Gabriele Blaeser-Kiel, Hamburg
Das Lennox-Gastaut-Syndrom (LGS) gehört zu den schwersten Epilepsiesyndromen. Der Beginn liegt in der Regel zwischen dem dritten und fünften Lebensjahr. Ursache ist in 70% der Fälle eine Enzephalopathie unterschiedlicher Genese, die in einem sehr hohen Prozentsatz mit Entwicklungsverzögerungen oder Störungen von Kognition, Affekt und Verhalten einhergeht. Charakteristisch ist die Vielfalt der auftretenden Anfälle. Für die Diagnosestellung obligat sind tonische Anfälle (vorwiegend im Schlaf) und so genannte „drop attacks“ (atonische, myoklonische, myoklonisch-astatische, tonisch-klonische Sturzanfälle). Etwa zwei Drittel der Patienten leiden zusätzlich an atypischen Absencen und/oder rezidivierenden Status epileptici.
Die LGS-Prävalenz ist niedrig. Schätzungen zufolge sind in der Europäischen Union zwischen 46000 bis 92000 Menschen betroffen, in Deutschland wahrscheinlich nicht mehr als 2000. Die Langzeitprognose ist schlecht. Zu einer Remission mit vollständiger Anfallsfreiheit und normaler geistiger Entwicklung kommt es sehr selten. Die Auswahl an zugelassenen Therapieoptionen ist schmal: Felbamat (Taloxa®) gilt als sehr wirksam, hat aber den Nachteil einer starken Hepato- und Hämotoxizität, weshalb engmaschige Laborkontrollen erforderlich sind. Alternativen sind Lamotrigin (z.B. Lamictal®) und Topiramat (Topamax®), wobei bei Topiramat das Risiko besteht, dass die bereits eingeschränkten kognitiven Fähigkeiten noch weiter verschlechtert werden. Ebenfalls häufig eingesetzt wird Valproinsäure (z.B. Orfiril®).
Mit Rufinamid (Abb. 1) steht jetzt eine Substanz zur Verfügung, die von der europäischen Arzneimittelbehörde EMEA die Zulassung exklusiv für die Indikation „Lennox-Gastaut-Syndrom“ erhalten hat. Der Einsatz ist ab dem vierten Lebensjahr im Add-on-Regime möglich. Der Wirkstoff ist ein Triazolderivat, das strukturell nicht mit anderen Antiepileptika verwandt ist. Es moduliert die Aktivität von Natriumkanälen und verlängert so deren inaktives Stadium. Man vermutet, dass dadurch eine Ausbreitung der Anfallsaktivität vom epileptogenen Fokus blockiert wird. Der genaue Wirkungsmechanismus ist jedoch unbekannt. Die Metabolisierung von Rufinamid erfolgt hauptsächlich durch Hydrolyse, also unabhängig von Cytochrom-P450-haltigen Enzymen oder Konjugationsreaktionen.
Abb. 1. Rufinamid
Wie jedes neue Antikonvulsivum wurde Rufinamid zunächst als Zusatztherapie bei Patienten mit refraktären fokalen Anfällen untersucht. In Hinblick auf den therapeutischen Effekt waren diese Studien mit insgesamt mehr als 1000 Patienten zwar weniger erfolgreich als erhofft, die Daten bieten aber eine solide Basis für Aussagen zur Sicherheit und Verträglichkeit. Die häufigsten unerwünschten Ereignisse waren passagere Schläfrigkeit und Übelkeit mit Erbrechen, die aber selten zum Abbruch der Studienmedikation führten. Klinisch sehr relevant war die Beobachtung, dass die Inzidenz psychiatrischer Nebenwirkungen inklusive kognitiver Beeinträchtigung auf Plazebo-Niveau lag.
Weniger Anfälle bei LGS
Die Zulassung von Rufinamid zur Behandlung von LGS beruht auf den Ergebnissen einer randomisierten Doppelblindstudie, die weltweit in spezialisierten Epilepsiezentren durchgeführt wurde. Eingeschlossen worden waren 138 LGS-Patienten im mitteren Alter von 14 Jahren (zwischen 4 und 30 Jahren), bei denen im Monat zuvor trotz stabiler Einstellung auf bis zu drei Antiepileptika mindestens 90 Anfälle in der für das Krankheitsbild typischen Vielfalt aufgetreten waren. Zusätzlich zu ihrer bisherigen Therapie erhielten die Patienten Rufinamid oder Plazebo. Bei den Patienten der Verum-Gruppe wurde die Rufinamid-Tagesdosis innerhalb von zwei Wochen auf eine Zieldosis von 45 mg/kg eingestellt.
Am Ende der zwölfwöchigen Doppelblindphase hatte im Rufinamid-Arm (versus Kontrollgruppe) im Median die Frequenz
l aller Anfälle um 32,7% vs. –11,7% (p=0,0015),
l der Sturzanfälle („drop attacks“) um 42,5% vs. +1,4% (p<0,0001),
l der typischen und atypischen Absen-cen um 56% vs. –36% (p=0,022)
abgenommen.
In der anschließenden Verlängerungsstudie wurde 123 Patienten offen bis zu drei Jahre (im Median 432 Tage) mit Rufinamid weiterbehandelt (Abb. 2). In dieser Zeit gab es keine Hinweise auf eine Toleranzentwicklung. Der Anteil der Responder (mindestens 50%ige Reduktion der Gesamtanfallsfrequenz) stieg sogar leicht von 31% nach drei Monaten auf 37% während der Fortsetzungsstudie.
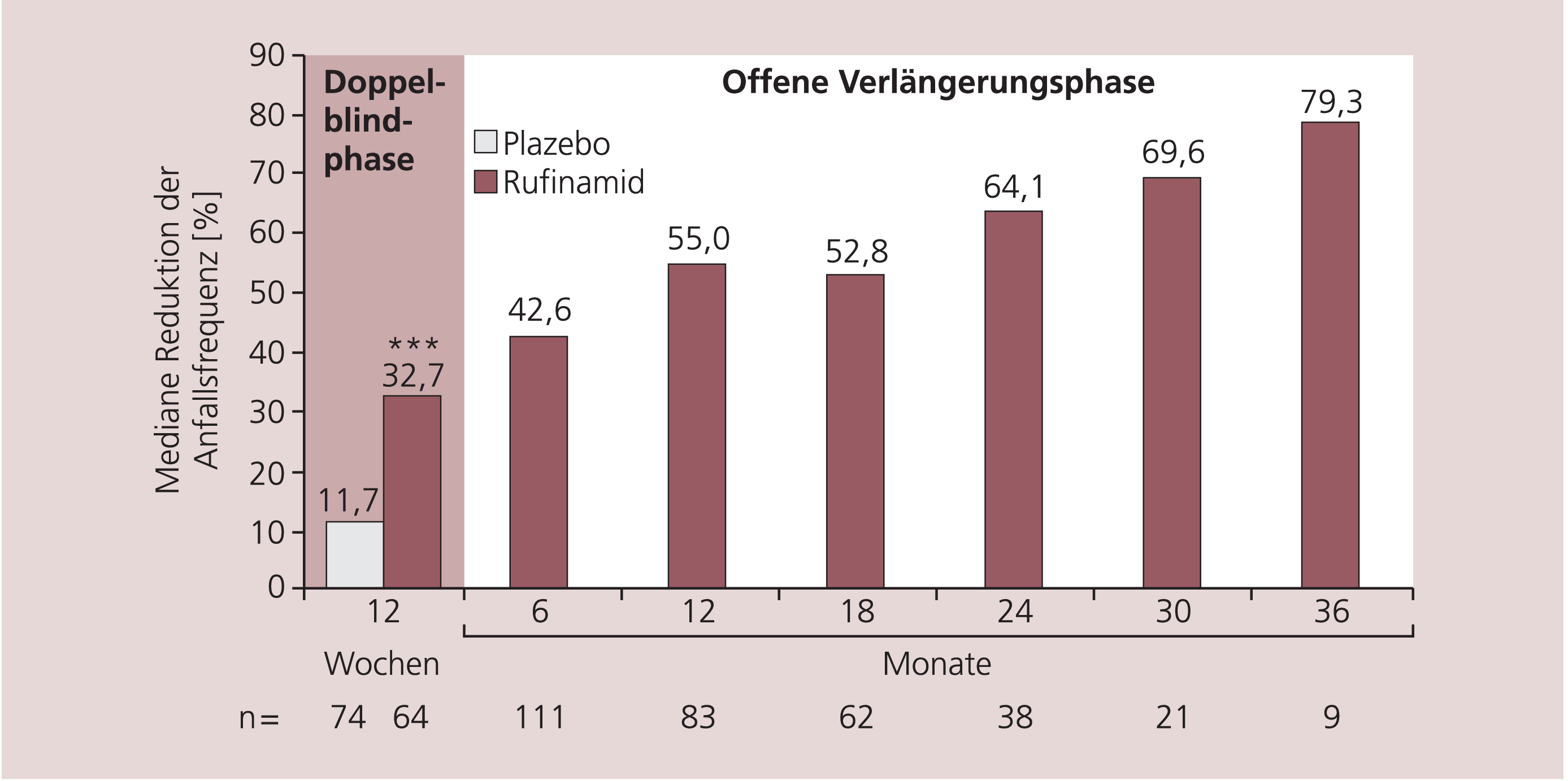
Abb. 2. Reduktion der Frequenz aller Anfälle bei Patienten mit Lennox-Gastaut-Syndrom nach Zusatzbehandlung mit Rufinamid zu einer Basistherapie mit einem bis drei Antiepileptika [nach Glauser]
Quellen
Prof. Dr. med. Martha Feucht, Wien, Dr. med. Günter Krämer, Zürich, „Meet the Expert Session“, veranstaltet von der Eisai GmbH bei der 5. Gemeinsamen Jahrestagung der Deutschen, Österreichischen und Schweizerischen Sektionen der Internationalen Liga gegen Epilepsie, Basel, 18. Mai 2007.
Glauser TA, et al. Efficacy and safety of rufenamide adjunctive therapy in patients with Lennox-Gastaut syndrome (LGS): a multicenter, randomized, double-blind, placebo-controlled, parallel trial. Neurology 2005;64:1862.
Glauser TA, et al. Open-label extension study of the efficacy and safety of rufenamide adjunctive therapy in patients with Lennox-Gastaut syndrome. Epilepsia 2005;46(Suppl 6):408.
Psychopharmakotherapie 2007; 14(05)