Walter K. Schwerdtfeger, Bonn
Über „innovative Arzneimittel“ wird in Kreisen der Fachöffentlichkeit und Fachpolitik häufig gesprochen und geschrieben. Bei der vergleichenden Betrachtung entsprechender Äußerungen und Beiträge wird jedoch deutlich, dass dieser Begriff keineswegs in einheitlicher Weise verwendet wird. Dies liegt nicht nur an einem Spektrum unterschiedlicher Sichtweisen, das sich von der uneingeschränkt positiven Erwartung größerer Behandlungserfolge einerseits bis zur Sorge über das Nebenwirkungsrisiko neuartiger Produkte andererseits erstreckt, sondern auch daran, dass die Vorstellungen davon, was den Neuheitswert eines Arzneimittels ausmacht, nicht einheitlich sind.
Versteht man unter „Arzneimittelinnovation“, dass ein Produkt beispielsweise mit bisher nicht bekannten Bestandteilen, in einer bis dahin nicht verfügbaren Zusammensetzung oder Form, auf der Grundlage eines neuartigen Herstellungsverfahrens oder für eine bisher nicht zugelassene Indikation auf den Markt gebracht wird, so ist dies eine plausible, neutrale und gleichzeitig wertfreie Definition. Nach einer Umfrage des Verbands Forschender Arzneimittelhersteller (VFA) von August 2005 bearbeiteten seine Mitgliedsunternehmen zu diesem Zeitpunkt 316 Arzneimittelprojekte (Abb. 1), die in diesem allgemeinen Sinne als innovativ gelten können und von denen erhofft wird, dass sie bis zum Jahr 2009 zu einer Neuzulassung oder zur Indikationserweiterung eines bereits zugelassenen Arzneimittels führen [20].
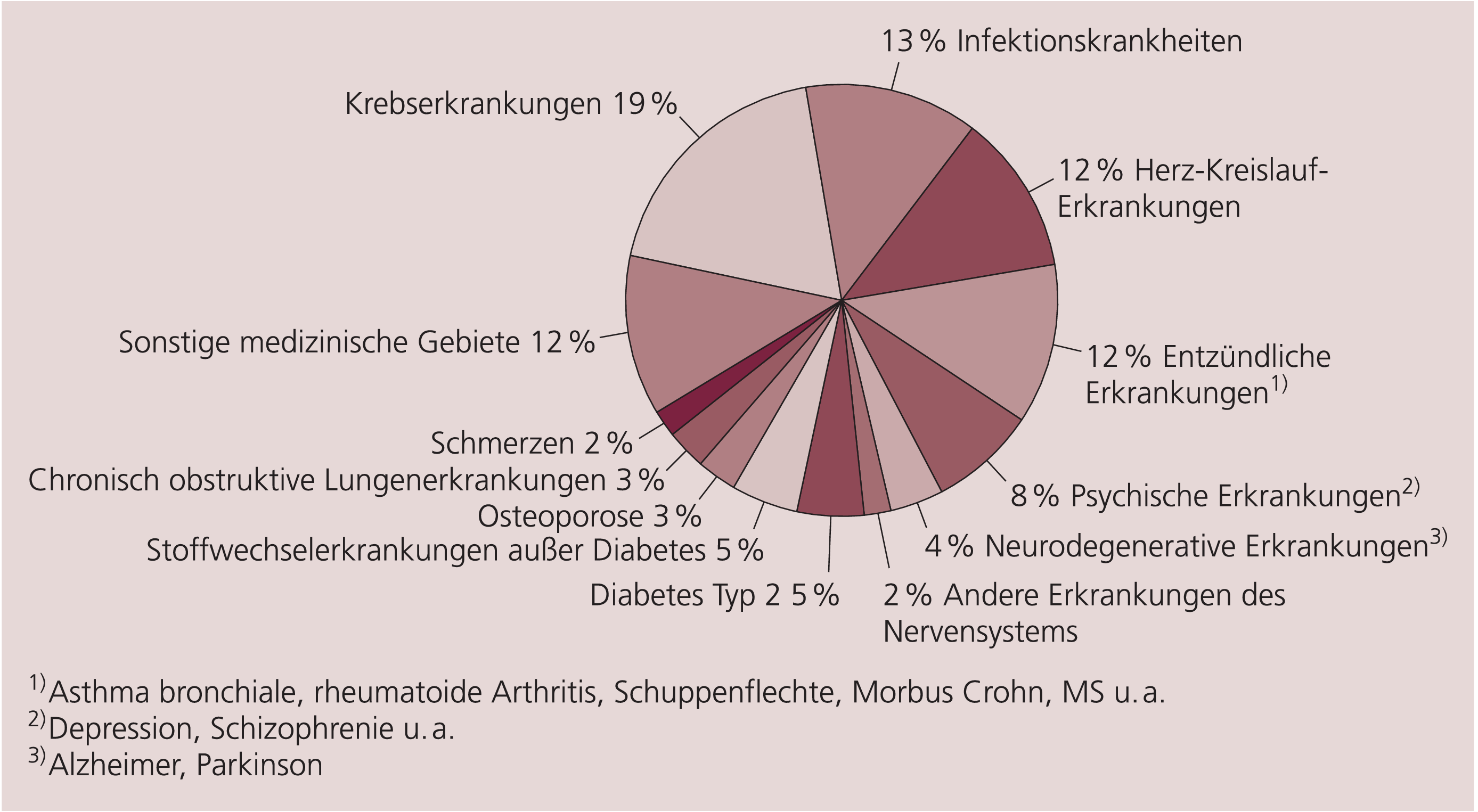
Abb. 1. Verteilung der vorgesehenen Indikationsgebiete fortgeschrittener Arzneimittelentwicklungen (316 Projekte) der Mitgliedsunternehmen des Verbands Forschender Arzneimittelhersteller e.V.; die behördliche Zulassung entsprechender Arzneimittel bis zum Jahr 2009 wird angestrebt [Quelle: VFA, Stand August 2005]
Im Folgenden soll auf den Umgang mit dem Innovationsbegriff, Nutzen und Risiken von Innovationen, einige ökonomische Hintergründe, die Akzeptanz in der Bevölkerung und mit diesen Fragen verbundene Erwartungen an Politik und Bundesregierung eingegangen werden.
Innovationen im Dienste der öffentlichen Gesundheit
Vor allem im politischen Raum wird der Begriff „Innovation“ differenzierter betrachtet. Die Förderung pharmazeutischer Innovationen ist ein erklärtes Ziel der Bundesregierung, das in die Koalitionsvereinbarung der Regierungsfraktionen aufgenommen worden ist. Als förderungswürdige, „echte“ Innovation betrachtet die Bundesregierung allerdings nur solche neuartigen Arzneimittel, Wirkstoffe oder Kombinationen, die nicht nur in irgendeiner Weise anders sind als die bisher auf dem Markt verfügbaren, sondern sich gegenüber diesen auch durch einen therapeutischen Zusatznutzen auszeichnen. Dem gegenüber steht wiederum der Begriff des „neuartigen“ Arzneimittels nach den Entscheidungsgrundlagen des Gemeinsamen Bundesausschusses zur Festbetragsgruppenbildung vom 15. November 2005 (www.g-ba.dewww.g-ba.de). Danach gelten Arzneimittel als „neuartig“, solange sie unter Patentschutz stehen.
Nicht als innovativ im Sinne der förderungswürdigen Arzneimittel gelten solche, die einem bereits verfügbaren in seinen Wirkungen und Nebenwirkungen gleichwertig sind und lediglich in einer neuen Molekülvariante oder nach einem anderen Verfahren hergestellt werden, selbst wenn dieses Verfahren an sich innovativ sein sollte und Patentschutz besteht. Ein Interesse von Politik und Bundesregierung, Anreize für die Entwicklung eines solchen Produkts zu setzen oder seine Anwendung durch geeignete Regelungen zur Gesetzlichen Krankenversicherung (GKV) zu unterstützen, könnte allenfalls darin liegen, dass das mit dem neuen Verfahren hergestellte Produkt merklich kostengünstiger wäre als die bisher verfügbaren und insoweit noch besser als diese das Gebot der Wirtschaftlichkeit der Versorgung im Sinne von §12 des Fünften Buchs Sozialgesetzbuch (SGB V) erfüllen würde.
Der erwünschte therapeutische Zusatznutzen dagegen kann sich in einer besseren Wirksamkeit, einer Verringerung der Nebenwirkungen oder anderen Kriterien wie beispielsweise der erleichterten Anwendung eines Arzneimittels bei der Behandlung eines Leidens äußern. Arzneimittel, die in diesem Sinne eine therapeutische Verbesserung darstellen, sind von Preisbegrenzungen aufgrund der Festbetragsgruppenbildung nach §35 Abs.1 SGB V ausgenommen (s. Entscheidungsgrundlagen des Gemeinsamen Bundesausschusses zur Festbetragsgruppenbildung vom 15. November 2005, www.g-ba.de).
Der jährlich erscheinende Arzneiverordnungs-Report des Wissenschaftlichen Instituts der Ortskrankenkassen gibt unter anderem Auskunft über Neuentwicklungen und die Bewertung ihres therapeutischen Nutzens (Tab. 1). Er hat beispielsweise von 33 im Jahr 2004 in Deutschland erstmals in die Therapie eingeführten Wirkstoffen 15 als „innovativ“, drei als „Verbesserungen“ und die verbleibenden 15 als Analogpräparate ohne relevanten Vorteil, also ohne therapeutischen Zusatznutzen gegenüber bekannten Arzneimitteln bezeichnet [8]. Der Anteil von Analogpräparaten in 33 untersuchten Arzneimittelgruppen am Gesamtmarkt stieg zwischen 1993 und 2003 von 5,5% auf 19,8% [16], sank aber im Folgejahr geringfügig auf 18,5% [17].
Tab. 1. Anzahl und Bewertung des Innovationsgehalts von in den Jahren 2000–2004 neu in den Markt eingeführten Wirkstoffen [nach Angaben im Arzneiverordnungsreport des Wissenschaftlichen Instituts der Ortskrankenkassen, Jahrgänge 2001 bis 2005]
|
2000 |
2001 |
2002 |
2003 |
2004 |
|
|
Innovationen |
13 |
15 |
10 |
7 |
15 |
|
Verbesserung |
2 |
9 |
7 |
5 |
3 |
|
Analogpräparate |
16 |
9 |
11 |
5 |
15 |
|
Neue Wirkstoffe insgesamt |
31 |
33 |
28 |
17 |
33 |
Neue Behandlungsmöglichkeiten
Die Hoffnung vieler Patienten richtet sich darauf, dass die gemeinsamen Anstrengungen von Grundlagenforschung, Medizin und Pharmaindustrie Möglichkeiten zur Therapie von bisher nicht wirksam behandelbaren Krankheiten erschließen. Nach Schätzungen ist von etwa 30000 definierten Krankheiten, davon bis rund 50% mit genetischem (Teil-)Bezug, höchstens ein Drittel wirksam behandelbar. Die Dimension des Bedarfs an therapeutischem Fortschritt bei häufigen, nicht hinreichend behandelbaren Erkrankungen wird schon anhand einiger weniger Zahlen erkennbar, wie zum Beispiel der Prävalenz des Diabetes mellitus (ca. 4 Mio.) [10], von Altersdemenzerkrankungen (ca. 1 Mio.) [21] und der Inzidenz von bösartigen Neubildungen (ca. 425000) [1] in Deutschland. Erst recht deutlich wird die Dringlichkeit von Innovationen bei der Betrachtung weltweit auftretender Krankheiten und ihrer Auswirkungen, vor allem Malaria, Tuberkulose und AIDS, deren Verbreitung bisher auch durch die vielfältigen Anstrengungen nationaler und internationaler Organisationen kaum gebremst werden konnte.
Vor allem Fortschritte in den Zukunftsdisziplinen Pharmakogenetik und Pharmakogenomik sowie die erwarteten neuen Ansätze aus der somatischen Zelltherapie, Gentherapie, dem „tissue engineering“ und anderen sollten mittel- bis langfristig dazu führen, dass bestimmte Erkrankungen grundsätzlich erfolgreicher und nebenwirkungsärmer behandelt werden können, ferner auch dazu, dass Therapiemaßnahmen immer individueller und damit treffsicherer auf den jeweiligen Patienten zugeschnitten werden können. Auf den einzelnen Patienten abgestimmte Behandlungsstrategien bestehen allerdings auch heute schon vereinzelt im Rahmen etablierter arzneimitteltherapeutischer Verfahren. Sie werden mit Erfolg eingesetzt, wie die Erfahrungen bei der Behandlung bestimmter, aggressiver Formen des Brustkrebses beispielhaft belegen.
Unerwünschte Arzneimittelwirkungen (UAW)
Der Begriff bezeichnet schädliche und unbeabsichtigte Reaktionen beim Patienten auf Arzneimittel, die bei Dosierungen auftreten, wie sie üblicherweise zur Prophylaxe, Diagnose und Therapie von Krankheiten oder zur Veränderung physiologischer Funktionen eingesetzt werden. Es wird geschätzt, dass etwa 5% der Arzneimittelanwendungen zu unerwünschten Wirkungen führen [18]. Krankenhauseinweisungen sollen zu etwa 3 bis 6% auf UAW zurückgehen und direkte Kosten von etwa 400 Mio. Euro in Deutschland [15] und 4 Mrd. Dollar in den USA [12] verursachen. Die betreffenden UAW werden zu etwa 40% als vermeidbar bewertet [4]. In Norwegen wurde in einer prospektiven Studie ermittelt, dass etwa 1% der in ein großes Krankenhaus der Inneren Medizin aufgenommenen Patienten infolge der dort erhaltenen Arzneimitteltherapie verstarben [5], wobei allerdings nicht sicher differenzierbar erscheint, in welchem Umfang diese Todesfälle im Rahmen des bestimmungsgemäßen Gebrauchs oder aufgrund von Behandlungsfehlern entstanden sind.
Aus diesen Zahlen wird deutlich, dass ein großer Bedarf an Neu- oder Weiterentwicklungen mit einem reduzierten Nebenwirkungsspektrum vor allem bei Arzneimittelgruppen, die häufig verordnet werden und deren Anwendung mit schwerwiegenden UAW verbunden sein kann, besteht. Dies gilt ungeachtet der Tatsache, dass auch das Verordnungsverhalten des Behandlers einen gewissen Einfluss auf die Häufigkeit von UAW hat, zumal diese auch zwischen Arzneimitteln derselben Gruppe variieren kann.
Erleichterte Anwendung
Die Darreichung von Arzneimitteln in einer Form, die eine möglichst einfache Anwendung beim Patienten oder durch den Patienten erlaubt, verspricht nicht nur bei diesem eine höhere Compliance, sondern auch eine Verringerung der Häufigkeit von Anwendungsfehlern. Aktuelle Beispiele für Arzneimittel mit entsprechenden Vorteilen sind das im vergangenen Jahr von der Europäischen Kommission zugelassene Exubera® (das erste Insulin, das auf dem Wege der Inhalation verabreicht werden kann), die neue galenische Zubereitung des Tumorwirkstoffs Cytarabin (der im Vergleich mit bisherigen Präparaten in einer deutlich verringerten Frequenz in den Rückenmarksliquor zu injizieren ist) oder auch der vor kurzem entwickelte orale Cholera-Impfstoff Dukoral®, dessen Wirksamkeit von der WHO anerkannt wurde. Hier handelt es sich zweifelsfrei um „echte“ Innovationen; ihre Verbreitung in der therapeutischen Praxis und ihre Bewertung aus Sicht der GKV werden allerdings von einer Gesamtbewertung unter Einbeziehung von Nebenwirkungen und Kosten abhängen. Dass die entsprechenden Hürden hoch sind, zeigen der Rapid Report des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) vom 4. Mai 2006 zu Exubera® (s. unter www.iqwig.de/publikationen-des-iqwig.114.html) und andere unabhängige Bewertungen [13].
Innovation erfordert Forschung und Entwicklung
Medizinische Innovationen entstehen als Produkt langjähriger und überaus kostenintensiver Forschungs- und Entwicklungsarbeiten. Der VFA beziffert den durchschnittlichen Zeitaufwand bis zur Marktreife eines Arzneimittels auf etwa 12 Jahre und die entsprechenden Kosten auf etwa 800 Mio. Euro [20]. (Zu Zahlen dieser Art gibt es allerdings, abhängig von den gewählten Berechnungsgrundlagen, durchaus abweichende Auffassungen, die zu erheblich niedrigeren Kostenschätzungen führen.) Die Zahl der jährlich am deutschen Markt eingeführten Arzneimittel mit neuen Wirkstoffen variiert deutlich (z.B.: 44 im Jahr 1997, 17 im Jahr 2003). Im Jahr 2005 waren es 23 Arzneimittel, bei denen es sich mehrheitlich um Medikamente gegen Krebserkrankungen, neurologische und psychische Erkrankungen, Herz-Kreislauf-Erkrankungen und Stoffwechselkrankheiten handelte [19].
An mehr als 80% der derzeit laufenden Neuentwicklungen, die von den überwiegend global tätigen, auch in Deutschland ansässigen forschenden Unternehmen finanziert werden, sind deutsche Kliniken und Praxen beteiligt. Dies spricht für eine nach wie vor wichtige Rolle der in Deutschland ansässigen klinischen Forschung. Offenbar hat sich daran auch durch das Inkrafttreten der Vorschriften der europäischen Richtlinie 2001/20/EG zur Durchführung klinischer Prüfungen [14] und ihre Umsetzung in nationales Recht (§§40–42a des Arzneimittelgesetzes [2] und Rechtsverordnung über die Durchführung klinischer Prüfungen [3]) nichts geändert, die in Teilen der Forschungslandschaft als erhebliche Verschlechterung der Rahmenbedingungen für klinische Prüfungen an Universitäten („investigator-initiated trials“) bezeichnet worden ist. Dazu ist anzumerken, dass die entsprechenden Vorschriften in erster Linie auf den Schutz der Probanden oder Patienten und die Steigerung der Forschungsqualität zielen. Es kann nicht bestritten werden, dass diese Ziele auch unter dem Gesichtspunkt der Arzneimittelsicherheit vorrangig sind gegenüber anderen Erwägungen. Nach Auffassung der Fördereinrichtungen Bundesministerium für Bildung und Forschung (BMBF) und Deutsche Forschungsgemeinschaft (DFG) wird die Anwendung der neuen Vorschriften die wissenschaftliche Qualität der in Deutschland durchgeführten klinischen Studien weiter erhöhen. Für den Bereich der „investigator-initiated trials“ werden ergänzende Regelungen in Form einer europäischen Leitlinie erwartet, die zurzeit noch zwischen den zuständigen Vertretern der EU-Mitgliedstaaten abgestimmt wird.
Als industrieller Forschungs- und Entwicklungs(FuE)-Standort wird Deutschland, vor allem im Inland selbst, häufig eher negativ bewertet. Ein aktuelles Expertengutachten bestätigt einerseits, dass der Anteil der FuE-Ausgaben der deutschen pharmazeutischen Industrie an den Gesamtausgaben der OECD-Länder für FuE gesunken ist, stellt andererseits jedoch fest, dass sich das Wachstum dieser Ausgaben in Deutschland seit der zweiten Hälfte der 90er Jahre beschleunigt hat und über dem europäischen Durchschnitt liegt. Zusammenfassend sieht das Gutachten Deutschland weiterhin als wichtigen Standort, in dem jedoch FuE-Potenzial und Wertschöpfungsmöglichkeiten auch seitens der Industrie weniger erfolgreich genutzt werden als in den wichtigsten Konkurrenzländern [11].
Unterstützende Maßnahmen des Bundes
Die Bundesregierung hat eine Reihe unterstützender Maßnahmen für die Forschung und Entwicklung im Arzneimittelbereich beschlossen. Das BMBF und die DFG konnten den Finanzrahmen ihres Förderprogramms für klinische Studien verdoppeln; zunächst bis zum Jahr 2008 stehen damit für die Förderung jährlich 20 Mio. Euro zur Verfügung [6]. Die von der Bundesministerin für Gesundheit eingerichtete Task Force Pharma verfolgt Bestrebungen, die rechtlichen und wirtschaftlichen Rahmenbedingungen für die Forschung im Bereich der Biotechnologie zu verbessern. Insgesamt sollen nach dem Koalitionsvertrag von CDU/CSU und SPD aus dem Jahr 2005 die Ausgaben des Bundes für Forschung und Entwicklung bis zum Jahre 2010 auf etwa 3% des Bruttoinlandsprodukts anwachsen. Dabei wurden Maßnahmen im Dienste der Prävention und Therapie geriatrischer und degenerativer Erkrankungen als besonderer fachlicher Schwerpunkt („Leuchtturmprojekt“) genannt.
Rund ein Viertel der zusätzlichen Investitionen soll unmittelbar als Prämieninstrument verwendet werden, um für erfolgreiche Wissenschaftler weitere Anreize zu bieten. Darüber hinaus hat die Bundesregierung eine „Exzellenzinitiative zur Förderung von Wissenschaft und Forschung an deutschen Hochschulen“ gestartet, der bis zum Jahr 2011 etwa 1,9 Mrd. Euro zur Verfügung stehen werden. Sie umfasst den Ausbau der Begabtenförderung, die Bildung von Schwerpunktregionen („Clustern“) zur Vernetzung anwendungsbezogener Forschung mit industrieller Entwicklung sowie die Entwicklung von Zukunftskonzepten der Spitzenforschung.
Staatliche Förderung könnte ferner unmittelbar dort ansetzen, wo medikamentöse Therapien nicht oder nur unzureichend verfügbar sind, weil ihre Entwicklung der pharmazeutischen Industrie keinen entsprechenden wirtschaftlichen Nutzen verspricht. Die Einführung entsprechender, wirksamer Fördermechanismen, die auch die Unterstützung marktnaher Entwicklungsarbeiten nicht aussparen dürfen, liegt im unmittelbaren Interesse der öffentlichen Gesundheit. Die verfügbaren, als Summe betrachtet nicht unerheblichen öffentlichen Fördermittel sollten möglichst aus einer Hand koordiniert und vergeben werden, außerdem vorzugsweise dort zum Einsatz kommen, wo sie zeitnahe Ergebnisse versprechen.
Eine entsprechende, zielgerichtete Förderung von Arzneimittelinnovationen kann sich in Form vermiedener Krankheitskosten (durch rascher und/oder nachhaltiger wirksame Behandlung sowie Vermeidung kostenträchtiger Nebenwirkungen) positiv auf die finanzielle Bilanz des Gesundheitswesens und die Volkswirtschaft insgesamt auswirken.
Ökonomische Vorteile von Arzneimittelinnovationen
Die Krankheitskostenrechnung des Statistischen Bundesamts (2002; siehe www.gbe-bund.de) weist für Deutschland einen Gesamtbetrag von 223,6 Mrd. Euro „direkter Krankheitskosten“ (medizinische Heilbehandlung, Prävention, Rehabilitation, Pflege einschließlich der Verwaltungskosten der Leistungserbringer und der die Gesundheitsleistungen finanzierenden Einrichtungen) aus. Davon entfielen etwa 16% auf Herz-Kreislauf-Erkrankungen, 14% auf Krankheiten des Verdauungssystems, 11% auf Krankheiten des Muskel-Skelett-Systems und 10% auf psychische Erkrankungen und Verhaltensstörungen. Der Umsatz mit verschreibungspflichtigen und verschreibungsfreien Arzneimitteln in Deutschland lag in demselben Jahr bei rund 30 Mrd. Euro (2004: 32,5 Mrd. Euro) und entsprach somit etwa 13% der Krankheitskosten.
Arzneimittelinnovationen, die zu tatsächlichen therapeutischen Fortschritten führen, können über ihre Bedeutung für die Gesundheit der Bevölkerung hinaus in großem Umfang zur Vermeidung direkter und indirekter Krankheitskosten (Verluste an Lebensarbeitszeit, hohe Krankenhaus- und Pflegekosten und andere negative Auswirkungen auf die Volkswirtschaft) beitragen. Die hohe wirtschaftliche Bedeutung von Arzneimittelinnovationen für die gesetzliche Krankenversicherung (GKV) lässt sich anhand einiger Entwicklungen der letzten Jahre eindrucksvoll belegen [9]:
Bei der Therapie des Magen- und Duodenalulkus führte schon die Anwendung von H2-Rezeptorantagonisten seit den 70er Jahren des vergangenen Jahrhunderts zu einer Halbierung der Anzahl von Operationen. Als noch wirksamer erwiesen sich dann die Protonenpumpenhemmer, und zu einem tatsächlichen Durchbruch im Sinne einer kausalen und heilenden Therapie kam es vor etwa 15 Jahren mit der Einführung der Kombinationsbehandlung unter Einschluss von Antibiotika, heute meist als Dreierkombination, zur Eradikation von Helicobacter pylori. Der sehr gute Behandlungserfolg dieses Verfahrens geht einher mit einer wesentlich geringeren 5-Jahres-Rezidivrate (5–10%) im Vergleich mit anderen medikamentösen Therapien. Die in der Regel überlegene Wirksamkeit der Eradikationsbehandlung gegenüber anderen verfügbaren Verfahren senkt die direkten Kosten der Behandlung eines Magen-Darm-Ulkus um etwa 70%. Hier dürften der Volkswirtschaft bereits Kosten in Milliardenhöhe erspart worden sein, gleichwohl sind für die Krankheitskostenrechnung noch weitere Einsparungen möglich, wenn die Anwendungshäufigkeit dieser innovativen Therapie noch weiter gesteigert werden kann.
Gut berechenbare wirtschaftliche Effekte erbringt beispielsweise auch die von einer nachsorgenden Immunsuppression gefolgte Nierentransplantation gegenüber der langjährigen Dialyse. Zu den Transplantationskosten von rund 50000 Euro kommen für die Kosten der Behandlung mit Immunsuppressiva etwa 10000 Euro jährlich, während die Dialyse allein jährlich etwa 45000 Euro erfordert. Hier sind Kostenvorteile bereits nach Ablauf des zweiten Jahrs zu erkennen, wobei einzuräumen ist, dass die Zahl der Transplantationen nicht beliebig gesteigert werden kann.
Die immer weiter verbesserten Therapiemöglichkeiten von HIV-Infektion/AIDS haben positive gesundheitliche und somit auch wirtschaftliche Auswirkungen gezeitigt. So sank während der vergangenen Jahre die Zahl der Klinikaufenthalte Erkrankter auf etwa ein Drittel, und die Häufigkeit der Mutter/Kind-Übertragungen verringerte sich von etwa 15% auf 1%.
Bei einigen Krankheitsgruppen besteht vor dem Hintergrund der demographischen Entwicklung besonderer Innovationsbedarf. Demenzerkrankungen und Depressionen beispielsweise verursachen in Deutschland jährliche Krankheitskosten von etwa 10 Mrd. Euro; von den für ihre Behandlung in den vergangenen fünf Jahren neu auf den Markt gekommenen Wirkstoffen wurde nur ein einziger als innovativ bewertet [7]. Die indirekten Kosten der psychischen Störungen überwiegen deutlich die direkten Kosten, von denen wiederum nur etwa 4% auf die medikamentöse Therapie entfallen [22].
Zahlen dieser Art verdeutlichen, dass die Entwicklung von Arzneimitteln mit therapeutischem Zusatznutzen trotz deren höherer Preise zu sehr erheblichen Einspareffekten im Gesundheitswesen führen könnte.
Verbreitung und Akzeptanz neuartiger Arzneimittel
Arzneimittel, die innerhalb der letzten fünf Jahre in Deutschland auf den Markt gekommen sind und neuartige Wirkstoffe enthalten, haben einen Anteil am Gesamtmarkt von etwa 8,5%, gemessen an der Zahl der Verordnungen (Stand 2004; Abb. 2). Im europäischen Vergleich ist dies ein eher niedriger Wert, der in den USA und den meisten EU-Staaten, insbesondere in Spanien (22,5%), Irland (21,0%), Belgien (18,5%) und Frankreich (18,3%), mehr oder weniger deutlich übertroffen wird [19]. Der Umsatzanteil dieser neuartigen Arzneimittel in Deutschland liegt allerdings deutlich höher, da es sich grundsätzlich um patentgeschützte, nicht den Festbetragsregelungen der GKV unterworfene Produkte handelt. Patentgeschützte, rezeptpflichtige Arzneimittel trugen aufgrund ihres in der Regel hohen Preises erheblich zum Anstieg der Arzneimittelkosten bei, der in den vergangenen Jahren außer 2004 kontinuierlich zu verzeichnen war. Dieser Preisfaktor ist Bestandteil der so genannten „Strukturkomponente“, zu der außerdem Veränderungen der Verordnung von Packungsgrößen, Darreichungsformen und Stärken identischer Arzneimittel beitragen. Dieser Kostenanstieg betrifft Arzneimittel mit therapeutischem Zusatznutzen ebenso wie Analogpräparate und spiegelt die ärztliche Verordnungspraxis wider, die wiederum von den Vertriebsstrategien der pharmazeutischen Industrie nicht unwesentlich beeinflusst werden dürfte.
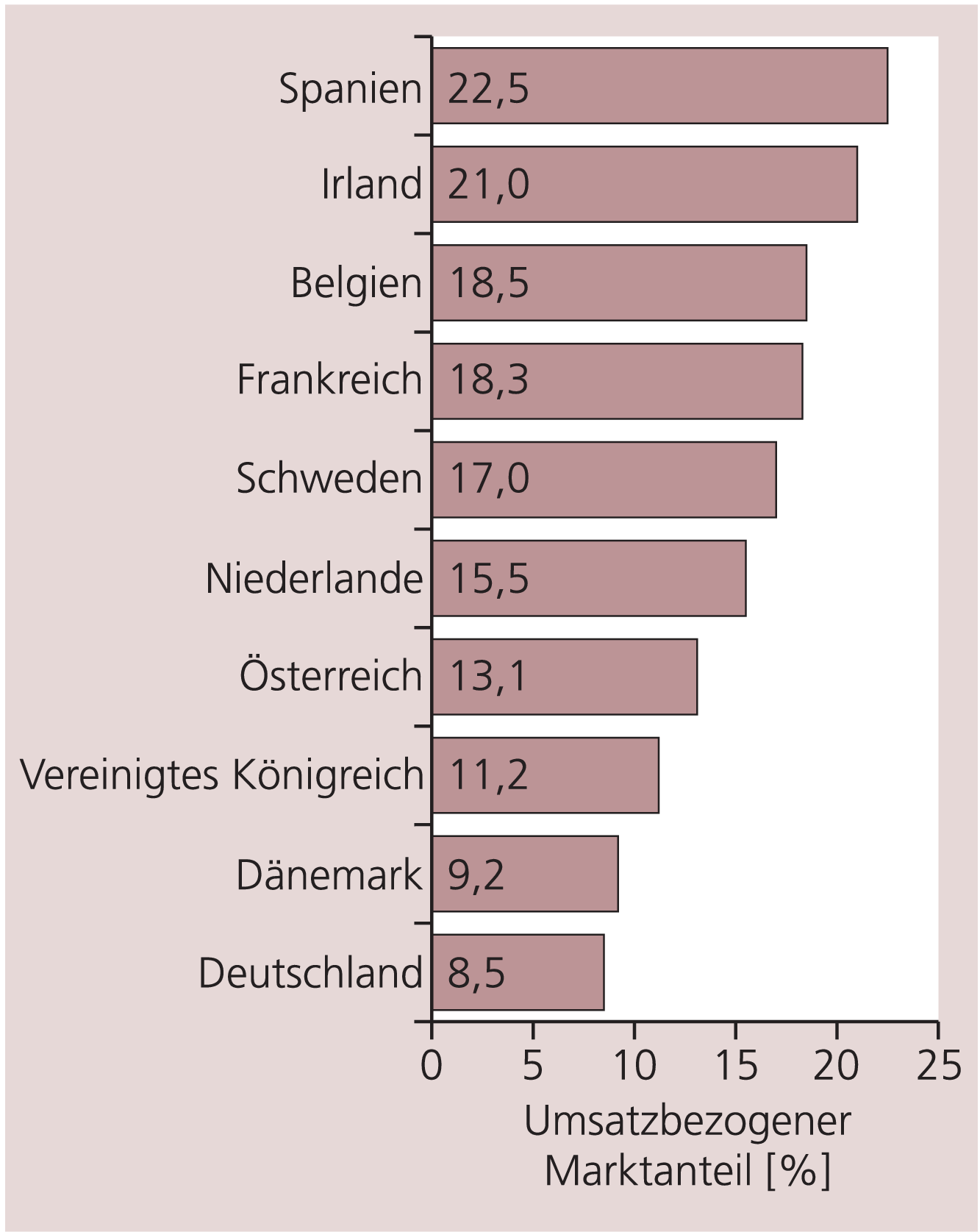
Abb. 2. Vergleichende Übersicht über den umsatzbezogenen Marktanteil neu (d.h. in den letzten fünf Jahren) eingeführter Arzneimittelwirkstoffe in verschiedenen Mitgliedstaaten der Europäischen Union (Stand 2004) [Quelle: VFA]
Das nachvollziehbare Interesse der Arzneimittelindustrie, die Amortisierung ihrer Ausgaben für Forschung und Entwicklung durch geeignete Marketingmaßnahmen zu beschleunigen, wird häufig kritisiert, kommt allerdings dem in der Bevölkerung überwiegenden Interesse an einer umgehenden therapeutischen Verfügbarkeit neuer Arzneimittel entgegen. Damit einher geht die Bereitschaft, einen höheren Preis zu akzeptieren, wenn mit dem betreffenden Arzneimittel entsprechende therapeutische Vorteile erzielt werden. Problematisch in diesem Zusammenhang ist jedoch, dass verlässliche Informationen in Form vergleichender und vor allem wissenschaftlich fundierter Wirksamkeitsbewertungen den Behandlern ebenso wie den Patienten nur in geringem Umfang zur Verfügung stehen.
Noch weniger können Arzt und Patient auf umfassendere Betrachtungen zurückgreifen, die über den Behandlungserfolg hinaus die Häufigkeit und Schwere unerwünschter Arzneimittelwirkungen eines neuartigen Arzneimittels einbeziehen. Denn fundierte Aussagen dieser Art setzen breite Anwendungserfahrungen voraus, die bei einem neuartigen Arzneimittel ebenso wenig vorliegen können wie Erkenntnisse über eventuelle, nachteilige Langzeitwirkungen. Der Arzt und der von diesem möglichst vollständig informierte Patient werden somit in jedem einzelnen Verordnungsfall entscheiden müssen, ob dem Gesichtspunkt der erhofften besseren Wirkung eines neuen Arzneimittels oder dem des besser bekannten Risikoprofils eines schon länger bekannten Produkts der Vorzug zu geben ist.
Neben diesen rein medizinischen Gesichtspunkten wird bei der Wahl des Arzneimittels auch weiterhin das Gebot der Wirtschaftlichkeit der Versorgung zu berücksichtigen sein. Mit dem GKV-Wettbewerbsstärkungsgesetz vom 26. März 2007 wurde der Auftrag des IQWiG nach §35b Abs.1 SGB V zu einer Kosten-Nutzen-Bewertung erweitert. Bewertungen dieser Art können in großem Umfang zu einer rationalen Therapie und zur Ressourcenschonung im Gesundheitswesen beitragen, wobei darauf zu achten sein wird, dass sie alle wesentlichen Kosten- und Nutzenfaktoren umfassen und international akzeptierte Forschungsstandards berücksichtigen.
Der vergleichsweise geringe Informationsstand führt auch dazu, dass bestimmte, verbreitete Wertungen weiterhin eher gefühlsmäßig als rational getroffen werden. So herrscht in Teilen der Bevölkerung eine deutliche Skepsis gegenüber bestimmten innovativen Behandlungsverfahren und Arzneimitteln, vor allem solchen, die mit gentechnischen Verfahren hergestellt worden sind oder gentechnisch veränderte Wirkstoffe enthalten. Der Umsatzanteil solcher Produkte – vor allem Insuline, Immunmodulatoren, Blutwachstumsfaktoren und Impfstoffe – am Markt rezeptpflichtiger Arzneimittel ist dennoch in den vergangenen Jahren kontinuierlich auf inzwischen etwa 10% gestiegen.
Die Unsicherheit gegenüber Ansätzen auf der Grundlage neuartiger Technologien geht nicht selten einher mit einem besonderen Vertrauen in naturheilkundliche, vor allem pflanzliche Produkte. Fast alle dieser Produkte bieten in der Tat den Vorteil sehr langjähriger Anwendungserfahrungen und insofern hinreichender Erkenntnisse über Wirksamkeit und Unbedenklichkeit. Jedoch mehren sich auch bei Phytotherapeutika, beispielsweise den in der Therapie weit verbreiteten Johanniskrautextrakten, Erkenntnisse über unerwartete Neben- und Wechselwirkungen, so dass auch in diesem Bereich ein möglichst hoher und aktueller Informationsstand bei behandelnden Ärzten und Patienten anzustreben ist.
Arzneimittelinformation
Ob neu entwickelte Behandlungsmethoden und Arzneimittel im Bewusstsein der Öffentlichkeit eher als Chance oder als Risiko wahrgenommen werden, wird neben dem jeweiligen individuellen Interesse sehr weitgehend davon abhängen, in welcher Weise Ärzte und andere Fachöffentlichkeit sowie Patienten über die Neuentwicklungen informiert werden. Der Informationsbedarf und die entsprechende Nachfrage seitens breiter Teile der Öffentlichkeit wächst, was grundsätzlich zu begrüßen ist. Die Bedeutung einer alle wesentlichen Aspekte umfassenden, verständlichen und objektiven Patienteninformation rückt zunehmend in das Blickfeld der Politik und hat sich auch auf die europäische Arzneimittelgesetzgebung ausgewirkt. Die Europäische Kommission hat dazu eine Arbeitsgruppe Patienteninformation im Rahmen des von ihr im vergangenen Jahr eingerichteten Pharmazeutischen Forums gegründet (Näheres unter http://europa.eu.int/comm/enterprise/phabiocom/comp_pip_commit_pf.htm).
Internetwerbung
Die intensivierten nationalen und europäischen Anstrengungen müssen darauf gerichtet sein, dass der Öffentlichkeit in den verschiedenen Medien verständliche, umfassende und vor allen Dingen zutreffende Informationen zur Verfügung gestellt werden. Vor allem bei der über das Internet verbreiteten Werbung, die eine wachsende Zahl von Nutzern erreicht, sind diese Kriterien nicht immer eingehalten und auch kaum durchsetzbar, da ihr Ursprung überwiegend außerhalb des Geltungsbereichs der stringenten Regelungen des europäischen und nationalen Heilmittelwerberechts liegt. Diese lassen eine Werbung für verschreibungspflichtige Arzneimittel außerhalb der Fachkreise nicht zu; das deutsche Heilmittelwerbegesetz enthält weitere Einschränkungen, vor allem das Verbot der Werbung für Arzneimittel, die gegen bestimmte schwerwiegende, im Anhang zum Gesetz gelistete Erkrankungen gerichtet sind.
Diese Sachverhalte betreffen auch ein hierzulande relativ neues Marketinginstrument auf dem Arzneimittelsektor, die so genannte Direct-to-Consumer-(DTC-)Werbung. Soweit sich diese auf verschreibungspflichtige Arzneimittel bezieht, stehen ihr innerhalb der Europäischen Union die genannten rechtlichen Regelungen entgegen. Die DTC-Werbung tritt an mit dem Anspruch, Informationsstand und Urteilsfähigkeit des Patienten zu stärken („patient empowerment“). Ihre Aussagen sind jedoch zumindest teilweise so gestaltet, dass der Anspruch eher vordergründig erscheint, wie am Beispiel der so genannten „Schulung zur Symptomerkennung“ besonders deutlich wird. Dieser Ansatz ist schon deshalb fragwürdig, weil er die Möglichkeiten einer sinnvollen Eigendiagnostik ohne Zuhilfenahme ärztlichen Rates überbewertet; dabei werden Symptome definiert und isoliert betrachtet, deren Aussagekraft nur im Kontext einer Gesamtsymptomatik beurteilt werden kann. Als Folge ist unter anderem zu erwarten, dass der so „informierte“ Interessent auf unspezifische, vorübergehende und zum Teil banale Einzelsymptome anders reagiert als zuvor und eine unnötig erhöhte Nachfrage nach Medikamenten generiert wird, die Abhilfe für diese Symptome versprechen.
Ergebnisse klinischer Prüfungen
Wesentliche Informationen für Fachkreise und Patienten, vor allem diejenigen, die an chronischen, nicht befriedigend behandelbaren Krankheiten leiden, können in den Ergebnissen klinischer Prüfungen liegen. Auch hier muss der Interessent sich auf die Seriosität der Informationsquellen verlassen können und sollte umfassend nicht nur über erzielte therapeutische Fortschritte, sondern auch über die gegebenenfalls aufgetretenen unerwünschten Arzneimittelwirkungen in Kenntnis gesetzt werden, ebenso über Abbrüche von klinischen Prüfungen unter Nennung der betreffenden Arzneimittel beziehungsweise Wirkstoffe. Erkenntnisse dieser Art erfordern eine klare Darstellung bereits in der wissenschaftlichen Publikation der Studienergebnisse.
Voraussetzung für eine lückenlose und inhaltlich zutreffende Information ist eine vollständige Erfassung und Registrierung sämtlicher Studien, einschließlich des Studienziels, des Teilnehmerkreises, aufgetretener unerwünschter Arzneimittelwirkungen sowie der erzielten Ergebnisse. Die auf der Grundlage von Artikel 11 der Richtlinie 2001/20/EG [14] eingerichtete europäische Datenbank enthält diese Informationen bis auf die Studienergebnisse, ist jedoch nur der Europäischen Kommission, der Europäischen Arzneimittelagentur und den Zulassungsbehörden der Mitgliedstaaten zugänglich. Zukünftig werden aufgrund der europäischen Kinderarzneimittelverordnung, deren Inkrafttreten bevorsteht, auch Studienergebnisse in diese Datenbank gemeldet, soweit die Studien bei Kindern durchgeführt wurden. In Deutschland verpflichtet darüber hinaus §13 Abs.9 der GCP-Verordnung [3] den Sponsor einer klinischen Prüfung, die wesentlichen Ergebnisse innerhalb eines Jahrs der zuständigen Bundesoberbehörde und Ethik-Kommission mitzuteilen.
Teile der Politik, auch der Wissenschaftsrat und ausgewiesene Fachinstitutionen wie die Arzneimittelkommission der deutschen Ärzteschaft und das IQWiG fordern den Ausbau öffentlich zugänglicher Informationen über klinische Forschungen. Die pharmazeutische Industrie reagiert zunehmend auf diesen Informationsbedarf. Die Pharmaverbände Europas, Japans und der USA haben gemeinsam mit dem Internationalen Pharmaverband IFPMA im Januar 2005 beschlossen, dass die Resultate sämtlicher von forschenden Arzneimittelherstellern gemeinsam mit Kliniken oder Arztpraxen durchgeführten Studien in öffentlich zugängliche Internet-Datenbanken gestellt werden sollen. Unter Hinweis auf den Schutz von Betriebsgeheimnissen bestehen jedoch nach wie vor Hindernisse, Erkenntnisse über klinische Prüfungen von solchen Arzneimitteln bekannt zu geben, die noch keine behördliche Zulassung erhalten haben. Dies betrifft somit auch diejenigen Prüfungen, die zu keinem befriedigenden Ergebnis geführt haben oder aus Sicherheitserwägungen abgebrochen werden mussten. Die Hersteller veröffentlichen jedoch im Internet Informationen zu laufenden Arzneimittelstudien mit wesentlichen Angaben zum Studienkonzept und den Orten der Durchführung (www.vfa.de/de/patienten/artikelpa/studienregister.html).
In diesem Zusammenhang soll nicht unerwähnt bleiben, dass der VFA und das IQWiG einen Mustervertrag geschlossen haben, der die Weitergabe der Ergebnisse unveröffentlichter Studien von Pharmafirmen an das Institut vorsieht und dabei den vertraulichen Umgang mit den Herstellerdaten regelt (www.iqwig.de/index.28.html?article[page]=3). Die Entscheidungen für die Nutzenbewertungen des IQWiG werden dadurch auf eine breitere Grundlage gestellt, so dass die betreffenden Ergebnisse mittelbar auch den Fachkreisen und der interessierten Öffentlichkeit zugute kommen.
Insgesamt ist in den vergangenen Jahren in der europäischen Rechtsetzung ein Trend zu mehr Transparenz und stärkeren Patientenrechten zu verzeichnen, der hinsichtlich der Ergebnisse klinischer Studien weitere Öffnungen erwarten lässt. Auch die Bundesregierung prüft vor diesem Hintergrund, ob und mit welchen Inhalten eine für die Fachkreise und Patienten offene und aussagekräftige Datenbank über klinische Studien in deutscher Sprache eingerichtet werden kann.
Innovation und Arzneimittelrisiko
Ungeachtet der Ergebnisse klinischer Prüfungen auch im Hinblick auf mögliche Anwendungsrisiken und weiterer relevanter Unterlagen, die der zuständigen Bundesoberbehörde im Rahmen eines Zulassungsantrags übermittelt werden müssen, kann die Frage nach der Sicherheit eines neuartigen Arzneimittels zum Zeitpunkt der behördlichen Zulassung nur mit erheblichen Einschränkungen beantwortet werden. Klinische Prüfungen werden bei einer begrenzten Zahl von Probanden beziehungsweise Patienten und über Zeiträume von meist nicht mehr als ein bis zwei Jahren durchgeführt, ferner in der Regel nur an einer nach bestimmten Kriterien vorselektierten Patientengruppe. Neben- oder Wechselwirkungen, die sehr selten sind, die erst nach langjähriger Anwendung des Arzneimittels oder nur bei bestimmten, beispielsweise genetisch entsprechend disponierten Patientengruppen auftreten, können daher im Rahmen einer üblichen klinischen Prüfung nicht erkannt werden. Studien mit wesentlich mehr Patienten und über deutlich längere Zeiträume wären jedoch kaum noch finanzierbar und würden den Zeitpunkt der breiten praktischen Anwendung des betreffenden Arzneimittels deutlich hinausschieben, was gerade bei Arzneimitteln mit therapeutischem Zusatznutzen nicht erwünscht ist.
Wegen dieser systembedingten Unsicherheit kann jedoch nicht auf die Einführung und Markterprobung neuer Arzneimittel verzichtet werden. Wenn diese hinreichenden Anlass geben zu der Erwartung, dass sie bestehende Behandlungsmöglichkeiten merkbar verbessern oder gar bestimmte Krankheiten überhaupt erst medikamentös therapierbar machen, ist auch aus Sicht betroffener Patientenkreise der therapeutische Einsatz gerechtfertigt. Das Risiko, dass sich unerwünschte Wirkungen erst zu einem Zeitpunkt nach der Zulassung herausstellen, ist weithin akzeptiert und muss auch eingegangen werden, damit die Erkenntnisse über Wirkungen und Nebenwirkungen mittel- bis langfristig eine gefestigte Grundlage erhalten.
Wichtigstes Instrument der Risikobeobachtung und -bewertung nach der Zulassung ist eine unabhängige, auf dem Stand von Wissenschaft und Technik arbeitende Pharmakovigilanz. Das Bundesministerium für Gesundheit beabsichtigt, den Wirkungsgrad der Pharmakovigilanz durch Ausbau des Netzes von Pharmakovigilanzzentren, Einrichtung pharmakoepidemiologischer Datenbanken sowie bessere Nutzung der Informationstechnologie weiter zu erhöhen. Die Einbeziehung fachkundiger Stellen im Inland, wie der Arzneimittelkommission der deutschen Ärzteschaft, ist unverzichtbar, ferner kann und sollte die internationale Zusammenarbeit, wie sie beispielsweise in Form behördlicher Fachgremien, Datenbanken über klinische Prüfungen (EudraCT) und Pharmakovigilanzereignisse (EudraVigilance), ferner anhand regelmäßiger Konsultationen zwischen der europäischen Arzneimittelagentur und der amerikanischen Food and Drug Administration besteht, weiter ausgebaut werden.
Nutzen-Risiko-Bewertung in besonderen Einzelfällen
Der gesamtgesellschaftliche Konsens mit dem Ziel, Kranken stets die medizinisch beste Behandlungsmöglichkeit verfügbar zu machen, ermöglicht in bestimmten Fällen die Anwendung von Arzneimitteln auch dann, wenn der Erkenntnisstand über ihre Wirkungen und Nebenwirkungen noch nicht in allen Teilen den Anforderungen an eine behördliche Zulassung entspricht. Die für die Zulassung zuständige Bundesoberbehörde kann beispielsweise die Zulassung mit bestimmten Auflagen erteilen, die zu einem späteren Zeitpunkt erfüllt werden müssen. Auch kann in Ausnahmefällen die Anwendung nicht zugelassener Arzneimittel bei solchen Patienten in Frage kommen, bei denen nach ärztlicher Erkenntnis die Heilungschancen durch andere Arzneimittel nicht oder nicht mehr verbessert werden („compassionate use“). Um dazu einheitliche Verfahrensregelungen zu schaffen, bereitet das Bundesministerium für Gesundheit eine Rechtsverordnung auf der Grundlage von §80 in Verbindung mit §21 Abs.2 Nr.6 AMG vor.
Ähnliche Erwägungen gelten, wenn Arzneimittel zwar zugelassen sind, jedoch außerhalb der zugelassenen Anwendungsgebiete eingesetzt werden („off-label use“). Die Off-Label-Verordnung eines Arzneimittels kann vor allem dann in Frage kommen, wenn sie einen größeren Behandlungserfolg erwarten lässt als zugelassene Alternativen. Die damit verbundene Unsicherheit über zu erwartende unerwünschte Wirkungen wird durch den Nutzen, der für den Betroffenen aus der Behandlung erwartet wird, zumindest kompensiert. Die praktische Anwendung stößt allerdings jenseits der therapeutischen Aspekte auf Schwierigkeiten wegen unklarer Haftungs- und Erstattungsfragen. Da es wegen der Vielfalt der einzelnen Behandlungsfälle aussichtslos ist, umfassende rechtliche Regelungen zum Off-Label-Use zu schaffen, bleibt zur Klärung häufig nur der Rechtsweg, wenn auch in den vergangenen Jahren einige höchstrichterliche Urteile ergangen sind, die einen plausiblen Rahmen gesetzt haben.
Das Bundessozialgericht hat in einer Entscheidung vom 19. März 2002 einschränkend entschieden, dass die Verordnung eines Medikaments zu Lasten der Gesetzlichen Krankenversicherung in einem von der Zulassung nicht umfassten Anwendungsgebiet nur dann in Betracht kommt, wenn dies der Behandlung einer schwerwiegenden Erkrankung dient, für die keine andere Therapie verfügbar ist, und wenn aufgrund der Datenlage die begründete Aussicht besteht, dass mit dem betreffenden Präparat ein Behandlungserfolg (kurativ oder palliativ) erzielt werden kann (AZ: B1 KR 37/00 R). Relativiert wurde dieses Urteil wiederum durch den Beschluss des Bundesverfassungsgerichts vom 6. Dezember 2005, in dem festgestellt wird, dass es mit den Grundrechten auf freie Entfaltung der Persönlichkeit sowie auf Leben und körperliche Unversehrtheit nicht vereinbar ist, einen gesetzlich Krankenversicherten, für dessen lebensbedrohliche oder regelmäßig tödliche Erkrankung eine allgemein anerkannte, dem medizinischem Standard entsprechende Behandlung nicht zur Verfügung steht, von der Leistung einer von ihm gewählten, ärztlich angewandten Behandlungsmethode auszuschließen, wenn eine nicht ganz entfernt liegende Aussicht auf Heilung oder auf eine spürbare positive Einwirkung auf den Krankheitsverlauf besteht (AZ: 1 BvR 347/98).
Maßgeblich für den Behandlungserfolg wie für die Vermeidung unerwünschter Arzneimittelwirkungen bleibt auch in diesen Fällen die ärztliche Erfahrung und fachliche Kompetenz. Gleichwohl bedarf es eines geeigneten Rahmens für solche Entscheidungen, damit Haftungs- und Erstattungsfragen einigermaßen berechenbar bleiben. Das Bundesministerium für Gesundheit hat daher zur Bewertung des Off-Label-Use Regelungen in §35b SGB V getroffen und Expertengruppen gegründet, die fachliche Bewertungen konkreter Arzneimittel erarbeiten und an den Gemeinsamen Bundesausschuss weiterleiten. Im Hinblick auf Arzneimittel, die ohne Prüfung entsprechender Eignung bei Kindern und Jugendlichen angewendet werden, wurde Ende 2006 eine Expertenkommission auf der Grundlage von §25 Abs.7 AMG eingerichtet. Angesichts der Vielzahl der zu bewertenden Produkte werden diese fachkundigen Gremien allerdings Prioritäten setzen müssen und in absehbarer Zeit nur die wichtigsten aktuellen Fragestellungen bearbeiten können.
Ökonomische Risiken
Arzneimittelrisiken, die erst aufgrund umfangreicherer Anwendungserfahrungen bekannt werden, haben unter Umständen neben medizinischen sehr erhebliche wirtschaftliche Auswirkungen. Ein Beispiel dafür bietet die vor allem in Deutschland über viele Jahre propagierte Hormonersatztherapie, die insbesondere der Vorbeugung der Osteoporose bei Frauen nach der Menopause dienen sollte. Nur nach und nach hat sich aufgrund der Erkenntnisse aus mehreren groß angelegten internationalen Studien die Erkenntnis durchgesetzt, dass die mit der Therapie verbundenen erhöhten Risiken von Brustkrebs und Herz-Kreislauf-Erkrankungen die Nutzen-Risiko-Bewertung gegenüber den früheren Annahmen deutlich ins Negative verschieben. Dies gilt analog für die durch unerwünschte Wirkungen entstehenden Folgekosten für die gesetzlichen und privaten Krankenversicherungen, die letztlich die Gesamtheit der Versicherten treffen.
Die jüngste Vergangenheit hat gezeigt, dass sich in unerwarteter Häufigkeit auftretende Arzneimittelrisiken massiv auf einzelne Produkte und das herstellende pharmazeutische Unternehmen auswirken können. Der 1997 behördlich zugelassene Cholesterol- und Lipidsenker Lipobay® (Wirkstoff Cerivastatin) hatte bereits im Jahre 2000 „block buster“-Status, das heißt einen Umsatz in der Größenordnung von 1 Mrd. Euro erreicht. Ein Jahr später musste er aufgrund des Bekanntwerdens schwerwiegender Muskeldegenerationen und einer Reihe von Todesfällen im Zusammenhang mit der Einnahme vom Markt genommen werden. Daran hat auch die Tatsache, dass in der weit überwiegenden Zahl der Fälle Behandlungsfehler in der Form ungeeigneter Kombinationsbehandlungen ursächlich oder zumindest mit ursächlich gewesen sind, nichts ändern können; ist das öffentliche Vertrauen in ein Arzneimittel erst einmal beeinträchtigt, kann es auch durch differenzierte Argumentation, selbst wenn diese zutreffend ist, praktisch nicht mehr zurückgewonnen werden.
Ähnliches gilt für das Schmerz- und Rheumamittel Vioxx® (Wirkstoff Rofecoxib), das schon im ersten Jahr nach der 1999 erteilten Zulassung etwa 1 Mrd. US-Dollar umsetzte, 2003 bereits mehr als 2,5 Mrd. US-Dollar, dann aber ebenfalls aufgrund der in einer Langzeitstudie nach der Zulassung auffällig gewordenen, erhöhten Herzinfarkt- und Schlaganfallrisiken vom Markt genommen wurde. Auch hier sind neben den Todesfällen und schwerwiegenden gesundheitlichen Schädigungen der Betroffenen mittelbare und unmittelbare wirtschaftliche Schäden auch für das verantwortliche pharmazeutische Unternehmen in sehr erheblichem Umfang entstanden.
Schlussbetrachtung
Es steht außer Frage, dass die Entwicklung neuartiger Arzneimittel mit dem Potenzial der Verbesserung bestehender therapeutischer Möglichkeiten im gemeinsamen Interesse von Patienten, Fachkreisen, Industrie und Behörden liegt und jede dieser Gruppen im Rahmen ihrer eigenen Möglichkeiten dazu beitragen muss, solche Entwicklungen zu verwirklichen. Das Risiko, dass neu entwickelte Arzneimittel nicht nur die erwünschten, sondern auch unter Umständen erhebliche unerwünschte Wirkungen zeigen, kann nie vollständig ausgeschlossen, sondern nur möglichst weitgehend minimiert werden. Dazu allerdings können die Eigenverantwortung der pharmazeutischen Unternehmer sowie der Sponsoren klinischer Prüfungen und die beratenden, zulassenden und überwachenden Behörden sehr weitgehend beitragen. Umfassende, wahrheitsgetreue Information des Arztes und Patienten, Qualitätssicherung der medizinischen Behandlung und angemessene rechtliche Regelungen des Gesetzgebers können dafür sorgen, dass das Gleichgewicht zwischen den Anforderungen an den medizinischen Fortschritt und den damit verbundenen Risiken eingehalten wird.
Literatur
1. Bertz J, Giersiepen K, Haberland J, Hentschel S, et al. Krebs in Deutschland. Häufigkeiten und Trends. Saarbrücken: Gesellschaft der epidemiologischen Krebsregister e.V., 2006.
2. Bundesministerium für Gesundheit und Soziale Sicherung. 12. Gesetz zur Änderung des Arzneimittelgesetzes. Bundesgesetzblatt 2004;I/41:2031–53.
3. Bundesministerium für Gesundheit und Soziale Sicherung. Verordnung über die Anwendung der Guten Klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Arzneimitteln zur Anwendung beim Menschen. Bundesgesetzblatt 2004;I/42:2081–91.
4. Dormann H, Neubert A, Criegee-Rieck M, Egger T, et al. Readmissions and adverse drug reactions in internal medicine: the economic impact. J Int Med 2004;255:653–63.
5. Ebbesen J, Buajordet I, Erikssen J, Brors O, et al. Drug-related deaths in a department of internal medicine. Arch Intern Med 2001;161:2317–23.
6. Forschungsförderung. Auskünfte zur Antragstellung erteilen für das BMBF: Projektträger im DLR, Gesundheitsforschung, Heinrich-Konen-Str. 1, 53227 Bonn; für die DFG: DFG-Geschäftsstelle Gruppe Lebenswissenschaften 1, Kennedyallee 40, 53175 Bonn.
7. Fricke U, Schwabe U. Neue Arzneimittel. In: Schwabe U, Paffrath D (editors). Arzneiverordnungsreport 2001. Berlin, Heidelberg: Springer, 2001:23–71.
8. Fricke U, Schwabe U. Neue Arzneimittel. In: Schwabe U, Paffrath D (editors). Arzneiverordnungsreport 2005. Heidelberg: Springer Medizin, 2006:37–107.
9. Gothe H, Höer A, Hagenmeyer EG, Häussler B. Magen- und Zwölffingerdarmgeschwür. Die Bedeutung von innovativen Arzneimitteln für die Gesundheit der Bevölkerung in Deutschland. Berlin: IGES, 2002:67–90.
10. Icks A, Rathmann W, Rosenbauer J, Giani G. Diabetes mellitus. In: Gesundheitsberichterstattung des Bundes, Heft 24. Berlin: Robert-Koch-Institut, 2005.
11. Institut für Gesundheits- und Sozialforschung (Häussler B, Albrecht M), Cassel D, Wille E, Wissenschaftliches Institut der AOK (Schröder H, Nink K, Lankers C). Steuerung der Arzneimittelausgaben und Stärkung des Forschungsstandortes für die pharmazeutische Industrie. Berlin: Institut für Gesundheits- und Sozialforschung, 2006.
12. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200–5.
13. Revue prescrire. Insuline humaine pour inhalation (Exubera). Rev Prescrire 2006;26:564–66.
14. Richtlinie des Europäischen Parlaments und des Rates vom 4. April 2001 zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln. Amtsblatt der Europäischen Gemeinschaften 2001;L121:34–44.
15. Schneeweiss S, Hasford J, Göttler M, Hoffmann A, et al. Admissions caused by adverse drug events to internal medicine and emergency departments in hospitals: a longitudinal population-based study. Eur J Clin Pharmacol 2002;58:285–91.
16. Schwabe U. Arzneiverordnungen 2003 im Überblick. In: Schwabe U, Paffrath D (editors). Arzneiverordnungsreport 2004. Berlin, Heidelberg: Springer, 2004:3–36.
17. Schwabe U. Arzneiverordnungen 2004 im Überblick. In: Schwabe U, Paffrath D (editors). Arzneiverordnungsreport 2005. Heidelberg: Springer Medizin, 2006:3–36.
18. Thürmann PA, Schmitt K. Erfassung und Bewertung unerwünschter Arzneimittelwirkungen. Med Klin 1998;93:687–92.
19. VFA. Die Arzneimittelindustrie in Deutschland. Berlin: VFA, 2006.
20. VFA. Forschung für das Leben. Berlin: VFA, 2005.
21. Weyerer S. Altersdemenz. In: Gesundheitsberichterstattung des Bundes, Heft 28. Berlin: Robert-Koch-Institut, 2005.
22. Wittchen HU, Jacobi F. Size and burden of mental disorders in Europe – a critical review and appraisal of 27 studies. Eur Neuropsychopharmacol 2005;15:357–76.
Priv.-Doz. Dr. Walter K. Schwerdtfeger, Bundesministerium für Gesundheit, Am Propsthof 78a, 53121 Bonn
Innovative drugs in the health care system – Progress and risk
The public is highly interested in the development and rapid availability of medicinal products with an added therapeutic value. Doctors as well as patients highly accept such innovations even though these are generally more expensive than alternative drugs, and a consolidated assessment of their usefulness will be available only after years of practical experience. Doctors and patients, however, need comprehensive and reliable information on the safety and efficacy of new drugs. Successful innovations might not only yield therapeutic progress but also help to reduce costs, especially from the treatment of chronic diseases. In the light of these goals, the German Federal Government supports research into and development of innovative drugs as well as appropriate measures to improve the quality of drug information.
Keywords: Innovative drugs, adverse drug reactions, drug information, cost of disease
Psychopharmakotherapie 2007; 14(05)