuronen unter qualitativen und quantitativen Gesichtspunkten erfolgen.
Dieses Beispiel macht deutlich, dass das Studiendesign zur Erfassung und Bewertung neurodegenerativer Arzneimittelwirkungen anhand der erläuterten Kriterien weiter optimiert werden kann. Dennoch haben diese Ergebnisse [27] und die nachfolgende Diskussion deutlich gezeigt, dass trotz einer über Jahrzehnte etablierten und bewährten Therapie letztlich keine endgültige Klarheit über die langfristigen toxischen Auswirkungen der Methylphenidat-Langzeitbehandlung auf das dopaminerge System im ZNS besteht. Tierexperimentelle Langzeitstudien (Applikationszeitraum ≥6 Monate), wie sie heute im Rahmen der präklinischen Arzneimittelentwicklung vor der chronischen Anwendung am Menschen vorgeschrieben sind, fehlen für Methylphenidat weitgehend. Die damaligen Standards zur Testung der Arzneimittelsicherheit sahen solche Studien nicht vor. Dennoch sollten die heutigen Prüfstandards – zumindest theoretisch – auf ihre Tauglichkeit überprüft werden, eine neurodegenerative Arzneimittelwirkung zuverlässig vorherzusagen. Zu diesem Zweck soll der gerade diskutierte Fall einer mesenzephalen dopaminergen Nervenzellschädigung – dem Morbus Parkinson vergleichbar – angenommen und zugrunde gelegt werden.
Erfasst die übliche Toxizitätsprüfung von Arzneimitteln neurodegenerative Veränderungen?
Einer neurodegenerativen Krankheit liegt ein chronisch fortschreitender pathologischer Prozess zugrunde, dessen Symptome erst spät sichtbar werden. Wenn arzneimittelbedingte neurodegenerative Veränderungen gezielt präklinisch untersucht werden sollen, kann das nur im Rahmen so genannter chronischer Toxizitätsstudien erfolgen. Deren wesentliche Prüfanforderungen sind in der Richtlinie CPMP/SWP/1042/99 [12] festgelegt. Die in diesem Dokument enthaltenen Empfehlungen zur ZNS-Untersuchung sind sehr allgemein. Es ist vorgesehen, das ZNS nach einer Standardfärbung histologisch zu untersuchen. Koronale Schnitte aus drei unterschiedlichen Bereichen sollen Aufschluss geben, ob Veränderungen und Schädigungen im Großhirn, im Kleinhirn und im Hirnstamm aufgetreten sind. Nur bei entsprechenden Hinweisen (z.B. Verhaltensauffälligkeiten, ZNS-Funktionsstörungen, histologische Befunde) sind gezielte Folgeuntersuchungen vorgesehen.
Mit dieser Standardprüfung würde eine vorhandene oder womöglich erst im Entstehen begriffene Degeneration mesenzephaler Nervenzellen angesichts der geringen Ausdehnung der betroffenen Kerngebiete mit hoher Wahrscheinlichkeit nicht festgestellt werden. Da vergleichbare neurodegenerative Krankheiten beim Tier nicht auftreten und nur gesunde Tiere für die Toxizitätsprüfungen verwendet werden, die keinen pathogenen Kofaktoren ausgesetzt sind, ist es fraglich, ob degenerative Veränderungen im erforderlichen Behandlungszeitraum von sechs oder neun Monaten überhaupt auftreten würden und ob sie ohne eine Spezialfärbung bei gezielter Untersuchung der Zielzellen auch festzustellen wären. Da eine arzneimittelbedingte Neurodegeneration in den meisten Fällen auf pharmakodynamisch bedingte Wirkungen zurückzuführen ist, könnte das neurodegenerative Potenzial einer Substanz trotz variabler Zielstrukturen und unterschiedlicher Schädigungsmechanismen durch die folgende Ergänzung der Richtlinie CPMP/SWP/1042/99 [12] gezielter als bisher erfasst werden: „Bei ZNS-aktiven Wirkstoffen sind die jeweiligen Zielzellen bzw. die ZNS-Regionen, die aufgrund des Rezeptorprofils dieser Substanz oder anderer substanzbedingter Effekte während einer Therapie direkt beeinflusst werden, in Ergänzung zu den unter Appendix 1 genannten Strukturen gezielt auf toxische Effekte zu untersuchen. Wenn es wissenschaftlich begründete Anhaltspunkte für eine spezifische Neurotoxizität gibt, sind ergänzende Studien – bevorzugt in Form gezielter histopathologischer Untersuchungen der betroffenen Hirngebiete – durchzuführen, die zum Nachweis und zur weiteren Charakterisierung dieser spezifischen Schädigung geeignet sind.“
Diese erweiterten Untersuchungen des Gehirns könnten – ebenfalls ohne die Notwendigkeit zusätzlicher Tierversuche – im Rahmen der gemäß Richtlinie CPMP/SWP/2877/00 [10] erforderlichen Karzinogenitätsstudien durchgeführt werden. Das hätte den Vorteil einer lebenslangen Wirkstoffexposition und würde mögliche weitere Einflussfaktoren im Rahmen der Alterungsprozesse im Organismus erfassen, die von erheblicher klinischer Relevanz sein können, im Rahmen der chronischen Toxizitätsstudien aber nicht ausreichend berücksichtigt sind. In Anlehnung an das vorgestellte Bewertungssystem ist festzustellen, dass diese über die gesamte natürliche Lebensspanne der verwendeten Nagetiere durchgeführten Untersuchungen das Bewertungskriterium „Ontogenese“, das für die Entwicklung einer Neurodegeneration beim Menschen von enormer Bedeutung ist, wesentlich besser berücksichtigen als Studien, die bei ausgewachsenen, aber bei Studienabschluss wesentlich jüngeren Tieren durchgeführt werden und nach einigen Wochen bis Monaten abgeschlossen sind. Natürlich gibt es weit mehr Einflussfaktoren auf die Entstehung und den Verlauf einer Neurodegeneration, daher ist es praktisch unmöglich, einen solchen multifaktoriellen Krankheitsprozess vollständig durch präklinische Modelle zu simulieren. Umfassende Sachkenntnis zur Krankheit und zum verwendeten Testmodell sowie eine geeignete Auswahl von relevanten Biomarkern können die Aussagekraft präklinischer Studien jedoch wesentlich erhöhen.
Schlussfolgerung
Im Zusammenhang mit den präklinischen Untersuchungen, die im Rahmen der Arzneimittelentwicklung durchgeführt werden, wurden einige Schwierigkeiten und Defizite dargestellt, die den Nachweis von neuroprotektiven und neurodegenerativen Substanzwirkungen erschweren oder sogar verhindern können. Diese Probleme sind einerseits den gegenwärtig nur begrenzt verfügbaren und nur bedingt geeigneten Krankheitsmodellen anzulasten, weil dadurch keine zuverlässige Vorhersage von klinisch-neuroprotektiven Wirkungen möglich ist, und sie sind andererseits auf spezifische Mängel im Bereich der standardisierten Sicherheitsprüfung zurückzuführen, die bei zu oberflächlicher Studienplanung und Interpretation von Daten und bei der Verwendung von ungeeigneten Biomarkern zu falsch positiven oder falsch negativen Ergebnissen führen können. Wissenschaftlicher Sachverstand, profunde und umfassende Kenntnis der Datenlage und eine interdisziplinäre Herangehensweise können helfen, die Planung und Durchführung von präklinischen Studien weiter zu optimieren und deren Ergebnisse im jeweils spezifischen Fall richtig zu interpretieren. Die vorgestellten Bewertungskriterien für präklinische Testmethoden, die Auswahl von adäquaten Biomarkern und eine bessere und flexiblere Planung und Vernetzung der experimentellen Untersuchungen zur Pharmakologie und Toxikologie im Einklang mit den klinischen Studien könnten dazu beitragen, die Vorhersage der neuroprotektiven und neurodegenerativen Wirkungen von Arzneimitteln beim Menschen anhand von präklinischen Untersuchungen weit zuverlässiger zu machen, als es gegenwärtig noch der Fall ist.
Erklärung
Diese Arbeit gibt ausschließlich die persönliche Auffassung des Autors wieder, konkrete Rückschlüsse auf die Position des BfArM zu bestimmten Fragen sind daraus nicht ableitbar.
Danksagung
Für die zahlreichen kritischen Anregungen und wertvollen Hinweise bei der Manuskripterstellung möchte ich mich bei allen Beteiligten herzlich bedanken. Mein besonderer Dank gilt Dr. Anke Blumberg und Dr. Ansgar Schulte.
Literatur
1. Albin RL, Young AB, Penney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci 1995;18:63–4.
2. Andreasen N. Brave new brain, Geist – Gehirn – Genom. Berlin, Heidelberg: Springer-Verlag, 2002.
3. Anon. Arznei-Telegramm 2002;33:16.
4. Arbuthnott GW, Fairbrother IS, Butcher SP. Dopamine release and metabolism in the rat striatum: an analysis by ‚in vivo‘ brain microdialysis. Pharmacol Ther 1990;48:281–93.
5. Browne RG, Segal DS. Metabolic and experimental factors in the behavioural response to repeated amphetamine. Pharmacol Biochem Behav 1977;6:545–52.
6. Cadet JL, Jayanthi S, McCoy MT, Vawter M, et al. Temporal profiling of methamphetamine-induced changes in gene expression in the mouse brain: Evidence from cDNA array. Synapse 2001;41:40–8.
7. Carman LS, Gage FH, Shults CW. Partial lesion of the substantia nigra: relation between extent of lesion and rotational behavior. Brain Res 1991;553:275–83.
8. Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: How relevant is the rat? Nature Rev Neurosci 2002;3: 574–9.
9. Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003;302:819–22.
10. European Medicines Agency. Note for guidance on carcinogenic potential (CPMP/SWP/2877/00, www.emea.eu.int/pdfs/human/swp/287700en.pdfhttp://www.emea.eu.int/pdfs/human/swp/287700en.pdf).
11. European Medicines Agency. Note for guidance on non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals (CPMP/ICH/286/95 modification, www.emea.eu.int/pdfs/human/swp/072895en.pdfhttp://www.emea.eu.int/pdfs/human/swp/072895en.pdf).
12. European Medicines Agency. Note for guidance on repeated dose toxicity (CPMP/SWP/1042/99, www.emea.eu.int/pdfs/human/swp/104299en.pdfhttp://www.emea.eu.int/pdfs/human/swp/104299en.pdf).
13. European Medicines Agency. Replacement of animal studies by in vitro models. CPMP/SWP/728/95, www.emea.eu.int/pdfs/human/swp/072895en.pdfhttp://www.emea.eu.int/pdfs/human/swp/072895en.pdf).
14. Giovanni A, Ping Liang L, Hastings TG, Zigmond MJ. Estimating hydroxyl radical content in rat brain using systemic and intraventricular salicylate: Impact of methamphetamine. J Neurochem 1995;64:1819–25.
15. Götz M, Riederer P. Advances in neuroprotection research for neurodegenerative diseases. Adv Exp Med Biol 2004;541:1–19.
16. Hanson GR, Sandoval V, Riddle E, Fleckenstein A. Psychostimulants and vesicle trafficking: A novel mechanism and therapeutic implications. Ann NY Acad Sci 2004;1025:146–50.
17. Ikonomidou C, Bosch F, Miksa M, Bittigau P, et al. al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999;283:70–4.
18. Jayanthi S, Deng X, Bordelon M, McCoy MT, et al. Methamphetamine causes differential regulation of pro-death and anti-death Bcl-2 genes. FASEB J 2001;15:1745–52.
19. Kokmen E, Beard CM, O’Brien PC, Kurland LT. Is the incidence of dementing illness changing? A 25 years time trend study in Rochester, Minnesota (1960–1984). Neurology 1993;43:1887–92.
20. Krause KH, Dresel SH, Krause J, La Fougere C, et al. The dopamine transporter and neuroimaging in attention deficit hyperactivity disorder. Neurosci Biobehav Rev 2003;27:605–13.
21. Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and noradrenaline: comparison with amphetamine. J Neurochem 1997;68:2032–7.
22. Larsen KE, Fon EA, Hastings TG, Edwards RH, et al. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci 2002;22:8951–60.
23. LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci 1999;19:1484–91.
24. Lohse MJ, Lorenzen A, Müller-Oerlinghausen B. Psychopharmaka. In: Paffrath D, Schwabe U (Hrsg.). Arzneiverordnungs-Report 2006. Heidelberg: Springer-Verlag, 2007.
25. Mandel S, Grünblatt E, Riederer P, Gerlach M, et al. Neuroprotective strategies in Parkinson’s disaese. CNS Drugs 2003;17:729–2.
26. Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci 1999;19:7699–710.
27. Moll GH, Hause S, Rüther E, Rothenberger A, et al. Early methylphenidate administration to young rats causes a persistent reduction in the density of striatal dopamine transporters. J Child Adolesc Psychopharmacol 2001;11:15–24.
28. Obata T, Yamanaka Y. Nitric oxide enhances MPP(+)-induced hydroxyl radical generation via depolarization activated nitric oxide synthase in rat striatum. Brain Res 2001;902: 223–8.
29. Os J, Krabbendam L, Myin-Germeys I, Delespaul P. The schizophrenia envirome. Curr Opin Psychiatry 2005;18:141–5.
30. Ravina BM, Fagan SC, Hart RG, Hovinga CA, et al. Neuroprotective agents for clinical trials in Parkinson’s disease. Neurology 2003;60:1234–40.
31. Reneman L, De Bruin K, Lavalaye J, Gunning WB, et al. Addition of a 5-HT receptor agonist to methylphenidate potentiates the reduction of [123I]FP-CIT binding to dopamine transporters in rat frontal cortex and hippocampus. Synapse 2001;39:193–200.
32. Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, et al. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res 1982;235:93–103.
33. Ricaurte GA, Seiden LS, Schuster CR. Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res 1984;303:359–64.
34. Ricaurte GA, Mechan AO, Yuan J, Hatzidimitriou G, et al. Amphetamine treatment similar to that used in the treatment of attention-deficit/hyperactivity disorder damages dopaminergic nerve endings in the striatum of adult nonhuman primates. J Pharmacol Exp Ther 2005;315:91–8.
35. Robertson ED, Mucke L. 100 years and counting: Prospects for defeating Alzheimer’s disease. Science 2006;314:781–4.
36. Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate redistributes vesicular monoamine transporter-2: Role of dopamine receptors. J Neurosci 2002;22:8705–10.
37. Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J Pharmacol Exp Ther 2003;304:1181–7.
38. Schapira AHV, Olanow CW. Neuroprotection in Parkinson disease. Mysteries, myths, and misconceptions. JAMA 2004;291:358–64.
39. Schneider CK, Kalinke U, Löwer J. TGN1412-A regulator’s perspective. Nat Biotechnol 2006;24:493–6.
40. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatry 2004;16:260–83.
41. Smythies J, Galzigna L. The oxidative metabolism of catecholamines in the brain: a review. Biochem Biophys Acta 1998;1380:159–62.
42. Sulzer D, Chen TK, Lau YY, Kristensen H, et al. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci 1995;15: 4102–8.
43. Verordnung (EG) Nr. 1901/2006 des Europäischen Parlaments und Rates vom 12.12.2006 über Kinderarzneimittel und zur Änderung der Verordnung (EWG) Nr. 1768/92, der Richtlinien 2001/20/EG und 2001/83/EG sowie der Verordnung (EG) Nr. 726/2004. Amtsblatt der Europäischen Union L 378/1-19 vom 27.12.2006.
44. Vles JS, Feron FH, Hendricksen JG, Jolles J, et al. Methylphenidate down-regulates the dopamine receptor and transporter system in children with attention deficit hyperkinetic disorder. Neuropediatrics 2003;34:77–80.
45. Wilber K. Integrale Psychologie. Freiamt: Arbor Verlag, 2001.
46. Wolf ME, Xue CJ, Li Y, Wavak D. Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem 2000;75:1634–44.
47. Xie T, Tong L, Barrett T, Yuan J, et al. Changes in gene expression linked to methamphetamine-induced dopaminergic neurotoxicity. J Neurosci 2002;22:274–83.
48. Yuan J, McCann U, Ricaurte G. Methylphenidate and brain dopamine neurotoxicity. Brain Res 1997;767:172–5.
Dr. med. Torsten Reum, Bundesinstitut für Arzneimittel und Medizinprodukte, Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, E-Mail: t.reum@bfarm.de
Neuroprotective and neurodegenerative drug effects – which role do preclinical data play for their detection?
Preclinical studies provide essential information about the effects and safety of drugs, and a battery of specific preclinical tests is required for the approval of clinical trials as well as for drug marketing authorization. However, validated preclinical tests to predict neuroprotection and neurodegeneration by drugs do not exist. The available experimental models are not capable of mimicking the progression of neurodegenerative diseases, and clinical neuroprotection or neurodegenerative drug effects are hardly predictable for this reason. Therefore, the present paper deals with the conditions under which preclinical studies are more suitable for detection and characterization of neuroprotective and neurodegenerative drug effects. Preclinical disease models should be checked and compared by certain assessment criteria, such as the degree of complexity, comparability with human physiology and pathophysiology and adequate consideration of ontogenic development for disease progression. Another essential criterion for the efficient interpretation of preclinical data is the selection of suitable biomarkers for the experimental question of concern. By a relevant example it should be demonstrated that these criteria allow a well-founded assessment of a drug’s desired effects and safety, and in the framework of such a preclinical safety testing specific neurodegenerative effects could be detected more reliably than it is common practice today.
Keywords: Preclinical drug development, animal models, neuroprotection, neurodegeneration
Psychopharmakotherapie 2007; 14(05)